Article Text

Abstract
Of the many palmoplantar keratoderma (PPK) conditions, only Papillon-Lefèvre syndrome (PLS) and Haim-Munk syndrome (HMS) are associated with premature periodontal destruction. Although both PLS and HMS share the cardinal features of PPK and severe periodontitis, a number of additional findings are reported in HMS including arachnodactyly, acro-osteolysis, atrophic changes of the nails, and a radiographic deformity of the fingers. While PLS cases have been identified throughout the world, HMS has only been described among descendants of a religious isolate originally from Cochin, India. Parental consanguinity is a characteristic of many cases of both conditions. Although autosomal recessive transmission of PLS is evident, a more “complex” autosomal recessive pattern of inheritance with phenotypic influences from a closely linked modifying locus has been hypothesised for HMS. Recently, mutations of the cathepsin C gene have been identified as the underlying genetic defect in PLS. To determine if a cathepsin C mutation is also responsible for HMS, we sequenced the gene in affected and unaffected subjects from the Cochin isolate in which both the PLS and HMS phenotypes appear. Here we report identification of a mutation of cathepsin C (exon 6, 2127A→ G) that changes a highly conserved amino acid in the cathepsin C peptide. This mutation segregates with HMS in four nuclear families. Additionally, the existence of a shared common haplotype for genetic loci flanking the cathepsin C gene suggests that affected subjects descended from the Cochin isolate are homozygous for a mutation inherited “identical by descent” from a common ancestor. This finding supports simple autosomal recessive inheritance for HMS in these families. We also report a mutation of the same exon 6CTSC codon (2126C→T) in a Turkish family with classical PLS. These findings provide evidence that PLS and HMS are allelic variants of cathepsin C gene mutations.
- Papillon-Lefèvre syndrome
- Haim-Munk syndrome
- cathepsin C mutation
- allelic mutations
Statistics from Altmetric.com
In 1965, Haim and Munk1described an unusual syndrome in four sibs of a Jewish religious isolate from Cochin, India. In addition to congenital palmoplantar keratosis and progressive early onset periodontal destruction, other clinical findings shared by these subjects included recurrent pyogenic skin infections, acro-osteolysis, atrophic changes of the nails, arachnodactyly, and a peculiar radiographic deformity of the fingers consisting of tapered, pointed phalangeal ends and a claw-like volar curve (fig 1B). Subsequently, pes planus was reported to be associated with the syndrome.2 This was the first reported association of these clinical findings, and the condition became known as Haim-Munk syndrome, or keratosis palmoplantaris with periodontopathia and onychogryposis (HMS MIM245010).3Although the palmoplantar findings and severe periodontitis were suggestive of Papillon-Lefèvre syndrome (PLS, MIM245000),4 the association of other clinical features, particularly nail deformities and arachnodactyly, argued that HMS was a distinct disorder. In contrast to PLS, the skin manifestations in HMS were reported to be more severe and extensive. In addition to a marked palmoplantar keratosis (fig 1C, D), affected subjects had scaly, erythematous, and circumscribed patches on the elbows, knees, forearms, shins, and dorsum of the hands. While the periodontium in HMS was reported to be less severely affected than in PLS, gingival inflammation and alveolar bone destruction are present and severe (fig1E, F). In a subsequent genetic study of this extended family, Hacham-Zadeh et al 5 concluded that the syndrome might not behave as a simple autosomal recessive trait. Based upon their estimate of the disease allele frequency in this population (0.1), the absence of the condition in other kindred of the Cochin isolate, and an inability to document consanguinity for many of the parents of affected subjects, they hypothesised that a “complex” autosomal recessive inheritance pattern with a closely linked dominant modifier locus may be responsible for the HMS phenotype.

Clinical and radiographic findings in Haim-Munk syndrome. (A) Dermal involvement of fingers in subject 34. (B) Subject 34: radiograph of terminal phalanges of the fingers showing marked thinning increasing towards the distal, tapering pointed ends and a claw-like volar bend. (C) Subject 17: palmar keratosis. (D) Subject 17: plantar keratosis. (E) Subject 17: gingival inflammation. (F) Subject 17: radiograph showing alveolar bone destruction associated with gingival inflammation shown in (D).
PLS and HMS are classified as type IV palmoplantar ectodermal keratodermas.6 The unique presence of severe, early onset periodontitis distinguishes PLS and HMS from other PPKs and raises the question of whether they result from the variable clinical expression of a common gene mutation, are allelic mutations at the same genetic locus, or result from expression of gene mutations at separate loci. Although the initial report of Haim and Munk1proposed that HMS was a distinct entity, Hacham-Zadehet al 5 referred to the disorder as Papillon-Lefèvre syndrome and cited the suggestion of Gorlinet al 7that HMS was a clinical variant of PLS. In his review of published cases of PLS, Haneke8 summarises an extensive list of clinical findings reported in PLS affected subjects, including increased susceptibility to infections, ectopic cranial calcifications, and nail anomalies.9 It is unclear if these additional clinical features are coincidental findings that may be segregating in a particular family or if they are aetiologically related to a syndrome with a very variable clinical expression. Because PLS is an uncommon condition, and generally occurs only in a single generation, it is difficult to determine if these occasional reports of associated clinical findings are aetiologically related to PLS. Additionally, consanguinity is common among parents of PLS cases and, therefore, it may be expected that an increased number of rare recessive conditions may be seen. This is probably the case for the reports of mental retardation associated with PLS.8
Three independent linkage studies have localised a gene of major effect for PLS to chromosome 11q14.10-12 Interestingly, there was no evidence for a common haplotype for the 20 consanguineous families reported. These findings would appear to suggest that the mutation responsible for PLS had arisen independently in these families. This hypothesis has been supported by the subsequent identification of at least four different mutations in the cathepsin C gene in PLS affected subjects.13 Although mutations in only five families have been reported, there does not appear to be a correlation between the four different CTSCmutations reported and the severity or variability of the PLS phenotype. The aim of the current study was to evaluate affected subjects from the Cochin isolate for mutations of the cathepsin C gene to determine if PLS and HMS are genetically related conditions.
Methods
FAMILY MATERIAL AND CLINICAL DIAGNOSIS.
Fifty members of the inbred Jewish isolate originally from Cochin, India were clinically examined and sampled as previously described.14 All subjects provided informed consent as required by Human Subjects protocols according to the Eastman Dental Center, Rochester, NY and Hebrew University-Hadassah, Jerusalem, Israel. Subjects with severe, early onset periodontitis and the clinical appearance of hyperkeratosis on the palmar and plantar surfaces were considered affected. In addition, four members of an inbred Turkish PLS family were clinically examined and sampled. They all provided informed consent as required by Human Subjects protocols according to the University of Istanbul, Istanbul, Turkey and the Wake Forest University School of Medicine, Winston Salem, NC, USA. A diagnosis of PLS was determined in subjects with severe early onset periodontitis as well as the clinical appearance of hyperkeratosis on the palmar and plantar surfaces. DNA was isolated from peripheral blood samples from available family members using standard techniques (Qiamp Blood Kit, Qiagen).
GENOTYPING
Family members were genotyped for polymorphic DNA markers (D11S1887, D11S1780, and D11S1367) that had been previously determined to flank the cathepsin C locus closely.15 These markers were genotyped using standard techniques for polymerase chain reaction (PCR) amplification with radioactively labelled [γ-32P] primers according to methods previously described.16(GenBank accession numbers: full length cDNA of cathepsin C (CTSC) (NM-001814) and full length genomic DNA of CTSC (U79415).)
SEQUENCING AND MUTATION ANALYSIS
The cathepsin C gene from affected and unaffected subjects was amplified and sequenced as previously described.13 Raw sequence data were analysed and consensus sequences and nucleotide/amino acid alignments generated using DNASIS V2.6 for Windows (Hitachi Software Engineering Co Ltd). Mutations were detected by creating nucleotide/amino acid alignments of reported cathepsin C sequence data versus affected (HMS or PLS) patients' sequence data using the Higgins-Sharpe UPGMA.
RESTRICTION ANALYSIS
The Q286R mutation creates anAvaI restriction site at position 2124. After amplification of a 465 bp fragment encompassing the 3′ end of exon 6 using primers: forward 5′-GTATGCTAGAAG CGAGAATCCGTAT-3′ and reverse 5′-CCAA TGCTAAAACTTGTTGAGACC-3′, the PCR products were purified using the Promega PCR kit according to the manufacturer's instructions. Purified products were eluted in 20 μl water. Approximately 5-10 μl of purified product was digested with 5 UAvaI (New England Biolabs) in a total volume of 15 μl for 1.5 hours at 37°C. Following digestion, the products were separated by electrophoresis through an 1.8 % agarose gel. Amplification of the wild type sequence results in a 465 bp product that is not cleaved by AvaI. Amplification of the mutated (2127A→G) sequence results in a 465 bp product that is cleaved by AvaI to yield products of 404 and 61 bp.
Results
COCHIN FAMILIES
Pedigrees of the reported familial relationships for the Cochin descendents are shown in fig 2A. Descendents of the Cochin isolate studied include sibships 2, 3, 4, and 5 in the pedigree originally described by Hacham-Zadeh et al.5

(A) Pedigree of Cochin descendants segregating Haim-Munk syndrome (HMS). Numbered subjects have been analysed for the current study. Shaded symbols=HMS affected patients. Subjects 10 and 11 are second cousins. Numbers inside circles, squares, and diamonds indicate the number of additional offspring not examined in this study. Sibships described in previous reports are indicated and referenced below the pedigrees. The subjects of the original report of Haim and Munk1 are Nos 33, 34, 35, and 36. Half shading indicates carriers based upon DNA sequencing/restriction enzyme analysis. Unshaded numbered subjects represent non-carriers based upon DNA analysis. (B) Pedigree of Turkish family segregating PLS. Numbered subjects were available for study. Half shading indicates carriers based upon DNA analysis. Subject 77 is a non-carrier based upon DNA sequencing.
COCHIN FAMILY GENOTYPING RESULTS
Irrespective of the HMS or PLS phenotype, affected subjects from the Cochin kindred were found to be homozygous for all three polymorphic DNA loci (D11S1887, D11S1780, and D11S1367) flanking the cathepsin C locus. Additionally, these subjects shared a common haplotype for these polymorphic markers. These findings are consistent with inheritance of both maternal and paternal copies of this genetic interval from a common familial ancestor (“identical by descent”).
ANALYSIS OF CATHEPSIN C IN COCHIN KINDRED
The genomic organisation of the cathepsin C gene consists of seven exons. Sequence analysis of exonic, intronic, and the 5′ regulatory regions of the cathepsin C gene showed that HMS affected subjects from the Cochin kindred were homozygous for a mutation in codon 286 of exon 6 (2127A→G) which results in substitution of a conserved glutamine residue at position 286 by an arginine, Q286R (fig 3). This glutamine residue is normally completely conserved in wild type cathepsin C from at least five species (fig 4). This was the only sequence change different from the reported wild type CTSCsequence (U79415). All available parents of affected subjects were found to be heterozygous for the mutated (2127A→G) allele and the wild type allele. None of the parents or sibs heterozygous for the mutated (2127A→G) allele and the wild type allele manifested clinically identifiable characteristics of PPK or had a history of severe, early onset periodontitis.
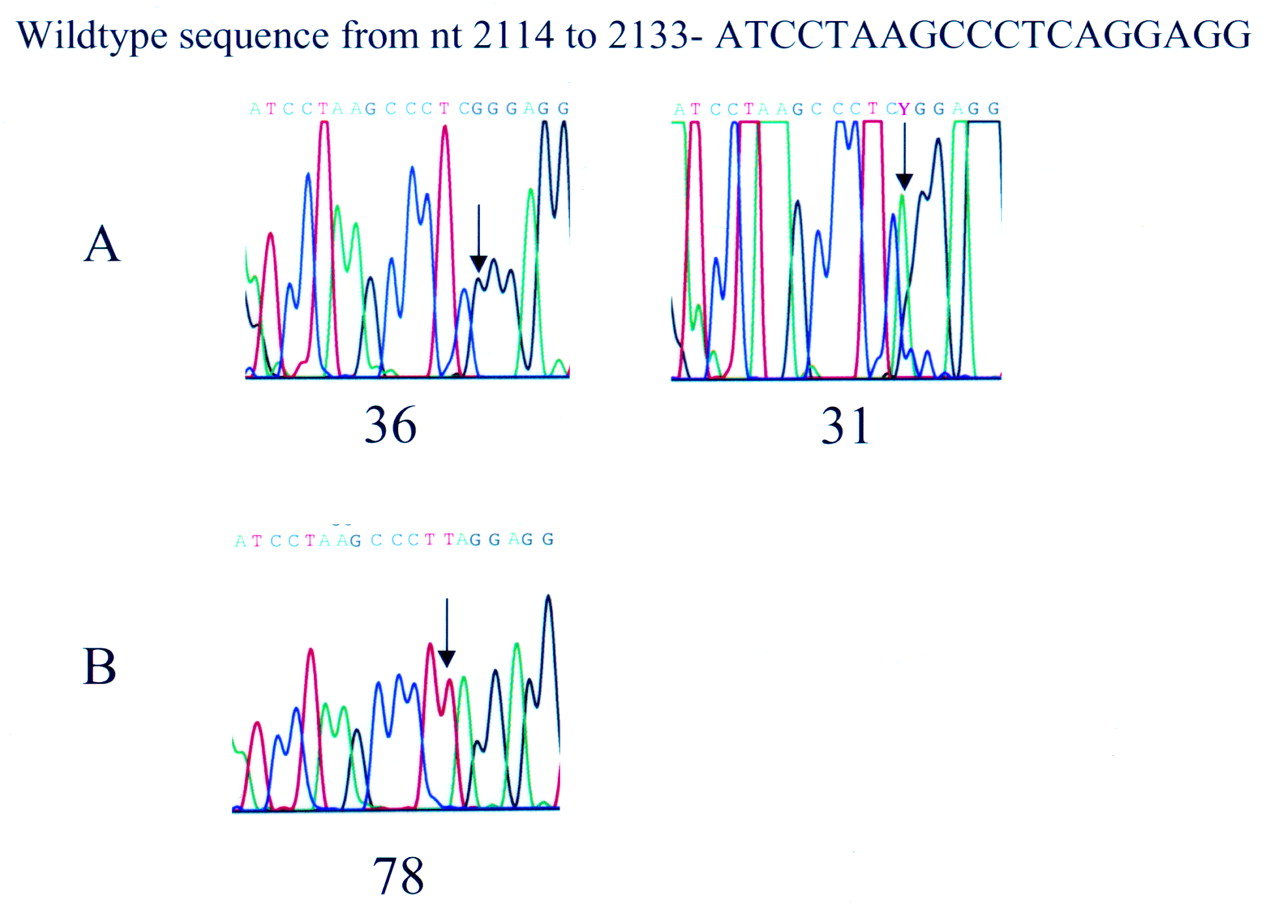
Sequence analysis of exon 6 of CTSC. The numbering of the wild type sequence is based upon the genomic sequence of CTSC (Accession U79415). (A) Family A (Cochin isolate diagnosed with Haim-Munk syndrome) from fig 1. Affected subjects are homozygous for a 2127A→G missense mutation which results in a conserved glutamine being changed to an arginine (Q286R). Representative sequences are shown for subjects 36 (affected) and 31 (carrier). (B) Family B from fig 1. Affected subjects are homozygous for a 2126C→T nonsense mutation which results in a premature stop codon at position 286 (Q286X). The Q286X has been previously reported in an inbred Turkish family.12

Alignment of the deduced amino acid sequence of exons 1−6 of human cathepsin C (Human CATC, GenBank accession NoX87212) with the cathepsin C of rat (Rat CATC, D90404), mouse (Mouse CATC, U74683), dog (Dog CATC, AF060171), Schistosoma japonicum (SchJp CATC, U77932), and S mansoni (SchMa CATC, Z32531). The asterisk indicates the active site cysteine. An open arrow indicates the putative site of the signal peptide cleavage. Note that the signal peptide from dogs was not reported in Genbank. A closed arrow indicates the conserved glutamine that is changed in the HMS kindred reported here.
RESTRICTION ANALYSIS
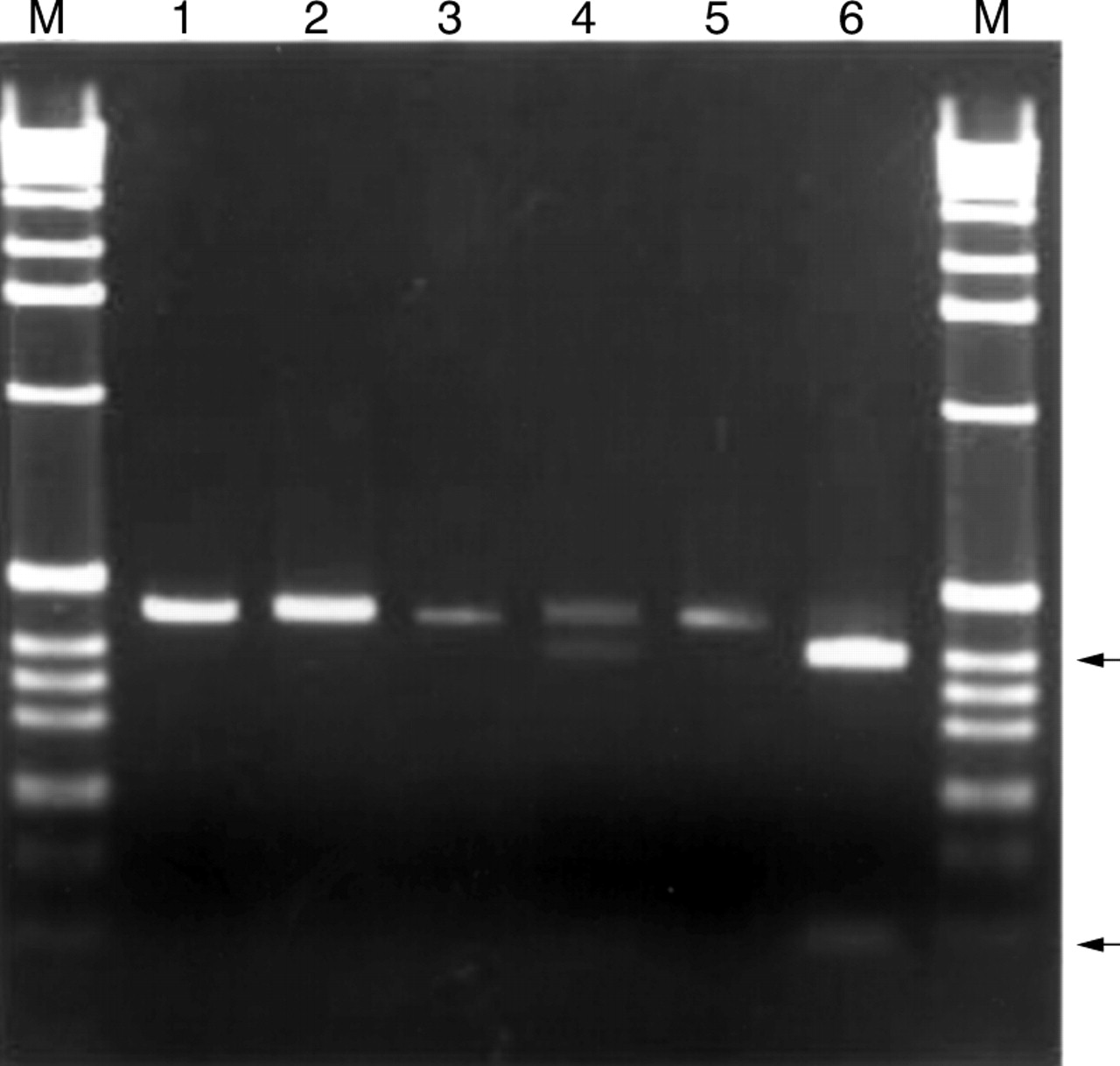
The Q286R mutation creates an AvaI restriction site at nucleotide 2124. We used this newly created restriction site to develop a rapid test to screen for the Q286R mutation. Amplification of nucleotides 2064-2529 using the primers listed under Methods results in a 465 bp fragment encompassing the 3′ end of exon 1. Subjects who are homozygous for the wild type sequence exhibit a 465 bp band. Heterozygotes exhibit three bands, 465, 404, and 61 bp. Homozygotes for the Q286R mutation exhibit bands of 404 and 61 bp. Restriction analysis confirmed the sequencing results of all subjects examined (fig 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Restriction enzyme analysis of Q286R mutation in family A. The 465 bp fragment of exon 6 was amplified and subjected to restriction digestion with AvaI as described under Methods. The Q286R mutation introduces a new AvaI site. After digestion and electrophoresis through 1.8% agarose gels, wild type subjects exhibit bands of 465 bp, affected subjects have bands of 404 and 61 bp, and carriers have bands of 465, 404, and 61 bp. M=1 kb ladder (Gibco). Lane 1, subject 5 uncut, showing 465 bp amplicon. Lane 2, subject 5 cut with AvaI. Only the 465 bp fragment is observed. Thus, subject 5 has the wild type sequence on both alleles. Lane 3, subject 31, uncut. Lane 4, subject 31 cut with AvaI. The 465 and 404 bp fragments are visible, confirming that subject 31 is a carrier of Q286R, consistent with the sequencing results shown in fig 3A. Lane 5, subject 34, uncut. Lane 6, subject 34 cut with AvaI. The 404 and 61 bp fragments are indicated by arrows.
TURKISH FAMILY
The pedigree of the Turkish PLS family is shown in fig 2B. The parents of the PLS affected subjects are consanguineous. Sequence analysis of exonic, intronic, and the 5′ regulatory regions of the cathepsin C gene showed that both PLS affected subjects from the Turkish family were homozygous for a mutation in codon 286 of exon 6 (2126C→T) that introduces a premature stop codon (Q286X). This was the only sequence change different from the reported wild typeCTSC sequence (U79415) found in this family.
Discussion
The PPKs represent a heterogeneous group of diseases that share the clinical characteristic of gross thickening of the palmoplantar skin. Traditional classifications of PPKs based upon histopathological findings, distribution, localisation of lesions within the skin of the palm and sole, and associated symptoms have not been helpful in understanding the molecular pathogenesis of PPKs.17 18 In their classification of the primary palmoplantar keratodermas, Stevens et al 6 proposed four categories for the PPKs, diffuse, focal, punctate, and palmoplantar ectodermal dysplasias.6Within this classification PLS and HMS are both listed as type IV palmoplantar ectodermal dysplasias. Although there are differences between the PLS and HMS phenotypes, the overlapping cardinal features of these conditions suggested they may be caused by allelic mutations of the same gene. Identification of cathepsin C mutations in PLS provided a strategy to test this possibility.13 For the current study, 50 descendants from the Cochin kindred were examined, including subjects initially described by Haim and Munk.1Detailed clinical descriptions of affected subjects from four of the sibships studied here have been previously reported by several investigators,1 19 as indicated in fig 2A. Members of a fifth affected sibship, described by Puliyel and Sridharan Iyer2 were not available for genetic analysis.
The Cochin Jews from India, constitute a well defined and extensively studied isolate that numbered approximately 2000 in 1954.20 In their initial report, Haim and Munk1 reported that parents of the affected subjects came from unrelated families with no consanguinity, and the authors were unable to determine whether the syndrome was transmitted as a dominant or recessive trait.1 In a study of immigrant Jews from Cochin, India, Cohen and Bloch20 reported that marriage is restricted to members of the community in this religious isolate, and estimated the rate of consanguinity at 40%.20 Yet the HMS phenotype has been reported to occur in only nine sibships in this population.2 In a genetic analysis that included five of the affected sibships from the Cochin kindred segregating the syndromic features first described by Haim and Munk,1 Hacham-Zadehet al 5 were able to document parental consanguinity in only one of these sibships. Based in part on this finding, they estimated the disease allele frequency to be 0.1 in the Cochin population. The discrepancy between the estimated high gene frequency for the disease allele in this inbred population (0.1), and the absence of affected subjects in other kindreds of the isolate, led Hacham-Zadeh et al 5 to suggest that the syndrome may not behave as a simple autosomal recessive trait. They hypothesised that that a “complex” autosomal recessive inheritance pattern with a closely linked dominant modifier locus may be responsible for the condition.5 They acknowledged that this was not the only possible explanation but that it was not possible to test other hypotheses. Identification of the genetic basis of PLS and the findings of this present study now permit clarification of the issue. All affected subjects tested from the four affected sibships are homozygous for the mutated (2127A→G) cathepsin C allele. All parents available for testing were found to be heterozygous carriers of one mutated (2127A →G) cathepsin C allele and one wild type cathepsin C allele. We found no instances of subjects homozygous for the wild type cathepsin C allele, or heterozygous carriers of one mutated (2127A→G) cathepsin C allele and one wild type cathepsin C allele showing PPK or severe, early onset periodontitis or both. Additionally, haplotype studies for genetic markers closely flanking the cathepsin C locus suggest that all affected subjects are homozygous for the same cathepsin C mutation, and are also homozygous for genetic markers closely flanking the cathepsin C locus. All parents of affected subjects are carriers of this haplotype and of the cathepsin C mutation. These data provide strong evidence that the parents of affected subjects are in fact consanguineous. As a whole, these findings are consistent with simple autosomal recessive inheritance for the syndrome affecting the Cochin kindred.
The presence of arachnodactyly, acro-osteolysis, pes planus, and nail deformities are the primary clinical findings that differentiate between HMS and PLS. Identification of cathepsin C mutations in both PLS and HMS confirms Gorlin's hypothesis that PLS and HMS are allelic variants, yet the basis for the varied clinical expression of these conditions is unknown. Variable clinical expression and pleiotropic effects of identical and allelic mutations are seen in other conditions such as the craniosynostoses.21 Cathepsin C (dipeptidyl-peptidase I) is a lysosomal cysteine proteinase that is important in intracellular degradation of proteins and appears to be a central coordinator for activation of many serine proteinases in immune inflammatory cells, particularly polymorphonuclear leucocytes and their precursors.22 Cathepsin C is normally expressed in palmar, plantar, and gingival epithelium, tissues affected by PPK and severe early onset periodontitis.13 In addition to being expressed in skin, cathepsin C is present in large amounts in osteoclasts.23 Osteoclasts are multinucleated giant cells that play an important role in bone resorption and hence in bone modelling and remodelling. Osteoclasts are considered to develop from haematopoietic stem cells. The formation of osteoclasts is regulated by many cytokines, which are known to be influenced by severe inflammatory states, as occurs in both PLS and HMS. It is unclear whether the additional clinical features seen in the HMS subjects, particularly the osseous findings, are the result of the specific cathepsin C (2127A→G) mutation present in HMS, or result from the epistatic effect of modifying genes present in the Cochin kindred.
Cathepsin C mutations of both exons 6 and 7 have been reported in PLS.13 In the Turkish family analysed in this report we identified a CTSC mutation (2126C→T) that introduces a premature stop codon (Q286X) in codon 286 of exon 6 in two subjects diagnosed with PLS. This same mutation has been reported in two PLS affected subjects from an unrelated Turkish family. Affected members of these two Turkish families do not share a common haplotype for genetic loci flanking the CTSC gene, suggesting that they have probably inherited independent mutations. The cathepsin C mutation in affected descendants of the Cochin kindred occurs in the same exon 6 codon (codon 286) mutated in the Turkish PLS family. This mutation occurs in the heavy chain region, the most conserved region of cathepsin C. However, in affected subjects of the Cochin kindred, the Q286R mutation results in the substitution of a charged arginine residue for a non-polar glutamine. This glutamine residue is highly conserved among cathepsin C proteins from at least five species, including mouse, rat, blood flukes, dog, and humans (fig4).24 25 It has been postulated that many, if not all, heavy chain residues are necessary for enzyme activity and specificity.25 The Q286R mutation may cause a conformational change, altering enzymatic activity and possibly specificity. This may be aetiologically important for the phenotypic features seen in the affected subjects.
Understanding the genetic basis of simple Mendelian conditions can provide valuable insight into the role of specific genes in complex developmental pathways. Such knowledge has significant implications for understanding both normal and pathological development. Inflammation and destruction of the oral gingiva occur only when teeth are present.8 26 Histologically, teeth are attached to the surrounding epithelium by a developmentally unique epithelium, the junctional epithelium.27 28 Junctional epithelium is not keratinised, is relatively thin, and is much more permeable that adjacent keratinised oral gingiva. Episodes of gingival inflammation and destruction occur in PLS and HMS only when the junctional epithelium is present. When teeth are exfoliated, junctional epithelium no longer exists. It is intriguing to speculate that the junctional epithelium and palmoplantar epithelium may share common developmental characteristics, and hence pathological effects of cathepsin C mutations are manifest in these tissues. Increasingly, genetic factors are proposed to play a significant role in host predisposition to periodontitis. Most existing paradigms focus upon the role of the host immune response as a risk factor for periodontitis. Identification of mutations in cathepsin C associated with severe periodontitis suggests that structural integrity of the epithelium may also play an important role in periodontal disease susceptibility. In PLS and HMS, the presence of intrinsic oral microbes, particularly gram negative species, may be sufficient to cause the severe periodontitis.14 29 The severe, early onset periodontitis that occurs in PLS and HMS is rapidly progressive and unfortunately responds poorly to conventional periodontal treatment strategies.30 Understanding the molecular basis for this dramatically increased propensity for periodontal destruction may provide insight into the genetic basis for periodontal disease susceptibility. The generality of cathepsin C gene mutations to other forms of periodontitis is unknown and should be investigated, particularly the early onset and rapidly progressive forms which appear to have a significant genetic basis.
Acknowledgments
The authors thank the members of families that have graciously participated in these studies and Tracie Stivason for manuscript preparation. These studies were supported by NIDCR grants DE10563 and DE12920. After acceptance of this paper, Toomeset al (Nat Genet)1999;23:421−4) published additionalCTSC mutations in PLS affected subjects.