


Abstract
Formyl peptide receptors (FPRs) are a small group of seven-transmembrane domain, G protein-coupled receptors that are expressed mainly by mammalian phagocytic leukocytes and are known to be important in host defense and inflammation. The three human FPRs (FPR1, FPR2/ALX, and FPR3) share significant sequence homology and are encoded by clustered genes. Collectively, these receptors bind an extraordinarily numerous and structurally diverse group of agonistic ligands, including N-formyl and nonformyl peptides of different composition, that chemoattract and activate phagocytes. N-formyl peptides, which are encoded in nature only by bacterial and mitochondrial genes and result from obligatory initiation of bacterial and mitochondrial protein synthesis with N-formylmethionine, is the only ligand class common to all three human receptors. Surprisingly, the endogenous anti-inflammatory peptide annexin 1 and its N-terminal fragments also bind human FPR1 and FPR2/ALX, and the anti-inflammatory eicosanoid lipoxin A4 is an agonist at FPR2/ALX. In comparison, fewer agonists have been identified for FPR3, the third member in this receptor family. Structural and functional studies of the FPRs have produced important information for understanding the general pharmacological principles governing all leukocyte chemoattractant receptors. This article aims to provide an overview of the discovery and pharmacological characterization of FPRs, to introduce an International Union of Basic and Clinical Pharmacology (IUPHAR)-recommended nomenclature, and to discuss unmet challenges, including the mechanisms used by these receptors to bind diverse ligands and mediate different biological functions.
I. Introduction and Historical Overview
A. Discovery of N-Formyl Peptides as Potent Chemoattractants for Phagocytes
The phenomenon of pus accumulation at sites of infection is part of the human condition and was described in the very earliest writings. Experimental evidence suggested that this might be due at least in part to bacterially derived phagocyte chemoattractants, as investigators injected live bacteria into tissues of experimental animals and observed accumulation of neutrophils at the injection site (for review, see Harris, 1954). In subsequent studies, Ward et al. (1968) found that filtrates from both Gram-positive and Gram-negative bacteria contain chemotactic activity for phagocytes in vitro. Further investigations resulted in the identification of these chemotactic factors as low-molecular-weight peptides with blocked amino termini (Schiffmann et al., 1975b). Because prokaryotes initiate protein synthesis with N-formyl methionine, Schiffmann et al. (1975b) chemically synthesized and tested short peptides starting with N-formyl methionine (CHO-Met). They found potent chemotactic activities for neutrophils in many of the synthetic N-formyl peptides tested, especially peptides containing N-formyl-methionyl-leucine and N-formyl-methionyl-phenylalanine (Schiffmann et al., 1975a). This first report of N-formyl peptides serving as chemoattractants for neutrophils spurred a series of studies by Showell et al. (1976) and Freer et al. (1980, 1982) using rabbit neutrophils on the structure-function relationship of the peptides. These studies resulted in identification of N-formyl-methionyl-leucyl-phenylalanine (fMet-Leu-Phe, or fMLF1)asthe most potent agonist among 24 synthetic peptides tested, with an ED50 of 7.0 ± 1.7 × 10-11 M in neutrophil chemotaxis assays (Showell et al., 1976). The high potency and efficacy of fMLF to induce chemotaxis of human blood neutrophils was subsequently confirmed in independent studies by many other laboratories (Wilkinson, 1979). At higher concentrations (ED50 of 3.2 ± 0.8 × 10-10 M), the same peptide could stimulate lysosomal enzyme release (Freer et al., 1980). Although the formylated Met at position 1 was optimal for its bioactivity, replacing the Met with norleucine only slightly impaired the potency of the peptide in enzyme release assays.
The above findings led Freer et al. (1980) to propose that the N-formyl group is essential for the bioactivity of these chemotactic peptides. Peptides of the same sequence lacking the N-formyl group (e.g., Met-Leu-Phe) display reduced potency by 2 to 5 orders of magnitude, as shown in Table 1. However, replacing the N-formyl group with a t-butyloxycarbonyl group (t-Boc), which provides a urethane linkage instead of a peptide linkage, converted the same peptide to an antagonist (Boc-Met-Leu-Phe, Boc-MLF, or Boc1) (Freer et al., 1980). A number of different sequence combinations were tested in addition to N-formylated and nonformylated peptides of the same sequence. Based on these experiments, a model for formyl peptide binding requirements was proposed at the time. In this model, the formyl group is necessary for both ligand binding to a receptor and biological activity, and the Met side chain of position 1 occupies a hydrophobic pocket in the receptor. The Leu side chain may interact with a hydrophobic area of the receptor, whereas the phenylalanine side chain resides in a hydrophilic area. The Phe carbonyl plays a role for the C=O interaction with the receptor (Freer et al., 1982). As discussed below, this working model was later tested on a molecularly defined and cloned formyl peptide receptor, and modifications to the model were made through several studies. For instance, the N-formyl group is not absolutely required for potency at the receptor, and an N-acetylated peptide, Ac-Met-Nle-Leu-Phe-Phe, was found to be highly potent at the human formyl peptide receptor (FPR1) (Gao et al., 1994). The effects of N-terminal modifications on the affinity and potency of these peptides are shown in Table 1.
N-terminal modifications of selected formyl peptides and effects on agonistic activity Most of the early studies were conducted using neutrophils from rabbits and humans. Note that the concentration of formyl peptides required for superoxide production and enzyme release is 10- to 50-fold higher than the concentration needed for maximal chemotaxis. Therefore, caution should be taken when comparing potency using different assays. Some variations may also exist between assays conducted in different laboratories.
Although synthetic formyl peptides were used in the initial studies, it was believed that these peptides mimicked the behavior of chemotactic peptides released from bacteria during infection (Bennett et al., 1980). In 1984, Marasco et al. reported the isolation of chemotactic peptides from Escherichia coli culture supernatants. Using a lysosomal enzyme release assay and competitive binding with [3H]fMLF, these investigators identified several distinct formyl peptides with bioactivity for rabbit neutrophils. Further characterization of these peptides led to the identification of fMLF as the major chemotactic factor in E. coli culture supernatants. It should be noted that secretion of chemotactic peptides is not limited to E. coli and is probably a general property of bacteria and that other bacterial species have also been experimentally shown to release phagocyte-chemotactic factors. For example, Rot et al. (1987, 1989) showed that culture supernatants of Staphylococcus aureus contain several peptides that are chemotactic for human monocytes. Two of these peptides, fMet-Ile-Phe-Leu (fMIFL) and fMet-Leu-Phe-Ile (fMLFI), displayed potent activities in chemotaxis and superoxide generation assays (Rot et al., 1987). More recently, Rabiet et al. (2005) reported that several peptides derived from Listeria monocytogenes, known to bind to the nonclassic MHC Class Ib molecule M2-H3 (Pamer et al., 1992), are potent chemoattractants for human leukocytes. These results demonstrate that bacteria release N-formyl peptides of various sequence compositions that can be recognized by human and rabbit neutrophils and monocytes, and that which can therefore be collectively considered as a pathogen-associated molecular pattern (PAMP). The PAMP concept has heretofore been mainly associated with Toll-like receptors (Medzhitov and Janeway, 2000). Inhibition of bacterial peptide deformylase (an enzyme that deformylates the methionyl group to generate mature proteins) leads to increased bacterial production of formylated peptides (Fu et al., 2003), suggesting a regulatory mechanism for the generation of formyl peptides. A list of selected formyl peptides derived from bacteria and mitochondria is shown in Table 2.
Binding affinity and potency of bacterial and mitochondrial formyl peptides pEC50 is defined as the negative logarithm of the EC50. HL-60 cells transfected to express FPR1 or FPR2/ALX, and Chinese hamster ovary cells transfected to express FPR1 were used in some studies
Unlike prokaryotes that initiate protein synthesis with an N-formylmethionine, the synthesis of eukaryotic proteins encoded by nuclear DNA is initiated with a nonformylated methionine. However, mitochondrial protein synthesis is initiated with N-formylmethionine. Carp (1982) reported that disrupted mitochondria from several types of cells that were tested could stimulate human neutrophil migration. Chemotaxis was also induced by purified proteins of the mitochondria respiratory chain. These activities were inhibited by t-Boc-Phe-d-Leu-Phe-d-Leu-Phe (Boc2), an antagonist of the fMLF receptor, suggesting that these mitochondrial chemotactic factors share the same receptor with fMLF. More recent studies of peptides of mitochondrial origin (Chiang et al., 2000; Rabiet et al., 2005) support the conclusion made by Carp, indicating that mitochondrial proteins bearing N-formyl modifications are endogenous ligands for formyl peptide receptors (Table 2). Attraction of phagocytes to sites of inflammation and tissue injury may be facilitated by these endogenous ligands.
B. Biochemical Characterization of Receptors That Bind and Internalize Formyl Peptides
The findings that N-formyl peptides could stimulate neutrophil chemotaxis (Schiffmann et al., 1975a; Zigmond, 1977) and lysosomal enzyme release (Showell et al., 1976) provided strong evidence for the presence of a functional receptor on target cells. Formyl peptide receptors were first defined at the biochemical level using radioisotope- and fluorescence-labeled peptide ligands. In 1977, Aswanikumar et al. demonstrated that rabbit neutrophils express specific and saturable binding sites for [3H]fNle-Leu-Leu-Phe, with a Kd of 1.5 × 10-9 M. A similar binding site for fMLF was reported by Williams et al. (1977). In both cases, the bound radiolabeled peptides could be effectively displaced by unlabeled peptides of the same or similar composition, and the concentrations of the peptides that effectively displaced the radiolabeled ligand matched the concentrations used in functional assays. In a series of binding experiments, Niedel et al. (1979a) studied neutrophil uptake of radiolabeled formyl peptides. They found that iodinated fNle-Leu-Phe-Nle-Tyr-Lys retained full biological activity with an EC50 of 0.4 nM in a human neutrophil chemotaxis assay. Reversibility of ligand binding was lost over time and at the appropriate temperature, suggesting receptor-mediated internalization of the formyl peptides. Because unlabeled formyl peptides of similar compositions could inhibit binding and uptake of the iodinated peptide, it was concluded that these peptides interact with the same receptor on neutrophils (Niedel et al., 1979a). Using tetramethylrhodamine-labeled fNle-Leu-Phe-Nle-Tyr-Lys and time-lapse video microscopy, Niedel et al. (1979b) observed rapid internalization and aggregation of the fluorescent peptide, suggesting receptor binding and receptor-mediated uptake of the labeled ligand. In other studies, the relationship between formyl peptide pre-exposure and cell responsiveness was investigated. Vitkauskas et al. (1980) reported that pretreatment of rabbit neutrophils with formyl peptides reduced the available binding sites for [3H]fNle-Leu-Phe as well as cell response to subsequent formyl peptide stimulation, a process that the authors termed deactivation. A study conducted by Donabedian and Gallin (1981) identified preferential and nonpreferential deactivation toward the same and different agonists, respectively. In preferential deactivation, incubation of human neutrophils with fMLF reduced the cell-surface binding sites for the same ligand, resulting in a decrease in chemotaxis toward subsequent fMLF stimulation. In nonpreferential deactivation, treatment of human neutrophils with a high concentration of the activated complement C5 fragment (C5a) caused reduced response of the cells to fMLF stimulation, without reducing (and actually increasing) the cell surface binding sites for fMLF. These published studies were among the earliest reports on G protein-coupled receptor (GPCR)-mediated internalization, although the identity of the formyl peptide receptor at the molecular level was still unknown at the time. In addition, what Donabedian and Gallin called preferential deactivation was in fact an early example of heterologous desensitization (Didsbury et al., 1991) and cross-desensitization of chemoattractant GPCRs (Richardson et al., 1995).
The study by Donabedian and Gallin (1981) also showed that agonist-induced decrease in the number of formyl peptide binding sites was transient, and these binding sites could return to the cell surface if the cells were kept at 37°C. The study demonstrated a recycling pool of formyl peptide receptors. When neutrophils were sonicated and fractionated on sucrose density gradients, fMLF binding sites were found in the fractions containing specific granules (Fletcher and Gallin, 1983). Therefore, neutrophils contain an intracellular pool of cryptic formyl peptide receptors that may be mobilized to the cell surface. Using time-resolved flow cytometry, Sklar and colleagues studied the dynamics of formyl peptide ligand interaction with its receptor in neutrophils (Sklar et al., 1981, 1984; Sklar and Finney, 1982; Finney and Sklar, 1983). These studies took advantage of the ability of cytometric and fluorimetric analyses to distinguish between receptor-bound and unbound ligands in real time to determine different states of the receptor. The results not only confirmed internalization of ligand-occupied receptors but also determined key parameters of formyl peptide association and dissociation, demonstrating that the ligand-receptor complex could undergo an alteration in affinity (Sklar et al., 1984). Jesaitis et al. (1984, 1985) initiated studies of formyl peptide receptor interaction with the cytoskeleton and found that a receptor-cytoskeleton complex was formed before receptor internalization and was resistant to Triton X-100. In this ternary complex, the formyl peptide ligand binds to its receptor with high affinity and then slowly dissociates from the receptor (Jesaitis et al., 1984). These studies demonstrate that the formyl peptide receptor interacts with intracellular proteins such as cytoskeleton proteins and this interaction can affect the binding properties of the receptor.
Early studies using radiolabeled fMLF identified one class of binding sites in intact neutrophils. Using membrane binding assays, Koo et al. (1982) reported that human neutrophils contain two classes of formyl peptide binding sites with dissociation constants of 0.53 and 24 nM, respectively. The heterogeneity of receptor binding to fMLF was not due to negative cooperativity, because the rate of dissociation was unaltered with increasing receptor occupancy. This result could be interpreted as evidence for the presence of two distinct, noninterconvertible populations of binding sites for formyl peptides, one responsible for neutrophil chemotaxis, which requires lower concentrations of formyl peptides, and the other mediating additional bactericidal functions such as lysosomal enzyme release and superoxide generation known to require higher agonist concentrations (Lehmeyer et al., 1979; Korchak et al., 1984). Alternatively, the different dissociation constants could indicate the presence of one class of receptors present in two affinity states that are interconvertible. A subsequent study conducted by the same authors found that a nonhydrolyzable derivative of GTP, when added to the membrane preparation in a binding assay, could convert a part of the high-affinity binding site to a low-affinity site without altering the total number of receptors (Koo et al., 1983). This effect was reverted by removal of the GTP analog. Similar guanine nucleotide regulation of receptor affinity was reported in other studies of receptors that couple to G proteins (Lad et al., 1977; De Lean et al., 1980). Thus, these biochemical studies were interpreted to mean that a single class of receptors for fMLF is present in two binding states, and that their affinities for binding formyl peptides are regulated by GTP in membrane binding assays in which the receptors are accessible to the GTP analog but the receptor-cytoskeleton interaction is disrupted. Under these experimental conditions, the G protein-bound receptors exhibit high affinity for fMLF, and the receptors uncoupled from G proteins display low binding affinity. This interpretation was later complicated by discovery at the molecular level of FPR1 and FPR2/ALX (see section II), binding fMLF with high and low affinity, respectively.
The biological activities of formyl peptides were extensively characterized after their initial discoveries. The prototypic formyl peptide, fMLF, possesses full agonistic activity that is similar to that of C5a in stimulating chemotaxis, lysosomal enzyme release, and superoxide generation (Schiffmann et al., 1975a; Showell et al., 1976; Lehmeyer et al., 1979). The ability of an activated receptor to mediate multiple cellular functions is intriguing, and the phenomenon spurred additional efforts to characterize the formyl peptide receptors at the biochemical level. Several radiolabeled formyl peptide derivatives were prepared for photoaffinity cross-linking to cell surface receptors. Using this approach, it was determined that the neutrophil formyl peptide receptor is a Mr 50,000∼70,000 glycoprotein (Niedel et al., 1980a; Dolmatch and Niedel, 1983; Allen et al., 1986). Treatment with endoglycosidase F reduces the size of the receptor to Mr 32,000, which retains the ability to bind formyl peptides (Malech et al., 1985). These results indicate that the formyl peptide receptor in neutrophils is an N-glycosylated cell surface protein, but ligand binding does not depend on glycosylation of the receptor. Additional efforts in purification of the formyl peptide receptor were met with difficulties. Because of the low abundance of the receptor in neutrophils, only partially purified proteins were obtained.
II. The Expanded Family of Formyl Peptide Receptors
A. Molecular Characterization of FPR1
Studies conducted throughout the 1980s led to the identification of the formyl peptide receptor as a GPCR. All major neutrophil functions stimulated by fMLF can be inhibited by treatment of the cells with pertussis toxin (Bokoch and Gilman, 1984; Bokoch et al., 1984; Lad et al., 1985; Sha'afi and Molski, 1988; Gierschik et al., 1989; Snyderman and Uhing, 1992), indicating that the G proteins that couple to the formyl peptide receptors belong to the Gi family of heterotrimeric G proteins (Simon et al., 1991). At that time, a number of G protein-coupled receptors from other tissues had been pharmacologically characterized, but only a few genes coding for these proteins, including rhodopsin, α- and β-adrenergic receptors, and two of the muscarinic receptors, had been cloned (Dixon et al., 1986; Kubo et al., 1986; Kobilka et al., 1987). The strategies used in cloning these receptor genes involved mostly purification of the receptor proteins and sequencing of the derived peptides to obtain partial sequence of the GPCR for the design of nucleotide probes, which were used for DNA hybridization with cDNA or gene libraries. In addition to DNA hybridization, an expression cloning approach using oocytes from Xenopus laevis was adopted to isolate the gene for a muscarinic receptor. A different approach was taken to clone the human formyl peptide receptor gene. Boulay et al. (1990b), with expertise in photoaffinity labeling techniques and protein chemistry, started a cloning project with the synthesis of a photoaffinity hetero-bifunctional derivative of N-formyl-Met-Leu-Phe-Lys (fMLFK), with one moiety that was photoactivatable and another that could bind to streptavidin. The rationale behind the synthesis of this derivative was to photolabel the receptor in dibutyryl cAMP-differentiated HL-60 cells, which express the fMLF receptor based on binding and internalization assays (Niedel et al., 1980b). The photoaffinity-labeled receptor was then purified through the interaction of its biotin moiety with streptavidin-coated beads. However, this initial effort to isolate the receptor gene was unsuccessful.
In addition to screening cDNA or genomic libraries using oligonucleotide probes and X. laevis oocyte-based expression cloning, the molecular cloning techniques then available included E. coli- or mammalian cell-based expression cloning with antibody detection of the recombinant protein on the cell surface or in cell lysate. The mammalian cell expression cloning approach was successfully used in the identification of genes for several cell surface molecules, including the T cell-specific surface protein (CD28), using antibodies against these proteins (Aruffo and Seed, 1987). Taking this approach, Boulay et al. (1990a,b) screened a cDNA expression library made with mRNA from differentiated HL-60 cells. Because no antibody against human formyl peptide receptor was available at that time, the approach described by Aruffo and Seed (1987) was not directly applicable. Therefore, the N-formyl peptide fMLFK was derivatized with a hydrophilic dodecapeptide (N-acetyl-SDQALSFLKDYC) that represents the N-terminal region of the bovine mitochondrial ADP/ATP carrier. The dodecapeptide was coupled via the C-terminal cysteine on the free ϵ-amino group of the lysine of fMLFK, yielding a water-soluble heterobifunctional ligand that retained its high affinity for FPR1. Because high-affinity antibodies against this peptide were available, it was anticipated that this ligand could be used as a bridge between the receptor and immunoglobulins coated on Petri dishes. However, this approach was also met with difficulties, prompting a switch to labeling the derivatized peptide with 125I and using this peptide for quantification of any expressed protein that bound the probe in COS cells transfected with fractions of the cDNA library. To improve the chance of detecting a specific receptor on the cell surface, the primary cDNA library was split into pools with approximately 700 independent bacterial colonies each, an approach used in the cloning of the erythropoietin receptor (D'Andrea et al., 1989). After screening 184 pools of the library, several pools yielded signals 5- to 20-fold higher than that of the background. The pools were further divided, and two cDNA variants were found to produce markedly increased signals, in part due to internalization and intracellular accumulation of the radiolabeled formyl peptide derivative. These two cDNA isolates (FPR-26 and FPR-98) represent the two allelic forms of human FPR1 in HL-60 cells (Boulay et al., 1990a,b), encoding a putative seven-transmembrane domain (7TM) receptor with 350 amino acids (Fig. 1).
Shortly after the completion of the initial FPR1 cloning work, Thomas et al. (1990) reported the isolation of a rabbit cDNA, known as F3R, encoding a protein that was first described as rabbit FPR1. This conclusion was based on the ability of the cDNA to encode a receptor capable of mobilizing calcium when expressed in X. laevis oocytes and stimulated with fMLF. Subsequent studies showed that F3R encodes a rabbit receptor for IL-8 (Thomas et al., 1991), and the high-affinity formyl peptide receptor in rabbits was found to be the product of another distantly related gene (Ye et al., 1993).
With an available cDNA for human FPR1, it became possible to conduct pharmacological characterization of the receptor in a defined cellular environment free of similar receptors. Functional expression of FPR1 was accomplished in transfected mammalian cells (Prossnitz et al., 1991) and X. laevis oocytes (Murphy et al., 1992). In these studies, the recombinant human FPR1 was shown to mediate fMLF-induced calcium mobilization in a pertussis toxin-sensitive manner. Subsequent studies demonstrated that the recombinant FPR1 is able to mediate fMLF-induced actin polymerization and chemotaxis in transfected HL-60 cells (Prossnitz et al., 1993) and release of granule contents in transfected rat basophilic leukemia (RBL) cells (Ali et al., 1993). More recently, FPR1-dependent production of superoxide was reconstituted in COSphox cells (He et al., 2004), a transgenic COS-7 cell line expressing the four essential phagocyte oxidase (phox) proteins (Price et al., 2002). In mice, loss of function assay has shown that Fpr1 is a major receptor to mediate fMLF-induced NADPH oxidase activation. Another receptor, possibly Fpr2, also contributes to oxidant production when mouse neutrophils are stimulated with fMLF at higher concentrations (e.g., 50 μM) (Lavigne et al., 2002). These results demonstrate the ability of FPR1 to activate multiple signaling pathways important for the microbicidal functions of phagocytes.
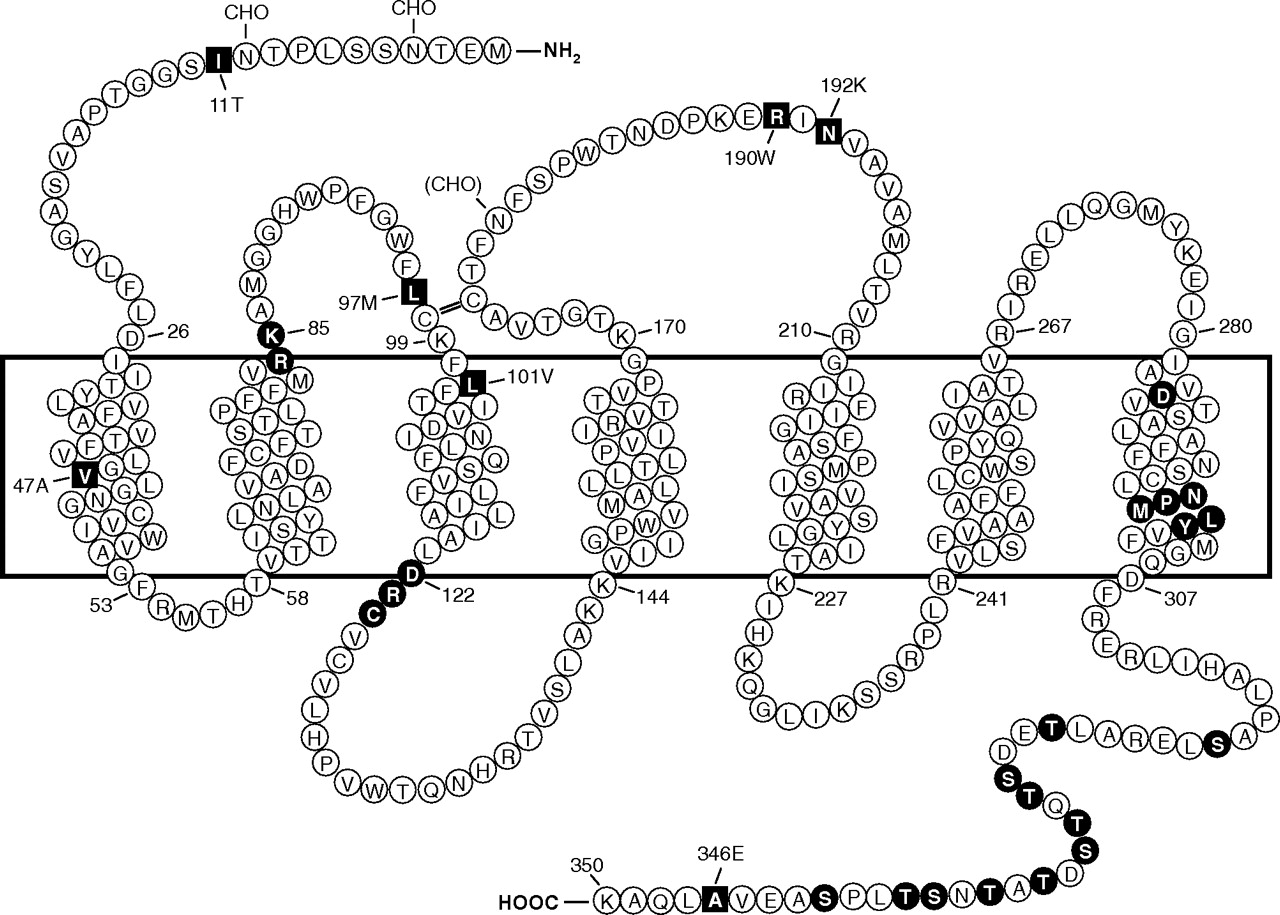
Predicted transmembrane disposition of the human FPR1. The protein sequence of the FPR-98 isoform (Leu110, Ala346) is shown (Boulay et al., 1990a). The transmembrane domains (TMs) are predicted based on hydrophobicity of the amino acid sequence and on similarities to the rhodopsin structure. The amino acids that form the boundaries of the transmembrane domains are numbered. One-letter amino acid code is used. The square blocks in reverce color represent positions at which amino acid substitutions result from polymorphisms, including amino acids 11 (Ile/Thr), 47 (Val/Ala), 101 (Leu/Val), 190 (Arg/Trp), 192 (Asn/Lys) and 346 (Ala/Glu). The circle blocks in reverse color indicate amino acids with known functions as follows. Arg84, Lys85, and Asp284 are critical for high-affinity binding of fMLF (Mills et al., 1998; Quehenberger et al., 1997). Asp122, Arg123, and Cys124 are the signature sequence for G protein interaction (DRY in many GPCRs). NPMLY in the TM7 are known signature sequence (NPXXY) for receptor internalization (Gripentrog et al., 2000; He et al., 2001). The 11 Ser and Thr residues in the cytoplasmic tail are potential phosphorylation sites for GRK2 and GRK3 (Prossnitz et al., 1995). CHO, carbohydrate, marks the identified and potential (in parenthesis) sites for N-glycosylation. The predicted disulfide bond between Cys98 and Cys176 is marked with double-line (=).
B. Identification of Additional Receptors of the FPR Gene Family
The availability of the FPR1 sequence not only facilitated molecular characterization of the receptor in heterologous expression systems but also prompted identification of receptors that share sequence homology with FPR1. Strategies were developed to use low-stringency DNA hybridization (under conditions of reduced temperature and/or increased salt concentration), with the cDNA of human FPR1 as a probe, for isolation of cDNAs or genes of similar sequences. In 1992, several laboratories reported the identification of a cDNA (Murphy et al., 1992; Perez et al., 1992; Ye et al., 1992) and gene (Bao et al., 1992) coding for a putative 7TM receptor that shares significant sequence homology to human FPR1. The gene product was given different names, including FPR2 (formyl peptide receptor 2) for its low-affinity binding of fMLF (Ye et al., 1992), FPRL1 (formyl peptide receptor-like 1) (Murphy et al., 1992), FPRH1 (formyl peptide receptor-homolog 1) (Bao et al., 1992), and “receptor related to formyl peptide receptor” (Perez et al., 1992) based on it sequence homology to the human FPR1. Other names used in the literature include HM63 (Nomura et al., 1993), and FMLP-related receptor II (GenBank accession no. M76672) (Table 3). Pharmacological characterization has led to the identification of the eicosanoids lipoxin A4 (LXA4) and aspirin-triggered lipoxins (Fiore et al., 1994; Chiang et al., 2006), and numerous peptides (Le et al., 2001b, 2002; Migeotte et al., 2006) as ligands for this receptor. Therefore, in addition to FPRL1, which appears frequently in the literature, the names LXA4R and ALX have been introduced to convey the ability of the receptor to interact with LXA4 and aspirin-triggered lipoxins (Brink et al., 2003). Given that IUPHAR nomenclature of a receptor is based on its agonists and N-formyl peptides are the only common ligands for this class of receptors, we will refer to the receptor as FPR2/ALX, a nomenclature now recommended by IUPHAR.
IUPHAR-recommended nomenclature for human FPRs and previously used names
FPR2/ALX is a 7TM receptor with 351 amino acids and shares 69% of its amino acids with human FPR1 (Fig. 2). Despite the relatively high level of sequence homology, FPR2/ALX is a low-affinity receptor for fMLF, with a Kd of 430 nM (Murphy et al., 1992; Ye et al., 1992; Quehenberger et al., 1993). It has been reported that mitochondria-derived formyl peptides are more potent agonists for FPR2/ALX than fMLF (Rabiet et al., 2005), suggesting that its primary function may be to recognize host-driven mitochondrial peptides or possibly other bacterially derived formyl peptides. As discussed in detail below, in addition to formyl peptides and LXA4, FPR2/ALX is also able to interact with nonformylated peptides.
A second gene with significant sequence homology to human FPR1 was identified using a similar cloning strategy. This gene, initially named FPRL2 (formyl peptide receptor-like 2) (Murphy et al., 1992) and FPRH2 (formyl peptide receptor homolog 2) (Bao et al., 1992) based on sequence homology to FPR1, encodes a putative 7TM receptor of 352 amino acids (Table 3). This receptor was also named RMLP-related receptor I (GenBank accession no. M76673). In this article, we will refer to it as FPR3, an IUPHAR-recommended name based on the ability of the receptor to bind certain N-formyl peptides. FPR3 shares 56% sequence identity with human FPR1 at the protein level. Although fMLF is inactive at FPR3 (Migeotte et al., 2005), fMMYALF, a peptide derived from a mitochondrial protein, is an agonist at FPR3 in calcium flux assays, with pEC50 of 6.00 (EC50 of 10-6 M; pEC50 is the negative logarithm of EC50) (Rabiet et al., 2005). Migeotte et al. (2005) recently reported that F2L, a naturally occurring endogenous acylated peptide derived from the N-terminal sequence of heme-binding protein, is a potent agonist for FPR3 with a pEC50 of 7.00 in a reporter assay. Additional ligands for FPR3 are discussed in Section III below. A survey of FPR3 expression using monoclonal antibodies found an intracellular pool of the receptor. It is noteworthy that, in one published study, one third of blood donors (8 of 24) had no detectable FPR3 expression on the surface of dendritic cells (Migeotte et al., 2005).
The three members of the human FPR gene family are clustered on chromosome 19q13.3–19q13.4, adjacent to the human C5a receptor gene (Bao et al., 1992; Murphy et al., 1992). This suggests that the genes arose by relatively recent duplication of a common ancestor and that the encoded proteins may have related biological functions.
C. Tissue and Cellular Distribution of the Human FPR Gene Family Members
FPR1 was initially found in human and rabbit neutrophils through functional characterization (Schiffmann et al., 1975b; Showell et al., 1976; Aswanikumar et al., 1977; Williams et al., 1977; Zigmond, 1977). More extensive investigations were conducted when key reagents such as cDNA probes and antibodies became available. The three receptors of the FPR gene family are primarily found in myeloid cells, but the distribution varies within myeloid cell subsets, as described in the next paragraph. The results of these studies also indicate the presence of formyl peptide receptors in nonmyeloid cells. Using an antibody recognizing the carboxyl terminal 11 amino acids of FPR1, Becker et al. (1998) found immunoreactivity at the immunohistochemical level in multiple organs and tissues that include epithelial cells in organs with secretary functions, endocrine cells (including follicular cells of the thyroid and cortical cells of the adrenal gland), liver hepatocytes and Kupffer cells, smooth muscle cells and endothelial cells, brain, spinal cord, and both motor and sensory neurons. In most cases, this has not yet been verified at the RNA level or by functional studies. Human FPR2/ALX has a tissue distribution pattern similar to that of FPR1. Because of significant sequence homology between FPR1 and FPR2/ALX in their carboxyl termini, it is possible that the rabbit polyclonal antibody used in the immunohistochemical analysis have detected both FPR1 and FPR2/ALX. Another study showed endothelial cell expression of formyl peptide binding sites (Rotrosen et al., 1987). The low affinity of the binding site for fMLF is consistent with the binding property of FPR2/ALX. Functional FPR1 has also been found in hepatocytes, glial cells, astrocytes, and platelets (Lacy et al., 1995; McCoy et al., 1995; Le et al., 2000a; Czapiga et al., 2005), suggesting that formyl peptide receptors may have more complex functions than are presently appreciated.

Alignment of the protein sequences of the human FPRs. The putative transmembrane domains (I to VII) are shaded. Sequence of the FPR-98 isoform is shown. Comparison of the three receptors has identified highly conserved regions, including most of TM-I and TM-II, and the short intracellular loop connecting TM-I and TM-II. The second intracellular loops from these receptors, known for G protein interaction, are nearly identical. TM-VII, including the NPXXY motif and a stretch of ∼25 amino acids extending toward the C-terminal tail, are also conserved among these receptors. Major differences are found in the extracellular domains between FPR1 and the other two receptors, especially in the amino termini (∼50% different), the second extracellular loops (56% different), and the third extracellular loops (∼50% different). The two putative N-glycosylation sites in the N-terminal domains are conserved among all three receptors. Most of the serines and threonines in the C-terminal tail, along with charged residues that constitute consensus GRK phosphorylation sites, are also conserved among these receptors.
Unlike FPR1 and FPR2/ALX, FPR3 transcripts are not found in neutrophils (Murphy et al., 1992). Instead, it can be detected together with transcripts for FPR1 and FPR2/ALX in monocytes, although the expression pattern changes with monocyte differentiation. In particular, in the process of monocyte differentiation into immature dendritic cells (DCs), the cellular expression of FPR2/ALX progressively declines (Yang et al., 2001a), whereas FPR2/ALX expression remains unchanged during monocyte differentiation into macrophages. There is a progressive loss of FPR1 during differentiation of immature DC to mature DC, such that FPR3 becomes the predominant human formyl peptide receptor in mature DC (Yang et al., 2002; Migeotte et al., 2005). The biological significance of differential expression of formyl peptide receptors in monocytes, macrophages, and DCs has not yet been clearly delineated.
D. Polymorphism of the Formyl Peptide Receptors
The initial cloning study resulted in the identification of two allelic forms of human FPR1 (FPR-26 and FPR-98) (Boulay et al., 1990a). The differences between the two cDNAs are Val101, Glu346 in FPR-26 and Leu101, Ala346 in FPR-98. In a subsequent study, additional single-nucleotide polymorphisms (SNPs) of FPR1 were identified (Sahagun-Ruiz et al., 2001), whereas no polymorphism has been identified for FPR2/ALX. The difference in polymorphism suggests that these two structurally similar receptors have undergone distinct processes of evolutionary selection. FPR1 SNPs identified to date are either synonymous or nonsynonymous, the latter resulting in amino acid changes at positions 11 (Thr/Ile), 47 (Val/Ala), 97 (Leu/Met), 101 (Leu/Val), 190 (Arg/Trp), 192 (Asn/Lys), and 346 (Ala/Glu) (Fig. 1). At least 30 variations have been identified at the nucleotide level, and these SNPs contribute to the different haplotypes of human FPR1 (Sahagun-Ruiz et al., 2001; Zhang et al., 2003; Gunji et al., 2007; Gripentrog et al., 2008).
There is a reported association between juvenile onset, localized periodontitis, and defects in neutrophil responses to formyl peptides (Clark et al., 1977; Van Dyke et al., 1986; Agarwal et al., 1989; Perez et al., 1991). Consequently, attempts have been made to establish a correlation between FPR1 SNPs and defective neutrophil functions in juvenile-localized periodontitis. Gwinn et al. (1999) used single-strand conformation polymorphism analysis to identify two base substitutions resulting in changes at positions 110 (F110S) and 126 (C126W) in FPR1. When tested as recombinant proteins, these FPR1 variants display defective Gαi protein coupling in reconstitution assays using Sf9-cell derived mutant receptors (Seifert and Wenzel-Seifert, 2001). The Trp126 form of FPR1 exhibits reduced function in chemotaxis and calcium mobilization assays (Jones et al., 2003; Nanamori et al., 2004b). One study also indicated failure of cell surface expression of the Ser110 variant (Jones et al., 2003), whereas in the other study, the Ser110 variant and Ser110/Trp126 variant of FPR1 were normally expressed on the cell surface, but their ability to take up fMLF was compromised (Nanamori et al., 2004b). Although these studies seem to have identified a structural basis for defective FPR1 in juvenile localized periodontitis, a subsequent and more extensive independent survey failed to identify the same base substitutions in 111 cases of juvenile localized periodontitis examined (Zhang et al., 2003). Therefore, it is still unclear whether the base substitutions resulting in amino acid changes at amino acids 110 and 126 are responsible for the defective neutrophil response to fMLF observed in localized juvenile periodontitis patients.
Mills (2007) and Gripentrog et al. (2008) found that some of the FPR1 haplotypes are indeed functionally distinct. In extracellular signal-regulated kinase (ERK) phosphorylation, chemotaxis, and receptor down-regulation assays, the three tested FPR1 haplotypes displayed similar responsiveness to fMLF; however, differences were found when these FPR1 haplotypes were activated by formyl peptides from other bacterial strains such as Mycobacterium avium. These findings suggest individual differences in FPR1-mediated detection of certain bacterial strains.
As discussed above, no SNP has been identified in the human FPR2/ALX gene. The only variant of FPR3 described to date involves an Asp to His substitution at position 338 (GenBank accession no. AAA58482). An examination of this SNP found no difference in receptor expression or functional response between the FPR3 variants in an F2L-induced cAMP inhibition assay (Migeotte et al., 2005).
E. Evolution of the Formyl Peptide Receptor Gene Family
Since the first reported cDNA cloning of human FPR1 (Boulay et al., 1990a,b), orthologs have been identified at the molecular level in other primates (Alvarez et al., 1996), rabbits (Ye et al., 1993), and mice (Gao and Murphy, 1993). Functional formyl peptide receptors have also been found in rat, guinea pig, and horse, and differences relative to human FPR1 have been noted (Oseas et al., 1980; Snyderman and Pike, 1980; Ward et al., 1984). For instance, horse neutrophils respond to fMLF with granule enzyme release but not chemotaxis (Snyderman and Pike, 1980). These cells produce superoxide only when primed with TNFα or platelet-activating factor (PAF) and then challenged with fMLF (Brazil et al., 1998). Neutrophils from other species such as dog were initially reported not to respond to fMLF (Redl et al., 1983) but were later found to respond to very high concentrations (100 μM) of fMLF in a chemotaxis assay (Linnekin et al., 1990). Because most studies have been conducted with fMLF, it will be interesting to determine whether neutrophils from dogs and other mammalian species can respond to the many other naturally occurring bioactive formyl peptides that have been described previously (Freer et al., 1980; Rot et al., 1987; Rabiet et al., 2005; Southgate et al., 2008).
The FPR gene family has a complex evolutionary history. The number of genes in the family can vary markedly in different mammalian species, indicating differential gene expansion or extinction and suggesting the presence of differential selective pressures. In particular, the mouse FPR gene family has at least eight members (Fig. 3) clustered on mouse chromosome 17A3.2, as opposed to only three in human on chromosome 19q13.3 in a region syntenic with the mouse cluster (Gao et al., 1998; for review, see Migeotte et al., 2006). The gene product of Fpr1 is the mouse ortholog of human FPR1. The gene products of both Fpr-rs1 and Fpr-rs2 (Fpr-related sequence 1 and 2) are structurally most similar to human FPR2/ALX (Hartt et al., 1999).
The gene product of mouse Fpr-rs2, referred to as mouse Fpr2 (mFpr2), is a low-affinity receptor for fMLF (Hartt et al., 1999). This receptor responds to several peptide agonists that activate human FPR2/ALX, including the amyloidogenic proteins serum amyloid A (Liang et al., 2000) and amyloid β(1–42) (Tiffany et al., 2001). An FPR2/ALX-specific antagonist inhibits activation of mouse neutrophils as well as cells stably expressing this receptor, suggesting that the receptor shares significant structural and pharmacological properties with human FPR2/ALX (Onnheim et al., 2008). A variant of mouse Fpr-rs1 has been cloned that encodes a protein with a four-residue insertion (ARNV, after Leu141 in the 4th transmembrane helix) relative to the reference sequence (Fig. 3); this variant has been named the mouse Lxa4 receptor (Takano et al., 1997). Another study showed that mouse Fpr2 can mediate phosphoinositide turnover when coupled to Gα16 and stimulated with LXA4 (Vaughn et al., 2002). It is noteworthy that mouse Fpr2 has been reported to be also a receptor for F2L (Gao et al., 2007), a potent agonist for human FPR3 (Migeotte et al., 2005). These results indicate that the gene products of Fpr-rs1 (including its variant, mouse Lxa4r) and mouse Fpr2 share some pharmacological properties with human FPR2/ALX, and mouse Fpr2 also has an overlap function with human FPR3 in the detection of F2L. The biological functions of other mouse Fpr gene family members have not been determined (Gao et al., 1998; Wang and Ye, 2002). Fpr-rs4 encodes a putative 7TM-spanning receptor with 323 amino acids. A stop codon at position 246 in the predicted transmembrane 6 causes an early termination of the Fpr-rs5 open reading frame, which makes it unlikely to encode a functional receptor, although it does not have the typical features of a pseudogene (Gao et al., 1998). Fpr-rs6 and Fpr-rs7 are other orphans in the family also identified by low-stringency DNA hybridization (Wang and Ye, 2002). The complex evolution of the FPR gene family is apparent from the high sequence divergence between species orthologs (∼ 25–30% between human and mouse) (Table 4), which is very high compared with most proteins but characteristic for immunoregulatory proteins, including other chemoattractant receptors, such as chemokine receptors (Murphy, 1993).
Sequence identity between the human and mouse FPRs Shown in the table are percent of identical amino acids between the receptors, based on pairwise comparison of the entire sequence of each receptor using the ALIGN program (Scientific & Educational Software, ver. 1.02). The FPR1–98 sequence (Boulay et al. 1990a) is used for the comparison. Note that the gene product of Fpr-rs5 used in sequence comparison is 246 amino acids. The size of each receptor is indicated in the first column
Although mouse Fpr1 shares a sequence identity of 77% with human FPR1 (Table 4), it displays low affinity for fMLF. In functional assays, fMLF concentrations of 100 nM or higher are required for activation of cell functions such as calcium mobilization (Gao and Murphy, 1993). Other neutrophil functions, including degranulation and superoxide production, require even higher fMLF concentrations (5–10 μM) in mice. It is notable that both mouse Fpr1 and human FPR2/ALX are low-affinity receptors for fMLF. The structural basis for the low-affinity binding is not entirely clear, but a comparison of human FPR1, FPR2/ALX, and mouse Fpr1 has identified major differences in key residues known to define the human FPR1 binding pocket for fMLF (Quehenberger et al., 1997; Mills et al., 1998). A pair of positively charged residues, Arg84 and Lys85 in human FPR1 (Fig. 1), is replaced with uncharged residues in both FPR2/ALX and mouse Fpr1. Moreover, the negatively charged Asp284 in human FPR1, known to stabilize the receptor structure, is also substituted with an uncharged residue in FPR2/ALX and a positively charged Lys in mouse Fpr1 (Fig. 3). Therefore, mouse Fpr1 shares certain structural features with human FPR2/ALX. Restoration of Arg84 and Lys85 in an FPR1-FPR2/ALX chimeric receptor displaying low affinity for fMLF (Quehenberger et al., 1993) has been shown to significantly improve the binding affinity for the formyl peptide from 105 to 1.6 nM (Quehenberger et al., 1997). In comparison, Lys85 and Asp284 are conserved in rabbit FPR1 that displays high binding affinity for fMLF (Ye et al., 1993). This finding suggests that small changes to key residues in formyl peptide receptors can profoundly affect their binding selectivity and may have interesting implications for the evolution of this receptor family.

Alignment of the predicted receptor sequence of the mouse Fpr genes. The putative transmembrane domains (TM-I–TM-VII) are shaded. Dashes indicate gaps in sequence created for alignment purposes. It is noteworthy that there is an eight-residue insertion in the N-terminal region of Fpr-1, just before TM-I. A three-residue insertion is found in the second extracellular loop in Fpr-1. The positively charged residues Arg84 and Lys85, found in human FPR1 and known for the interaction with fMLF, are missing from Fpr-1 and other mouse Fpr-related sequences. In its place are the noncharged residues Ser92 and Met93. The predicted Fpr-rs5 sequence is truncated at amino acid 246, resulting in a putative protein with only five TMs. Fpr-rs4 encodes a protein of 323 residues with a short C-terminal tail. In Fpr-rs1, there is a four-residue deletion in TM-IV, whereas the cloned mouse LXA4 receptor gene encodes a protein with the sequence of ARNV in its place. Polymorphisms exist in the Fpr-rs1 gene that result in amino acid substitutions at positions 3 (Thr/Ser), 8 (Pro/His), 13 (Asp/Glu), 16 (Ile/Val), 222 (Thr/Tyr), 236 (Phe/Ser), 296 (Ile/Met), and 318 (Gln/Pro) (Takano et al., 1997; Gao et al., 1998; Wang et al., 2002). The highest sequence identity (81%) is found between Fpr-rs1 and Fpr-rs2, and between Fpr-rs3 and Fpr-rs4.
The transcripts of Fpr1 and Fpr-rs2 are most abundant in mouse neutrophils, suggesting that these two receptors might be primarily responsible for detection of formyl peptides. Mouse Fpr2 is 81% identical in protein sequence to mouse Fpr1 and 58% to human FPR1 (Gao et al., 1998) (Fig. 4). It responds even more weakly to fMLF than mouse Fpr1 and contributes to the second concentration optimum in mouse neutrophil chemotaxis assays using fMLF at micromolar concentrations (Hartt et al., 1999). As a result, mouse neutrophils exhibit a binding affinity for fMLF that is 100- to 500-fold lower than that of human neutrophils. The absence of high affinity interaction with fMLF in mouse neutrophils raises the question of whether mice can effectively detect bacterially derived formyl peptides and whether FPR1 is important for host defense in mice. To address these questions, Gao et al. (1999) generated Fpr1 knockout mice and examined the ability of these mice to clear L. monocytogenes. They found defective chemotaxis of Fpr1(-/-) neutrophils toward fMLF. Moreover, the Fpr1(-/-) mice were more susceptible to L. monocytogenes infection, showing an increased mortality rate (Gao et al., 1999). These results indicate an important function of mouse Fpr1 in host defense. More recently, Southgate et al. (2008) examined mouse neutrophil responses to formyl peptides derived from L. monocytogenes (with the sequence of fMIVIL) and Staphylococcus aureus (with the sequence of fMIFL) and found that these peptides were at least 100-fold more potent than fMLF in stimulating mouse neutrophil chemotaxis and superoxide production. Using transfected cell lines that individually express mFpr1 and mFpr2, the authors demonstrated that mFpr1 can respond to low nanomolar concentrations of fMIVIL and fMIFL, whereas mFpr2 requires micromolar concentrations of fMIVIL and fMIFL in calcium mobilization assay. Targeted deletion of Fpr1 results in compromised neutrophil responses to both of these peptides in assays measuring chemotaxis, degranulation, and superoxide generation (Southgate et al., 2008). Therefore, although mouse neutrophils are inefficient in responding to fMLF, a major chemoattractant from E. coli culture (Marasco et al., 1984), these innate immune cells are able to detect low nanomolar concentrations of the two peptides from L. monocytogenes and S. aureus. It is possible that the increased susceptibility of Fpr1(-/-)mice to L. monocytogenes infection may result from defective neutrophil detection of the L. monocytogenes-derived formyl peptides such as fMIVIL. Listeria fMIVIL is also known to bind the mouse Class Ib MHC molecule H2-M3 (Gulden et al., 1996) and to activate human FPR2/ALX (Rabiet et al., 2005). S. aureus fMIFL is one of the six oligopeptides purified from an S. aureus culture and known to activate human neutrophils and monocytes with high potency (Rot et al., 1987). Differential detection of these peptides by mouse Fpr1 may reflect the environment in which mice live. E. coli infection can be lethal in humans but is not known to be a natural mouse pathogen. Thus, it is interesting to speculate that selection pressure has favored the acquisition of additional binding properties for more sensitive detection of the E. coli-derived fMLF by human FPR1.
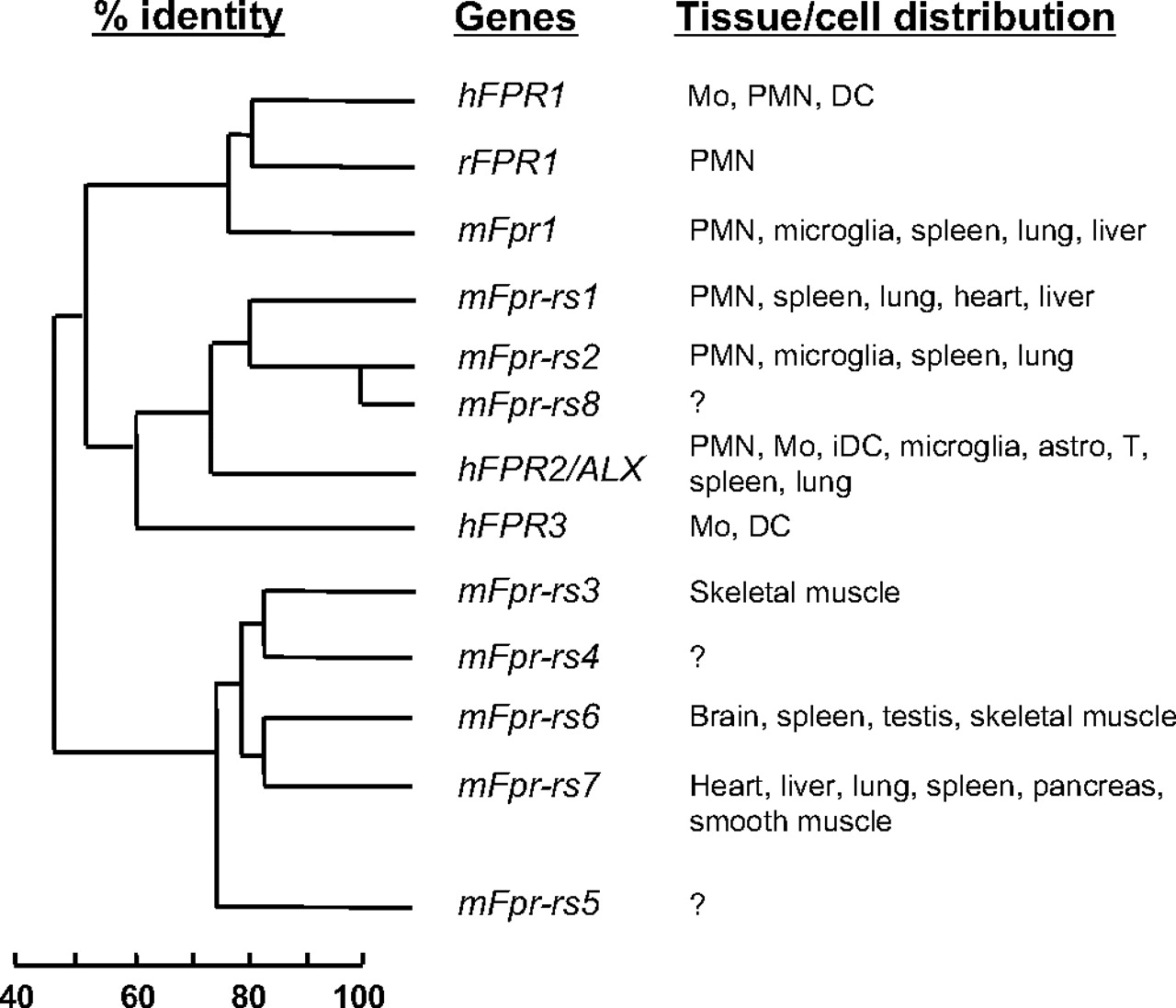
Sequence homology between the FPR family members and their tissue distribution. The predicted protein sequences of the three human (h) FPR genes, the eight mouse (m) Fpr genes, and the rabbit (r) FPR1 gene were compared (Boulay et al., 1990a,b; Ye et al., 1993; Gao et al., 1998; Wang et al., 2002). Based on sequence homology, the hFPR1, mFpr1, and rFPR1 are in the same cluster. The mFpr-rs1, mFpr-rs2 (also termed mFpr2), and mFpr-rs8 are in another cluster closely related to hFPR2/ALX and hFPR3. The mFpr-rs3, mFpr-rs4, mFpr-rs6, and mFpr-rs7 (and, to a lesser extent, mFpr-rs5) are closely related based on their protein sequences (see Table 4 for sequence identity between the gene products). Note that some of these genes are not expressed in neutrophils and monocytes. The tissue expression profiles for mFpr-rs4, mFpr-rs5, and mFpr-rs8 have not been determined. Mo, monocytes; PMN, polymorphnuclear leukocytes; iDC, immature dendritic cells; astro, astrocytes; T, T lymphocytes.

Chemical structures of selected ligands for the formyl peptide receptors. Despite their abilities to bind to FPR1 and/or FPR2/ALX, these ligands have quite different structures. Of the ligands shown, t-Boc-FLFLF and CsH are antagonists and others are agonists. Note that the N-formyl group that defines agonistic activities in peptides such as fMLF is replaced with a bulky t-butyloxycarbonyl group that defines antagonistic activities in peptides such as t-Boc-FLFLF. LXA4, Quin-C1, and compound 43 are highly selective agonists for FPR2/ALX, whereas AG-14 and fMLF are selective for FPR1. t-Boc-FLFLF is selective for FPR1 at low concentrations but the selectivity is lost at high micromolar concentrations (e.g., 100 μM).
III. Ligands for the Formyl Peptide Receptor Family of Receptors
Ligand diversity is a prominent and unusual feature of the FPR family of receptors. With the exception of the eicosanoid LXA4, all known FPR family ligands are peptides. More recently, synthetic small-molecular-weight ligands for the formyl peptide receptors have emerged from a number of compound library screens. The structural diversity of these ligands is illustrated with selected agonists and antagonists shown in Fig. 5. The reader is also referred to other recent reviews for a discussion on the agonists and antagonists of FPRs (Le et al., 2001b, 2002; Migeotte et al., 2006). In this article, agonists for the FPRs are listed in separate groups in Table 5, based on their chemical constituents and origins. Whereas many of the agonists for FPRs are identified and purified from living organisms (marked by asterisks in Table 5), a number of peptides are synthesized based on the sequences of known proteins of microbe and host origins. Whether these peptides are present in vivo and have physiological functions has yet to be determined.
Agonists for the human FPRs The agonists are listed in the order of their potency within each group. The mitochondrial N-formylated peptides, listed in the first group, are also host-derived peptides. A more detailed list of N-formyl peptides is given in Table 2. Ligands that have been isolated from living organisms in the forms listed, and those generated by the actions of physiologically relevant enzymes, are indicated with an asterisk (*)
A. Agonists for the Formyl Peptide Receptor Family of Receptors
1. N-Formyl Peptides.
The E. coli-derived tripeptide fMLF is the most widely used chemotactic peptide for several reasons. It was one of the first characterized synthetic chemotactic peptides and has been extensively studied by the phagocyte research community since its initial discovery in 1975. fMLF is the smallest formyl peptide that displays full agonistic activities. Its potency and efficacy in activating major bactericidal functions of neutrophils equals that of C5a, yet fMLF is readily available from most peptide companies at a fraction of the cost of C5a. fMLF can be easily radiolabeled with tritium, and the radiolabeled fMLF serves as an excellent ligand in direct binding assays.2
Whereas fMLF is by far the most frequently used chemotactic peptide in studies of neutrophil functions, this prototypic formyl peptide should not be taken as the sole standard in judging the presence of functional formyl peptide receptors. Because bacterial protein synthesis starts with an N-formyl methionine, formyl peptides released from bacteria can be considered a type of microbe-associated molecular pattern, recognizable by specialized receptors in the innate immune cells of the host, such as the Toll-like receptors (TLRs). These non-rearranging innate receptors have evolved to aid the host in detecting nonself such as bacterial products (Medzhitov and Janeway, 2000). Ample evidence shows that the formyl peptide receptors can detect not only the E. coli-derived fMLF but also formyl peptides from other bacteria strains and from mitochondria of the host cells. Table 2 lists selected bacterial and mitochondrial formyl peptides that have been characterized for their bioactivities.
Given the variety of formyl peptides from both bacteria and mitochondria, it is worthwhile to revisit some previous studies conducted with the use of fMLF and determine whether the receptors of interest actually are more selective for formyl peptides of different sequences. For instance, FPR2/ALX was first identified as a low affinity receptor for fMLF (Kd = 430 nM), raising the question of whether its true ligand is a formyl peptide. Rabiet et al. (2005) recently conducted an extensive survey of formyl peptides from various sources, including bacteria and mitochondria of mammalian cells (Rabiet et al., 2005). The results from this study demonstrate the ability of FPR2/ALX to respond to mitochondria-derived formyl peptides (fMYFINILTL, fMLKLIV, and fMMYALF) in calcium mobilization assays, with EC50 values of 10 to 160 nM, showing that these peptides are similarly potent on both FPR1 and FPR2/ALX. Moreover, the study also demonstrates the ability of FPR2/ALX to detect and respond to several L. monocytogenes-derived formyl peptides, including fMIVIL (Gulden et al., 1996). These results indicate that FPR2/ALX is able to discriminate between N-formyl peptides of different sizes, hydrophobicities, and charges. The biological relevance of this property of the receptor is not yet entirely understood.
Two studies discussed in the previous sections provide additional examples that N-formyl peptides other than fMLF are more selective for certain receptors. Mouse Fpr1, for example, prefers fMIVIL and fMIFL, peptides derived from L. monocytogenes and S. aureus, respectively, over the E. coli-derived fMLF (Southgate et al., 2008). One of the FPR1 haplotypes tested in the study by Gripentrog et al. showed reduced ability to respond to a M. avium-derived formyl peptide but normal response to fMFADRW, a fragment of cytochrome c oxidase subunit I (Gripentrog and Miettinen, 2008). These studies demonstrate that although N-formyl peptides are a class of ligands representing a pattern recognized by the FPRs, as has been the case since the initial discoveries by Schiffmann et al. (1975a), there are important differences in potency and receptor selectivity among the individual peptides.
There are other examples in which addition of an N-formyl group increases agonistic activity of the peptides. MMWLL (Met-Met-Trp-Leu-Leu), a synthetic peptide isolated from a library screen, becomes more potent at FPR1 with the addition of an N-formyl group (Chen et al., 1995). Humanin is an endogenous peptide (MAPRGFSCLLLLTSEIDLPVKRRA) with neuroprotective activity (Hashimoto et al., 2001) that also binds to FPR2/ALX and FPR3 (Ying et al., 2004). If humanin is N-formylated, it becomes a more potent agonist than nonformylated humanin for these receptors (Harada et al., 2004). In the latter example, although the primary sequences of FPR2/ALX and FPR3 differ considerably from those of FPR1, especially in the ligand binding domains (Mills et al., 1998), these two receptors seem to have retained the ability to preferentially interact with formylated peptides. Whether these receptors mediate the neuroprotective effects of humanin is not yet known.
It has long been hypothesized that N-formyl peptides derived from mitochondrial proteins may attract leukocytes to sites of inflammation and tissue damage. Consistent with this, N-formylated hexapeptides corresponding to the N terminus of mitochondrial NADH dehydrogenase subunits 4 (fMLKLIV) and 6 (fMMYALF) and cytochrome c oxidase subunit I (fMFADRW) are equally potent at FPR1 and FPR2/ALX (Rabiet et al., 2005). fMMYALF is also a low-affinity agonist at FPR3 (Rabiet et al., 2005). A nonformylated peptide fragment (MYFINILTL) derived from mouse NADH dehydrogenase subunit 1 is also an agonist at FPR2/ALX (Chiang et al., 2000). It is not yet known whether these peptides are produced in vivo and whether they modulate inflammation.
2. Microbe-Derived Nonformyl Peptides.
A cecropin-like peptide (with the sequence AKKVFKRLEKLFSKIQNDK) from Helicobacter pylori, Hp(2–20), was found to attract monocytes and basophils to the gastric mucosa in response to H. pylori infection. Hp(2–20) was identified as an agonist at FPR2/ALX and FPR3 (Betten et al., 2001; de Paulis et al., 2004b). Despite the absence of an N-formyl group in this case, Hp(2–20) is a full agonist capable of stimulating superoxide production.
HIV-1 envelope proteins contain peptide sequences capable of interacting with either or both FPR1 and FPR2/ALX, including at least three sequences in gp41 and two in gp120. Although T20/DP178 from gp41 (Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2) specifically activates human FPR1 in vitro (Su et al., 1999c) and mouse Fpr1 in vivo (Hartt et al., 2000), T21/DP107 from gp41 (Ac-NNLLRAIEAQQHLLQLTVWGIKQLQARILAVERYLKDQ-NH2) uses both FPR1 and FPR2/ALX with higher efficacy at FPR2/ALX (Su et al., 1999a). N36 from gp41 (Ac-SGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL-NH2), which partially overlaps with T21/DP107, signals only through FPR2/ALX (Le et al., 2000b). Two peptide domains in HIV-1 gp120 are potent chemoattractants and activators for FPR2/ALX, but not for FPR1, in human phagocytic leukocytes. One of the peptide domains, F peptide, consists of 20 amino acids (Ac-EGSDTITLPCRIKQFINMWQE-NH2) located in the C4-V4 region of gp120 of the HIV-1 Bru strain (Deng et al., 1999). Another peptide (V3 peptide, Ac-RIHIGPGRAFYTTKN-NH2) was derived from a linear sequence of the V3 region of the HIV-1 MN strain (Shen et al., 2000). The existence of these peptides in vivo and their biological significance are not known at present. The peptide T20/DP178 is a licensed antiretroviral agent (pentafuside) administered subcutaneously that acts at the level of HIV-target cell fusion.
In addition to HIV-1 proteins, other viral proteins contain sequences that can serve as ligands for formyl peptide receptors when tested in the form of synthetic peptides. In Herpes simplex virus type 2, a gG-2p20 peptide corresponding to amino acids 190 to 205 of the secreted glycoprotein sgG-2 (GLLWVEVGGEGPGPT) activates neutrophils and monocytes via FPR1 (Bellner et al., 2005). The gG-2p20–induced activation of phagocytes releases reactive oxygen species that inhibits NK cell cytotoxicity and accelerates apoptotic cell death. Therefore, gG-2p20 peptide may contribute to the reduced function and viability of NK cells during HSV-2 infection by activating phagocytic cells. In a more recent study, Mills (2006) identified additional peptides from coronavirus 229E and Ebola virus as FPR1 ligands. Most of these peptides are antagonists, including the coronavirus 229E peptide MYVKWPWYVWL. It is noteworthy that an N-formylated form of this peptide becomes a potent agonist for FPR1. It is important to determine in future studies 1) whether and how these peptides are generated during viral infection and 2) the functional consequences of phagocyte response to these peptides.
3. Host-Derived Peptides.
In addition to mitochondrial formyl peptides discussed above, a large number of endogenous peptides of various compositions, often without an N-formyl group, have been identified as agonists for the formyl peptide receptors, especially FPR2/ALX (Table 5). Of particular interest are peptides associated with amyloidogenic diseases, peptides associated with inflammatory and antibacterial responses, and a peptide derived from heme-binding protein that serves as a potent endogenous agonist at FPR3.
a. Peptides Associated with Amyloidogenic Diseases.
At least three amyloidogenic polypeptides, associated with chronic inflammation and amyloidosis, have been identified as agonists for FPR2/ALX. Serum amyloid A (SAA) is an acute-phase protein the serum concentration of which is increased by as much as 1000-fold in response to trauma, acute infection, and other environmental stress causing acute-phase responses (Kushner and Rzewnicki, 1999). Studies with recombinant human SAA identified it as the first mammalian host-derived chemotactic peptide ligand for FPR2/ALX (Su et al., 1999b). SAA, acting through FPR2/ALX, is chemotactic for monocytes, neutrophils, mast cells, and T lymphocytes; stimulates production of metalloproteases and cytokines; and increases expression of cytokine receptors. In neutrophils, SAA activates FPR2/ALX and induces CXCL8 (IL-8) secretion (He et al., 2003). In monocytes, SAA shows a peculiar pattern of cytokine induction via FPR2/ALX; i.e., the cells respond to low concentrations of SAA by producing TNF-α while releasing IL-10 in response to high concentrations of SAA (Lee et al., 2006a). The synovial tissues of patients with inflammatory arthritis express high levels of SAA and FPR2/ALX, and SAA induces the expression of matrix metalloproteinase-1 and -3 in fibroblast-like synoviocytes (O'Hara et al., 2004; Sodin-Semrl et al., 2004). Furthermore, SAA promotes synovial hyperplasia and angiogenesis through activation of FPR2/ALX (Lee et al., 2006b). In addition to using the formyl peptide receptors, SAA activates neutrophil NADPH oxidase through a different receptor (Björkman et al., 2008). More recent studies have identified additional receptors for SAA, including the class B type I scavenger receptor (mouse) and its human ortholog CLA-1 (CD36 and LIMPII Analogous-1) (Baranova et al., 2005; Cai et al., 2005), the receptor for advanced glycation end product (RAGE) (Cai et al., 2007), Toll-like receptor 2 (Cheng et al., 2008), and possibly Toll-like receptor 4 (Sandri et al., 2008). These SAA receptors are involved in SAA regulation of cholesterol metabolism and production of selected cytokines.
Another peptide, the 42-amino acid form of β amyloid peptide (Aβ42, DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA), which is a cleavage product of the amyloid precursor protein in the brain and a pathologic protein in Alzheimer's disease, was also found to activate FPR2/ALX (Le et al., 2001a; Tiffany et al., 2001). An additional amyloidogenic disease-associated FPR2/ALX agonist is prion protein fragment PrP(106–126), which is produced in human brains with prion disease (Le et al., 2001c). FPR2/ALX mediates the migration and activation of monocytic phagocytes, including macrophages and brain microglia, induced by β amyloid (Aβ) (Le et al., 2001a; Chen et al., 2006). Moreover, FPR2/ALX promotes the endocytosis of Aβ by macrophages and microglia in vitro in the form of receptor and ligand complexes (Yazawa et al., 2001; Chen et al., 2006). If the exposure of macrophages to Aβ42 is transient, the internalized Aβ42 is degraded, and FPR2/ALX is rapidly recycled back to the cell surface. But prolonged exposure results in accumulation of the Aβ42 and FPR2/ALX complex in macrophages, culminating in progressive fibrillary aggregation of Aβ42 and macrophage death (Yazawa et al., 2001). Therefore, FPR2/ALX not only mediates the proinflammatory activity of the peptide agonists associated with amyloidogenic diseases, it may also participate in the regulation of fibrillary peptide formation and deposition, which are pathologic features of the diseases that contribute to tissue and organ destruction (Cui et al., 2002). The in vivo significance of this to the pathogenesis of Alzheimer's disease is not yet known.
Humanin is a peptide encoded by a cDNA cloned from a relatively healthy region of an Alzheimer's disease brain (Hashimoto et al., 2001). Both secreted and synthetic humanin peptides protect neuronal cells from damage by Aβ42. Humanin uses FPR2/ALX, FPR3 and mouse Fpr2 as functional receptors to induce chemotaxis of mononuclear phagocytes (Harada et al., 2004; Ying et al., 2004). In addition, humanin reduces aggregation and fibrillary formation by suppressing the interaction of Aβ42 with mononuclear phagocytes through FPR2/ALX. Human neuroblastoma cell lines express functional FPR2/ALX but not FPR1. In these cells, although humanin and Aβ42 both activate FPR2/ALX, only Aβ42 causes apoptotic death of the cells, a process blocked by humanin. These observations suggest that humanin may exert its neuroprotective effects by competitively inhibiting the access of Aβ42 (Ying et al., 2004).
b. Peptides Associated with Inflammatory and Antibacterial Responses.
Urokinase-type plasminogen activator (uPA) is a serine protease known for its ability to regulate fibrinolysis. uPA binds to a specific high affinity surface receptor (uPAR). In addition to regulating fibrinolysis, the uPA-uPAR system is crucial for cell adhesion and migration and tissue repair. uPAR contains three domains: D1 (binding uPA), D2, and D3. A D3-bound glycosylphosphatidylinositol anchor links uPAR to the plasma membrane. uPAR can be cleaved by different proteases, including uPA, in the D1-D2 linker region. The cleaved soluble uPAR D2D388–274 binds and activates FPR2/ALX in monocytes, inducing cell migration (Resnati et al., 2002). The ability of cleaved soluble uPAR (c-suPAR) to activate other members of the FPR family has been reported. For instance, SRSRY, a peptide corresponding to residues 88 to 92 of uPAR, binds to and activates FPR1 (Gargiulo et al., 2005). uPAR84–95 induces basophil migration by activating both FPR2/ALX and FPR3 (de Paulis et al., 2004a). Recent studies show that uPAR is involved in granulocyte-colony-stimulating factor (G-CSF)-induced mobilization of CD34+ hematopoietic stem cells. G-CSF increases the expression of uPAR on circulating CD34+ and CD14+ cells. This is associated with increased uPAR shedding, leading to the appearance of serum c-suPAR. c-suPAR mobilizes hematopoietic stem cells by promoting their FPR1-mediated migration and by desensitization of the chemokine receptor CXCR4 (Selleri et al., 2005). Additional studies showed that pretreatment of monocytes with the FPR2/ALX agonist D2D388–274 markedly decreases chemokine-induced integrin-dependent rapid cell adhesion (Furlan et al., 2004), indicating that the FPR family receptors regulate leukocyte chemotaxis at multiple levels, i.e., in addition to being direct mediators of cell migration, they may suppress cell responses to chemokines by desensitizing chemokine receptors.
Formyl peptide receptors interact with several bactericidal peptides contained in human neutrophil granules. LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES), an enzymatic cleavage fragment of the neutrophil granule protein cathelicidin (Yang et al., 2000), and its mouse homolog CRAMP (GLLRKGGEKIGEKLKKIGQKIKNFFQKLVPQPEQ) (Kurosaka et al., 2005) are agonists for FPR2/ALX. LL-37 is expressed by leukocytes and epithelial cells and secreted into wounds and onto the airway surface. In addition to its microbicidal activity, LL-37 induces directional migration of human monocytes, neutrophils, and T lymphocytes, a function mediated by FPR2/ALX (Yang et al., 2000). Recent studies showed that LL-37-induced angiogenesis is mediated by FPR2/ALX in vascular endothelial cells. Decreased vascularization during wound repair observed in mice deficient for CRAMP indicates that cathelicidin-mediated angiogenesis is important for cutaneous wound neovascularization in vivo (Koczulla et al., 2003). LL-37 seems to be a multifunctional peptide with a central role in innate immunity against bacterial infection and in the induction of arteriogenesis important for angiogenesis. Another antibacterial granule protein, cathepsin G, which is a serine protease and participates in wound healing, is identified as a specific agonist for FPR1 (Sun et al., 2004).
FPR2/ALX is also known to interact with a chemokine variant that potently activates phagocytic leukocytes. CCL23/MPIF-1 (CKβ8–1) uses a typical G protein-coupled receptor CCR1 for its leukocyte chemotactic activity. However, an N-terminally truncated form of the CCL23 splice variant CCL23β (amino acids 22–137) activates myeloid cells and FPR2/ALX transfected cell lines at low nanomolar concentration range, making it one of the most potent FPR2/ALX agonists identified so far (Elagoz et al., 2004). A more recent study identified a mechanism of CCL23β processing that involves sequential cleavage of the chemokine in vitro with proinflammatory proteases, generating CCR1-specific chemokine and an 18-amino acid peptide, termed SHAAGtide (MLWRRKIGPQMTLSHAAG), that activates FPR2/ALX at nanomolar concentrations (Miao et al., 2007). This study illustrates a novel mechanism by which protease cleavage of a chemokine produces two peptides acting on two different receptors. It will be interesting to determine whether the cleavage product is generated in vivo and how the simultaneous activation of two receptors influences the course of inflammation.
c. Annexin A1 and Its N-terminal Peptides.
ANXA1 and its N-terminal peptides have interesting properties in activating formyl peptide receptors by playing dual roles in inflammatory host responses. ANXA1 (also termed lipocortin I) is a glucocorticoid-regulated, phospholipid-binding protein of 37 kDa that possesses both pro- and anti-inflammatory activity, mediated in part by activating FPR1 (Ernst et al., 2004a). Expressed in a variety of cell types, ANXA1 is particularly abundant in neutrophils. The protein is primarily cytosolic, but it may also be secreted through a nonclassic secretory process and found on the outer cell surface, causing leukocyte detachment and thereby inhibiting their transendothelial migration (for review, see Perretti, 2003). At low concentrations, both ANXA1 holoprotein and its N-terminal peptides (Ac2–26, Ac-AMVSEFLKQAWFIENEEQEYVQTVK, and Ac9–25) elicit Ca2+ transients through FPR1 without fully activating the MAPK pathway (Ernst et al., 2004a), causing neutrophil desensitization and inhibition of transendothelial migration induced by other chemoattractants such as the chemokine IL-8 (CXCL8). In contrast, at high concentrations, the ANXA1 peptides fully activate neutrophils in vitro and become potent proinflammatory stimulants. The antimigratory activity of exogenous and endogenous ANXA1 has been shown in both acute and chronic models of inflammation (Perretti, 2003). Fpr1 knockout mice exhibit normal neutrophil accumulation during thioglycolate-elicited peritonitis (Perretti et al., 2001). However, a significant part of the effect in reducing intraperitoneal neutrophil infiltration, observed in ANXA1-treated wild type mice, was abolished in the Fpr1(-/-) mice. Other studies have shown that the ANXA1 N-terminal peptides use FPR2/ALX for its anti-inflammatory actions (Perretti et al., 2002). The peptides are also ligands for FPR3 (Ernst et al., 2004a). The ANXA1 core-derived peptide antiinflammin 2 (amino acids 246–254; HDMNKVDK) activates FPR2/ALX (Kamal et al., 2006). The utilization of the formyl peptide receptors by ANXA1 and its amino terminal peptides for their various functions is a complex issue. One published report demonstrates that Ac9–25 stimulates neutrophil NADPH oxidase activation through FPR1, but its inhibitory effect is mediated through a receptor other than FPR1 or FPR2/ALX (Karlsson et al., 2005), suggesting the presence of additional receptors for ANXA1 and its peptides. ANXA1 has been shown to bind to the α4β1 integrin on undifferentiated U937 cells that do not express FPR1 or FPR2/ALX and inhibits U937 adhesion to microvascular endothelial cells (Solito et al., 2000).
d. F2L, a Potent Endogenous Agonist for FPR3.
In 2005, Migeotte et al. (2005) reported the purification of a highly potent and efficacious agonist peptide for human FPR3, termed F2L. F2L is an amino-terminally acetylated peptide (Ac-MLGMIKNSLFFGSVETWPWQVL-NH2) resulting from the natural cleavage of human heme-binding protein, an intracellular tetrapyrole-binding protein. The peptide binds and activates FPR3 in the low nanomolar range, triggering typical G protein-mediated intracellular calcium release, inhibition of cAMP accumulation, and phosphorylation of the ERK 1/2 MAPKs. F2L also chemoattracts and activates monocyte-derived DCs. Thus, F2L seems to be a novel and unique natural chemotactic peptide for FPR3 in DCs and monocytes, in agreement with the selective expression of FPR3 in these cells (Yang et al., 2002). F2L may play a role in linking innate and adaptive immune responses by activating antigen-presenting FPR3+ DCs, which express little FPR1 and FPR2/ALX. It is interesting to note that in mice, F2L activates the FPR2/ALX homolog mFpr2 (Gao et al., 2007). In bone marrow-derived neutrophils deficient in Fpr2, the activity of F2L was totally lost, suggesting that in mice this function of human FPR3 is carried out by Fpr2. The overlapping function of mouse Fpr2 with human FPR3 in binding F2L provides another example for the complexity in studying the FPR family receptors using genetically altered mice.
e. Other Host-Derived Peptides.
Pituitary adenylate cyclase-activating polypeptide 27 (HSDGIFTDSYSRFRKQMAVKKYLAAVL-NH2) is an agonist for FPR2/ALX, stimulating chemotaxis, up-regulation of CD11b, and activation of phagocytes (Kim et al., 2006). Temporin A (TA; FLPLIGRVLSGIL-NH2) is a frog-derived antimicrobial peptide found to induce the migration of human monocytes, macrophages, and neutrophils (Chen et al., 2004). Characterization of the signaling properties of TA in monocytes and the use of receptor-transfected human embryonic kidney (HEK) 293 cells revealed that this peptide uses FPR2/ALX as a receptor. TA is also chemotactic in vivo because it elicited infiltration of neutrophils and monocytes into the injection site in mice. Another temporin peptide, Ranatuerin-6 (Rana-6; FISAIASMLGKFL) also uses FPR2/ALX for chemotaxis of human phagocytes (Chen et al., 2004). The biological and evolutionary significance of these findings is still unclear because, so far, formyl peptide receptors have not been characterized in frogs, and TA homologs have not been characterized in mammals.
4. Host-Derived, Nonpeptide Agonists.
LXA4 (5S,6R,15S-trihydroxy-7,9,13-trans-11-eicosatetraenoic acid) is a potent mediator biosynthesized from arachidonic acid. It is a small molecule with physical chemical properties that differ from most lipids: it has a unique structure and belongs to a class of conjugated tetraene-containing eicosanoids that display stereoselective and highly potent anti-inflammatory and pro-resolving activity in vivo in many mammalian systems (for review, see Serhan, 2005) and articles contained in the special issue). In this regard, LXA4 is unusual in that most other eicosanoids are pro-inflammatory. As an endogenous mediator, LXA4 displays multilevel control of processes relevant in acute inflammation via specific and selective actions on multiple cell types via specific receptors (Serhan, 2007). In particular, LXA4 has been reported to interact directly with both human FPR2/ALX and CysLT1. It also induces signals that regulate BLT1, and production of chemokines, cytokines (e.g., TNF), and growth factor receptors (e.g., vascular endothelial growth factor receptor) in human leukocytes, vascular cell types, and mucosal epithelial cells, each contributing to regulate the resolution of inflammation (Serhan, 2005).
LXA4 is the first identified endogenous ligand for FPR2/ALX. Using freshly isolated radioligand for each experiment, specific LXA4 binding sites were initially characterized with isolated human neutrophils (Fiore et al., 1992) and demonstrated to be responsible for the specific LXA4 functions on neutrophils. LXA4 stimulates rapid (within seconds) phospholipase activation in these cells that directly correlates with the induction time course of specific LXA4 binding (Nigam et al., 1990; Fiore et al., 1993). In parallel experiments, a second approach was undertaken with GPCRs that were known to be induced within the same time frame in the promyelocytic cell line HL-60 when differentiated into neutrophils by treatment with either retinoic acid or dimethylsulfoxide. These cells were screened for [3H]LXA4 binding and ligand-dependent increases in GTPase activity. Labeled LXA4 gives high-affinity binding with FPR2/ALX as well as ligand selectivity, compared with other eicosanoids including LXB4, leukotriene B4, leukotriene D4, and PGE2. FPR2/ALX expressed in Chinese hamster ovary cells transfected with the FPR2 cDNA pINF114 gave a Kd of ∼1.7 nM for [3H]LXA4 determined by Scatchard plot analysis (Fiore et al., 1994). This value for recombinant FPR2/ALX is comparable with those obtained for endogenous LXA4 specific binding sites present on peripheral blood neutrophils (e.g., Kd values of 0.7 and 0.8 nM obtained with isolated plasma membrane fractions and granule membrane-enriched fractions, respectively). In Chinese hamster ovary cells expressing the recombinant receptor, LXA4 stimulates both GTPase activity and the release of esterified arachidonic acid, which is inhibited by pertussis toxin (Fiore et al., 1994).
Each of the actions of LXA4 proved to be stereoselective in that changes in potencies are associated with double bond isomerization and alcohol chirality (R or S) as well as dehydrogenation of alcohols and reduction of double bonds. Elimination of the carbon 15 position alcohol from LXA4, denoted 15-deoxy-LXA4, is essentially inactive in vivo and does not stop either neutrophil transmigration or reduce adhesion (Serhan et al., 1995). In nature, LXA4 is enzymatically inactivated by conversion to 15-oxo-LXA4 and 13,14-dihydro-LXA4 (Serhan et al., 1993). The biologically inactive metabolic products and synthetic compounds tested, including 15-oxo-LXA4, 15-deoxy-LXA4, 11-trans-LXA4, and 13,14-dihydro-LXA4, do not effectively bind to FPR2/ALX. This contrasts with the active lipoxin ligands that include native LXA4, which carries a 15S configuration alcohol that originates from the lipoxygenase biosynthetic pathway and the aspirin-triggered 15R- or 15-epi-LXA4 (also denoted ATL or aspirin-triggered LXA4) biosynthesized in the presence of aspirin, each exhibiting stereospecific receptor binding to both human FPR2/ALX and the murine counterpart encoded by Fpr-rs1 (Takano et al., 1997).
Evidence supporting the anti-inflammatory and proresolving functions of LXA4 comes from a number of in vivo studies, as summarized in several recent reviews (Serhan, 2005; Chiang et al., 2006; O'Meara et al., 2008). These in vivo studies have shown that, in nanogram amounts, LXA4 stops neutrophil infiltration (Takano et al., 1997, 1998; Hachicha et al., 1999) and blocks human neutrophil transmigration across mucosal epithelial cells and vascular endothelial cells (Colgan et al., 1993; Kucharzik et al., 2003). One of the mechanisms by which LXA4 and ATLs inhibit neutrophil infiltration is through induction of NO production, which suppresses leukocyte-endothelial cell interaction (Paul-Clark et al., 2004). LXA4 treatment inhibits proinflammatory cytokine production by synoviocytes (Sodin-Semrl et al., 2000), intestinal epithelial cells (Goh et al., 2001; Kucharzik et al., 2003), and bronchial epithelial cells (Bonnans et al., 2007). Several studies have shown that LXA4 inhibits TNFα-induced production of cytokines such as IL-1β and IL-6 (Wu et al., 2005) and chemokines such as IL-8 (Bonnans et al., 2007), as well as LPS-induced secretion of IL-1β, IL-6, and IL-8 (Wu et al., 2008). Dendritic cell production of the immunomodulatory cytokine IL-12 is also regulated by LXA4, as shown in a model of Toxoplasma gondii infection (Aliberti et al., 2002a,b). Part of the anti-inflammatory effect of LXA4 is known to involve the inhibition of transcription factors, including NF-κB and activator protein 1, that are responsible for the expression of many proinflammatory cytokines and chemokines (Gewirtz et al., 2002; József et al., 2002; Sodin-Semrl et al., 2004; Wu et al., 2008). Whereas LXA4 suppresses the expression of these proinflammatory cytokines, it stimulates the expression of the anti-inflammatory molecule IL-10 (Souza et al., 2007) and the antioxidant molecule heme oxygenase-1 (Nascimento-Silva et al., 2005). In synovial fibroblasts, LXA4 inhibits the expression of matrix metalloproteinase-3 but enhances the synthesis of tissue inhibitors of metalloproteinases (Sodin-Semrl et al., 2000). LXA4 has also shown antifibrotic effects as it regulates PDGF-induced genes such as TGF-β in renal mesangial cells (Rodgers et al., 2005). In addition to its inhibition of proinflammatory cytokine production, LXA4 plays a role in regulating inflammation-induced pain (Svensson et al., 2007). This function is probably mediated through alteration of spinal nociceptive processing through astrocyte activation.
Besides its anti-inflammatory functions, LXA4 and its stable analog exhibit pro-resolution properties (Serhan, 2007). One of the mechanisms involves overriding the apoptosis-delaying effect of proinflammatory factors such as SAA, thereby promoting neutrophil apoptosis (El Kebir et al., 2007). LXA4 also stimulates nonphlogistic phagocytosis of apoptotic neutrophils, which accelerates the clearance of neutrophils and promotes resolution of inflammation (Godson et al., 2000). These examples show that, in general terms, LXA4 actively inhibits many endogenous processes that can amplify local acute inflammation, leading to potent anti-inflammatory as well as pro-resolving actions in vivo.
The multilevel control by LXA4 on processes relevant to acute inflammation raises the intriguing question of how LXA4 binding to FPR2/ALX might translate into the anti-inflammatory and pro-resolving activities and whether other receptors contribute to these activities. Evidence supporting a role of FPR2/ALX in mediating the anti-inflammatory effects of LXA4 came from studies using Boc2, stereoselective analogs, antibodies, and transgenic approaches. Boc2 is a peptide antagonist for both FPR1 and FPR2/ALX as detailed in the following section. Its effect on blocking FPR2/ALX in inflammation models was first reported by Gavins et al. (2003) and confirmed by others (von der Weid et al., 2004; Nascimento-Silva et al., 2005; Machado et al., 2006; Grumbach et al., 2009). Transgenic overexpression of FPR2/ALX resulted in mice that display reduced neutrophil infiltration when challenged with LTB4 and PGE2. The transgene also enhanced the response to aspirin-triggered lipoxins in a mouse model of zymosan-induced peritonitis (Devchand et al., 2003). Binding of LXA4 to FPR2/ALX is stereo-selective, because other hydroxyl-containing eicosanoids, including LXB4, LTB4, and LXA4 stereoisomers, did not compete for [3H]LXA4 binding specifically (Fiore et al., 1992). However, the finding that LXB4 also exhibits anti-inflammatory functions (Maddox and Serhan, 1996) suggest the possible presence of another functional receptor that differs from FPR2/ALX for the lipoxins. One observed difference between the two receptors is that LXB4 does not induce calcium mobilization in the monocytic cell line THP-1, whereas LXA4 does (Romano et al., 1996). It is noteworthy that the aryl hydrocarbon receptor (AhR) has also been identified as a functional receptor for LXA4 (Schaldach et al., 1999). AhR is not a GPCR and is located intracellularly, whereas FPR2/ALX is found on the plasma membrane. AhR binding of LXA4 renders the receptor capable of interacting with a dioxin responsive element, which can direct expression of a reporter gene with an EC50 of 50 nM for LXA4. It was recently shown that LXA4 acts on both FPR2/ALX and AhR to exert its anti-inflammatory effect in dendritic cells through induction of suppressor of cytokine signaling-2 (SOCS-2) (Machado et al., 2006, 2008). Whether AhR is a receptor for LXB4 and the relative contribution of each of these receptors to the anti-inflammatory activities of the lipoxins has yet to be determined.
5. Agonists from Peptide Library.
Using combinatorial peptide library screens, a number of peptides have been identified as potent agonists for the formyl peptide receptors. Trp-Lys-Tyr-Met-Val-d-Met-NH2 (WKYMVm), a hexapeptide representing a modified sequence isolated from a random peptide library, was found to be a highly efficacious stimulant for human B lymphocytes, monocytic cell lines, and blood neutrophils (Baek et al., 1996). WKYMVm uses FPR1, FPR2/ALX, and FPR3 for activation of human phagocytic cells (Le et al., 1999; Kang et al., 2005). WKYMVm is by far the most potent peptide agonist for FPR2/ALX, with an EC50 well within the picomolar range in chemotaxis assays. WKYMVM, a derivative of WKYMVm with an l-methionine at the carboxyl terminus, is a highly selective agonist for FPR2/ALX and is also a weaker activator of FPR3 (Christophe et al., 2001). A recent study investigated the relationship between FPR1 and FPR2/ALX, both found in neutrophils, in mediating the WKYMVm-stimulated cellular functions. It was found that WKMYVm activates neutrophils through FPR1 only when the signal through FPR2/ALX is blocked (Karlsson et al., 2006), suggesting its preference for one type of receptor despite the presence of two different types of receptors in neutrophils. WKYMVm also inhibits LPS-induced maturation of human monocyte-derived dendritic cells via FPR1 and FPR3, presumably through interference with signaling pathways activated by Toll-like receptor 4 (Kang et al., 2005).
The peptide MMK-1 (LESIFRSLLFRVM) was identified from a library screen in genetically engineered Saccharomyces cerevisiae cells designed to couple receptor (FPR2/ALX) activation to histidine prototrophy (Klein et al., 1998). Selection for histidine prototrophs among transformants obtained with a plasmid-based random peptide library identified several agonists, each of which yielded autocrine stimulation of the receptor expressed in yeast. MMK-1 was found to be a highly selective chemotactic agonist for FPR2/ALX (Klein et al., 1998; Hu et al., 2001). In another study, a tethered library was screened based on the activation mechanism of the thrombin receptor PAR-1 (Chen et al., 1995). A peptide with the sequence MMWLL was identified as an FPR1 agonist. This peptide becomes 1000 times more potent when N-formylated, consistent with the preferential recognition of N-formylmethionine-containing peptides by FPR1.
A recent report shows that a peptide library-derived ligand of 21 amino acids, CGEN-855A (TIPMFVPESTSKLQKFTSWFM-amide), exhibits anti-inflammatory properties (Hecht et al., 2009). CGEN-855A displaced 125I-WKMYVm binding to FPR2/ALX with an IC50 value of 189 nM (pIC50 = 6.72) and a Ki value of 54.1 nM (pKi = 7.27). CGEN-855A does not affect monocyte secretion of cytokines. It has anti-inflammatory activity in the zymosan-triggered air pouch inflammation model, and displays cardioprotection in a mouse model of ischemia/reperfusion-induced myocardial infarction (Hecht et al., 2009). The study indicates that peptides such as ANXA1 N-terminal fragments and CGEN-855A have functional properties similar to those of LXA4.
6. Agonists from Nonpeptide Library.
Several laboratories have recently identified ligands for the formyl peptide receptors through screening of combinatorial libraries consisting of synthetic, nonpeptide compounds. These synthetic small molecules are of different structures (see Fig. 5) and are highly selective for either FPR1 or FPR2/ALX, providing useful tools for the characterization of formyl peptide receptors. The pharmacological properties of these compounds also make them potentially useful for therapeutic intervention.
Nanamori et al. (2004a) first reported the identification of a quinazolinone derivative named Quin-C1 as a highly selective agonist for FPR2/ALX. Quin-C1 (4-butoxy-N-[2-(4-methoxy-phenyl)-4-oxo-1,4-dihydro-2H-quinazolin-3-yl]-benzamide) induces chemotaxis and secretion of β-glucuronidase in peripheral blood neutrophils with a potency of approximately 1/1000 that of the peptide agonist WKYMVm, which is the most potent agonist for FPR2/ALX identified so far. In studies using RBL cells expressing either FPR1 or FPR2/ALX, Quin-C1 induced enzyme release only from cells expressing the latter. Quin-C1 selectively stimulates calcium mobilization in RBL cells expressing recombinant FPR2/ALX, induces phosphorylation of ERK1/2, and promotes internalization of FPR2/ALX fused to an enhanced green fluorescent protein. However, unlike most peptide agonists of FPR2/ALX, Quin-C1 did not induce substantial neutrophil superoxide generation, even at concentrations up to 100 μM. The structural basis for this biased agonistic activity is still unknown but may be beneficial for its potential use as a therapeutic agent.
Bürli et al. (2006) characterized and subsequently modified a series of compounds initially identified from a cell-based assay for high-throughput screening. These ligands are also highly selective for FPR2/ALX and exhibit anti-inflammatory properties in a mouse ear swelling assay. Two of the pyrazolones (compounds 24 and 43) tested in their study were able to stimulate calcium flux in transfected cells expressing FPR2/ALX, whereas other structural analogs were ineffective. Compound 43 was formulated for oral administration and was found to significantly reduce inflammation in an ear swelling model in mice.
Using a strategy combining computer model-based virtual screening and high-throughput, no-wash cytometry screening, Edwards et al. (2005) identified 30 lead compounds (from an initial pool of 480,000) that are partial agonists or antagonists for FPR1. The in silico virtual screening, based on a bovine rhodopsin crystal structure, effectively boosted the physical screen hit rate by 12-fold, eliminating most of the compounds before physical screen. The pharmacophore model for FPR1 developed in this study may be useful in future identification of agonists and antagonists for this class of receptors.
Schepetkin et al. (2007) used a different approach for compound screening. They started in a neutrophil superoxide production assay, combined with substructure screen, fragment focusing, and structure-activity relationship analyses to identify t-butyl benzene and thiophene-2-amide-3-carboxylic ester derivatives as potential agonists for neutrophil chemoattractant receptors. Eleven of the compounds were further analyzed in functional assays and one (AG-14) was found to activate neutrophils at nanomolar concentrations. Based on desensitization and antagonist inhibition data, the investigators concluded that AG-14 is an agonist for FPR1. These investigators also identified arylcarboxylic acid hydrazide derivatives as agonists for FPR2/ALX that induce de novo production of TNFα through activation of macrophages (Schepetkin et al., 2008).
B. Antagonists for the Formyl Peptide Receptor Family of Receptors
Early studies showed that replacing the formyl group of fMLF with tertiary butyloxycarbonyl group (t-Boc) renders the peptide antagonistic (Freer et al., 1980). t-Boc-Met-Leu-Phe (Boc1) and t-Boc-Phe-d-Leu-Phe-d-Leu-Phe (Boc2) are two frequently used antagonists for FPR1, with pIC50 values of 6.19 and 6.59, respectively (Freer et al., 1980) (Table 6). The N-ureido-substituted Phe-d-Leu-Phe-d-Leu-Phe was found to also have antagonistic activity (Higgins et al., 1996). It is noteworthy that whereas t-Boc-Phe-d-Leu-Phe-d-Leu-Phe-OMe is a full antagonist, the peptide with the l-Leu, t-Boc-Phe-Leu-Phe-Leu-Phe-OMe, has agonistic activity (Toniolo et al., 1990). In several recent studies, Boc2 was used at high concentrations (e.g., 100 μM) for inhibition of FPR2/ALX (Gavins et al., 2003; Machado et al., 2006). A recent study has shown that, when used at low micromolar concentrations, both Boc1 and Boc2 are selective antagonists for FPR1; at high micromolar concentrations, Boc2 partially inhibits FPR2/ALX in addition to FPR1 (Stenfeldt et al., 2007). Therefore, the antagonistic effect of Boc2 at high concentrations is not specific for FPR2/ALX. In another ligand screen, the peptide WRWWWW was identified as a highly selective antagonist for FPR2/ALX (Bae et al., 2004), with a pIC50 of 6.64 in calcium flux assay. As an antagonist, WRWWWW is more selective at FPR2/ALX than Boc2 (Stenfeldt et al., 2007).
Antagonists for the human formyl peptide receptors Antagonists for the FPRs are listed in the order of their approximate potency, except that antagonists of same types are listed together.
Cyclosporin H (CsH) is a cyclic undecapeptide produced by fungi. It is an optical isomer of the immunosuppressant CsA and contains at its position 11 the amino acid residue N-methyl-d-valine, in place of the N-methyl-l-valine as seen in CsA (von Wartburg and Traber, 1986). CsH lacks immunosuppressive activity but displays selective antagonistic activity at human FPR1 (Wenzel-Seifert et al., 1991). Studies have shown that CsH is 14-fold more potent than the tertiary butyloxycarbonyl analogs of formyl peptides such as Boc2 in FPR1 binding assays, and approximately 5-fold more potent than Boc2 in the inhibition of fMLF-induced calcium flux and enzyme release (Wenzel-Seifert and Seifert, 1993). CsH is an inverse agonist (negative antagonist) that suppresses the constitutive activity of FPR1 in a GTPase activity assay (Wenzel-Seifert et al., 1998). The biological significance of constitutive activity for FPR is not established. Both Boc2 and CsH competitively displace FPR1-bound [3H]fMLF, indicating that its antagonistic activity is mediated through inhibition of fMLF binding.
A study of CsA on differentiated HL-60 myeloid cells has led to the finding that CsA also inhibits fMLF-induced degranulation, although the inhibitory effect is less potent than CsH (Loor et al., 2002). In a more recent study, Yan et al. (2006) reported that CsA dose-dependently suppresses fMLF- and WKYMVm-induced cell activation and inhibits their binding to FPR1. It has not been determined whether CsA is an inverse agonist. Neither CsA nor CsH displays any detectable inhibitory effect on FPR2/ALX-mediated cellular functions.
Two types of endogenous FPR1 antagonists have been identified. Spinorphin (LVVYPWT), an opioid, is an endogenous peptide antagonist for FPR1 with a pIC50 of 4.30 (Yamamoto et al., 1997; Liang et al., 2001). The bile acids deoxycholic acid (DCA) and chenodeoxycholic acid (CDCA) are two other identified antagonists for FPR1 (Chen et al., 2000, 2002). The pIC50 values for DCA and CDCA are 4 and 3.76, respectively. The physiological functions of DCA- and CDCA-mediated FPR1 antagonism have not been fully understood.
Chemotaxis inhibitory protein of S. aureus (CHIPS) is a bacteria-derived protein of 14.1 kDa found in more than half of the clinical strains of S. aureus. CHIPS has antagonistic activity for FPR1 and C5aR. An N-terminal peptide (FTFEPFPTNEEIESN) derived from CHIPS is a selective antagonist for FPR1 but not C5aR (Haas et al., 2004). Iodinated CHIPS exhibits a pKd of 7.46 at FPR1, which is by far the most potent peptide antagonist for this receptor. The identification of a bacteria-derived FPR1 antagonist suggests a mechanism used by microorganisms to thwart host defenses.
In a subsequent study, the same group reported the identification from S. aureus of a 105-amino acid protein, termed FPRL1 inhibitory protein (FLIPr), that selectively inhibits the binding of and activation by FPR2/ALX agonists including MMK-1, WKYMVM, the prion fragement PrP106–126, and amyloid peptide Aβ1–42 (Prat et al., 2006). At higher concentrations, FLIPr also inhibits fMLF binding to FPR1. FLIPr was found to bind directly to FPR2/ALX and FPR1, but not to FPR3 and C5aR. It does not interfere with LXA4 activity on LTB4. The biological function of this inhibition has not been identified.
Quin-C7 is a synthetic, nonpeptide antagonist of FPR2/ALX, developed through chemical modification of the FPR2/ALX agonist Quin-C1 (Zhou et al., 2007). In binding assays, Quin-C7 inhibited iodinated WKYMVm binding to FPR2/ALX with a pKi of 5.18. This antagonist is highly selective for FPR2/ALX, as it does not affect the binding of [3H]fMLF to transfected cells expressing FPR1 (Zhou et al., 2007).
In summary, several FPR1 and FPR2/ALX antagonists have been identified and characterized. It is notable that, in most cases, these antagonists differ considerably from the identified FPR1 and FPR2/ALX agonists. The observation that t-Boc peptides are antagonistic while N-formyl peptides of the same or similar composition are agonistic may be helpful to define the binding pocket of these peptides in FPR1. The synthetic, nonpeptide antagonist, Quin-C7 differs from the agonist Quin-C1 only in the para position of the phenyl ring (Zhou et al., 2007). Its antagonistic activity on FPR2/ALX provides a potentially useful tool in study the binding properties of FPR2/ALX.
IV. Structure-Function Relationship of the Formyl Peptide Receptor Family of Receptors
A. Ligand-Binding Domains
The spectrofluorimetric and flow cytometric techniques developed by Sklar et al. (1981, 1987) and Posner et al. (1994) vastly expanded our knowledge of the dynamics of FPR1 and the properties of its ligand binding pocket. These studies are based on N-formylated hexapeptides, modified with fluorescein, and used in conjunction with a high-affinity antibody directed against the fluorescein moiety. The antibody possessed the property to capture free fluoresceinated peptide within a few seconds; the peptide-antibody complex neither stimulates the cells nor inhibits subsequent stimulation by an unlabeled formyl peptide. Using a series of fluoresceinated N-formyl peptides with four, five, or six amino acids, Sklar et al. (1990) showed that the chromophore of peptides containing four and five amino acids was not exposed to the extracellular environment, whereas the fluorescein moiety of the hexapeptides was accessible to high concentrations of antibodies. These results indicate that the binding pocket can accommodate no more than five amino acids and, like many GPCRs, that interact with small ligands, the binding pocket for fMLF is believed to involve amino acids inserted into the transmembrane domains.
With the cloning of FPR1 and other chemoattractant receptors, such as human FPR2/ALX, murine and rabbit FPR1, and the C5a receptor C5aR, it became theoretically possible to discriminate residues that are critical for the formation of a high-affinity binding site for these peptides. A number of groups have used chimeric receptor approaches to define the molecular determinants involved in ligand recognition. After the observation that FPR2/ALX binds fMLF with an affinity that is 400-fold lower than that of FPR1 (Ye et al., 1992; Quehenberger et al., 1993), receptor chimeras were constructed by sequential replacements of FPR1 segments with the corresponding regions in FPR2/ALX or vice versa (Gao and Murphy, 1993; Quehenberger et al., 1993). Comparison of the functional differences of the FPR1-FPR2/ALX chimeras has allowed the identification of domains that may be essential for binding of fMLF (Quehenberger et al., 1993) and for functions induced by fMLF (Le et al., 2005). The replacement of the first and third extracellular loops of FPR with those of FPR2/ALX resulted in a dramatic decrease in the affinity of fMLF for the chimeras. Conversely, simultaneous replacement of the corresponding loops in FPR2/ALX by those of FPR1 resulted in a significant increase in ligand-binding affinity with a Kd that shifted from 400 to 18 nM (Quehenberger et al., 1993). Thus, the first and the third extracellular loops and their adjacent transmembrane domains seem to be essential for high-affinity binding of fMLF. Using a similar approach of domain swapping between FPR1 and C5aR, Perez et al. (1993) proposed a ligand binding model in which the second, third, and fourth extracellular domains and/or their adjacent transmembrane domains together with the first transmembrane domain form a binding pocket for N-formyl peptides. The extracellular N- and C-terminal domains were found to affect ligand binding. These authors proposed that the extracellular N-terminal region provide a “lid” to the pocket. However, the role of the extracellular amino terminus of FPR1 is questionable, because other studies have indicated that the N-terminal region is dispensable for high-affinity binding of fMLF (Malech et al., 1985; Mery and Boulay, 1994).
Based on amino acid differences between the transmembrane domains of FPR1 and FPR2/ALX, Miettinen et al. (1997) prepared FPR1 mutants to identify residues responsible for high-affinity ligand binding. Their study led to the identification of 10 amino acid residues in α helices (transmembrane domains, TM) II to VII that may participate in fMLF binding. The mutations L78A (TM II), D106N, L109A (TM III), T157A (TM IV), R201A, I204Y, R205A (TM V), W254A, Y257A (TM VI), and F291A (TM VII) resulted in reduced affinities for fMLF (Kd = 30–128 nM). Several of these mutations, (i.e., D106N, R201A, and R205A) were found to affect G protein coupling, suggesting that these residues may also be involved in signal transduction and/or are essential for proper folding of the receptor. Quehenberger et al. (1997) used a “gain-of-function” strategy by selectively replacing the nonconserved region of FPR2/ALX with those of FPR1. This approach has allowed the identification of three clusters of amino acids that are able to restore high-affinity fMLF binding to two distinct chimeras (Quehenberger et al., 1993) that exhibited low-affinity fMLF binding. Introduction of two positively charged amino acids, Arg84 and Lys85, markedly improved binding affinity of one chimeric receptor (Kd shifted from 105 to 1.6 nM). Likewise, restoration of either Gly89/His90 or Phe102/Thr103 improved the binding affinity of another chimeric receptor from a Kd of 275 nM to 2.3 or 3.3 nM, respectively. Because a high-affinity binding site for fMLF most likely involves multiple noncontiguous residues that must be positioned by the proper folding of all extracellular and transmembrane domains, it is difficult to locate the ligand binding site precisely or to conclude that these three clusters of amino acids are directly involved in the interaction with fMLF without a crystal structure of the receptor. However, it is worth noting that the identification of amino acids at positions 84 and 85 as key residues for the formation of a high-affinity fMLF binding site is consistent with data derived from an independent photoaffinity labeling approach. Mills et al. (1998) developed an elegant strategy that combined the photolabeling properties of CHO-Met-p-benzoyl-l-phenylalanine-Phe-Tyr-Nϵ-(fluorescein)-Lys-OH, the ability of an anti-fluorescein antibody to immunoprecipitate the cross-linked fluorescein-labeled peptides, and matrix-assisted laser desorption ionization mass spectroscopy for characterization of the fMLF binding pocket in FPR1. They identified a major photolabeled cyanogen bromide peptide, Val-Arg-Lys-Ala-Hse, corresponding to residues 83 to 87 of FPR1, as the photolabeled region. In the current three-dimensional model, this peptide lies at the interface between the second transmembrane domain and the first extracellular loop. A model has been proposed in which the NH2 terminus of an N-formyl peptide is hydrogen-bonded to both Asp106 and Arg201, the leucine side chain of fMLF is close to Val-Arg-Lys85, and the COOH-terminal group of fMLF is ion-paired with Arg205 (Mills et al., 2000).
To evaluate the structural contributions of the major domains from the receptor side via interactions with LXA4 or anti-inflammatory and proinflammatory peptide ligands, chimeric receptors were constructed from FPR2/ALX and BLT1 receptors with opposing functions. These chimeras demonstrated that TM7 and adjacent regions of FPR2/ALX are essential for LXA4 recognition, and additional regions, including extracellular loops, are required for high-affinity binding for peptide ligands such as the MMK-1 and MHC peptides. A single GPCR can recognize and function with specific chemotactic peptides as well as nonpeptide ligands such as LXA4. Clearly, however, these ligands act with different affinities and/or at separate interaction sites within the receptor (Chiang et al., 2000). It is noteworthy that conserved N-glycosylation sites are present on Asn4 and Asn179 of human FPR2/ALX, and bacterial and viral infection are known to interfere with normal N-glycosylation of the host cells (Olofsson et al., 1980; Kim and Cunningham, 1993; Villanueva et al., 1994). In this regard, deglycosylation of FPR2/ALX does not dramatically alter LXA4 recognition but significantly lowers the affinity for peptide ligands (Chiang et al., 2000). Thus, N-glycosylation is essential for ligand specificity of this receptor and may play an important role in switching or changing this receptor's functional role at local host defense sites.
B. G Protein-Coupling Specificities
A series of studies initiated in the mid-1980s revealed that the biochemical and the functional responses of neutrophils to fMLF are largely inhibited by pertussis toxin (PTX), a bacterial toxin that ADP-ribosylates the Gi class of heterotrimeric G protein α subunits, thereby preventing their interaction with GPCRs (Bokoch and Gilman, 1984; Bokoch et al., 1984; Lad et al., 1985; Sha'afi and Molski, 1988; Gierschik et al., 1989; Snyderman and Uhing, 1992). With the cloning of FPR1, studies of the structure-function relationships of FPR1 with respect to G protein specificities and G protein coupling became possible. The first such studies involved injection of crude or purified mRNA preparations into X. laevis oocytes. This system was tentatively used for the functional cloning of FPR1 (Murphy et al., 1990) before the expression cloning of FPR1 using COS cells (Boulay et al., 1990a,b). Although X. laevis oocytes had been used to demonstrate the functional coupling of a variety of GPCRs, no functional coupling could be detected after the injection of FPR1 mRNA despite appropriate expression of the receptor at the surface of the oocytes (Schultz et al., 1992). This result was unexpected, because all nonchemoattractant GPCRs have been capable of functional coupling in X. laevis oocytes. Furthermore, a functional FPR1 could be reconstituted after the injection of crude mRNA preparations from differentiated HL-60 cells but not from undifferentiated cells (Murphy et al., 1990). Surprisingly, X. laevis oocytes exhibited a functional response to fMLF when FPR1 mRNA was coinjected with an mRNA fraction of 3 to 3.5 kb from undifferentiated HL-60 cells, which by itself did not confer responsiveness to fMLF (Murphy and McDermott, 1991; Schultz et al., 1992). From these results, it had been concluded that a cofactor was required for FPR1 to function properly in X. laevis oocytes. The nature of the cofactor(s) remained unknown until Amatruda et al. (1991) demonstrated that the cofactor might be Gα16, a pertussis toxin-insensitive G protein of the Gq class in which expression is restricted to a subset of hematopoietic cells in the early stage of differentiation; coinjection of FPR1 mRNA with the transcript for Gα16 was found to be sufficient to enable fMLF-induced ion flux in X. laevis oocytes (Burg et al., 1995). The restoration of functional coupling by Gα16 is most likely due to the fact that Gα16 is a promiscuous G protein that couples to a wide variety of GPCRs (Offermanns and Simon, 1995; Wu et al., 1995), including FPR1 in transfected mammalian cells (Yang et al., 2001b). However, an additional factor may exist because the size of the mRNA for Gα16 (2.2 kb) differs from the size of the transcript (3–3.5 kb) for the complementary factor present in the undifferentiated HL-60 cells (Murphy and McDermott, 1991; Schultz et al., 1992).
Another approach used in the study of FPR1 signaling involves the measurement of fMLF-mediated increase in the intracellular concentration of calcium in L-cell mouse fibroblasts (Prossnitz et al., 1991) and human HEK 293 cells (Didsbury et al., 1992) that transiently or stably express FPR1. In contrast to X. laevis oocytes, transfected L cells or HEK293 cells responded to fMLF with a rapid and transient calcium flux without any additional cofactor(s) from differentiated HL-60 cells. In contrast to L-cells and HEK293 cells, transfected COS-7 cells were unable to elicit fMLF-stimulated phospholipase C activation unless they were cotransfected with Gα16 (Amatruda et al., 1991). The deficiency in signaling may be attributable to the fact that COS-7 cells express PI-PLCβ1 but not the myeloid-specific PI-PLCβ2 isotype. PI-PLCβ1 is activated by the α subunits of PTX-resistant G proteins, such as Gαq/11 and Gα16 (Kozasa et al., 1993), whereas PI-PLCβ2 is activated by the βγ subunits of G proteins, such as Gαi2 (Camps et al., 1992a,b). The α subunits of PTX-sensitive G proteins cannot activate any isoforms of PI-PLC (Hepler et al., 1993), but Gαq and Gα11, which are resistant to PTX, are able to activate PI-PLCβ2.
In leukocytes, the role of Gα16 seems to be marginal for several reasons. First, the fMLF-mediated functional responses are largely inhibited by treatment with PTX. Second, Gα16 transcript abundance is very low in mature neutrophils, and induction of HL-60 cell differentiation is associated with a progressive decrease in expression of Gα16 and an increase in the expression of Giα2 (Amatruda et al., 1991). Third, a large body of evidence indicates that the fMLF-induced responses require physical association of FPR1 with PTX-sensitive Giα2 protein in leukocytes (Bommakanti et al., 1992, 1993, 1994, 1995; Schreiber et al., 1993). However, residual molecules of Gα16 may account for a small PTX-resistant activity that has been observed in neutrophils, monocytes, and differentiated HL-60 cells stimulated with fMLF (Verghese et al., 1987). It is noteworthy that mouse Fpr1, along with a number of chemokine receptors known to couple to the Gi proteins, can also couple to Gαq (Shi et al., 2007). This property is responsible for an alternative pathway for the activation of chemoattractant and chemokine receptors that leads to chemotaxis.
A quantitative analysis of FPR1 coupling to the three Gαi isoforms, Gαi1,Gαi2, and Gαi3, was performed with FPR1 fused to the different Gαi isoforms to obtain a defined 1:1 stoichiometry of the signaling partners. After expression in Sf9 cells, high-affinity agonist binding, GTPγS binding kinetics, and GTP hydrolysis were analyzed. The results indicate that 1) FPR1 couples to all three Gαi isoforms with similar efficiency, 2) FPR1 can couple efficiently to these G proteins even in the low-affinity state for agonist binding, and 3) in contrast to what was previously thought, FPR1 activates the Gαi proteins linearly and not catalytically (Wenzel-Seifert et al., 1999).
C. G Protein-Coupling Domains
A variety of approaches have been taken to define the contact sites responsible for G protein-FPR1 coupling, including site-directed mutagenesis, construction of chimeric receptors, inhibition of high-affinity G protein-dependent fMLF binding, and peptide “walking” to map the regions of the intracellular loops that are in contact with the G protein (for review, see Savarese and Fraser, 1992). The latter approach is based on the use of synthetic receptor-mimetic peptides to block the receptor-G protein interactions. A competition assay based on the sedimentation of a GTP-sensitive FPR1-Gαi2 complex on sucrose gradients in the presence of octyl glucoside was developed by Bommakanti et al. (1992, 1994) to assess the effect of FPR1-derived peptides on the conversion of FPR1 from a 7S (FPR1-Gi complex) to a 4S species (FPR1 only). Synthetic FPR1-derived peptides were also used by Schreiber et al. (1994) in a competitive enzyme-linked immunosorbent assay. This assay measures the ability of FPR1-derived peptides to inhibit the binding of an anti-Gαi2 antibody to soluble Gαi2 (Weingarten et al., 1990).
The results from these assays excluded the third intracellular loop of FPR1 as a significant determinant for G protein coupling. The second intracellular loop (amino acids D122RCVCVLHPQNHRTVSLAKK144) and the entire C-terminal region were identified as the major sites of contact between FPR1 and Gαi proteins. It is notable that the DRY (Asp-Arg-Tyr) motif conserved in many GPCRs (Rovati et al., 2007) becomes DRC in the FPR family of receptors. Peptides corresponding to different regions of the carboxyl-terminal tail produced variable effects. The sequence extending from Arg322 to Thr336 had the best blocking activity and seemed to be the major structural determinant for the carboxyl-terminal tail to interact with Gαi. Although several studies on other receptors, namely rhodopsin and the β-adrenergic receptors (König et al., 1989; Münch et al., 1991), have suggested no role for the first intracellular loop in Gαi interaction, Bommakanti et al. (1995) found that a peptide corresponding to the first intracellular loop of FPR1 was able to disrupt FPR1-Gαi complex formation. A role for the first intracellular loop in G protein coupling was further supported by a study showing that its replacement by the counterpart from C5aR resulted in a constitutively active chimeric receptor (Amatruda et al., 1995). This latter observation may either suggest that the short stretch of amino acids that constitutes the first intracellular loop help to maintain an inactivated state of the receptor or reflect an unexpected conformational change resulting from substitution with the C5aR sequence.
Detailed information on the mechanisms of coupling between FPR1 and G proteins were obtained by site-directed mutagenesis studies that targeted amino acids conserved among the different GPCRs (Prossnitz et al., 1995b; Miettinen et al., 1999). Replacement of a cluster of three amino acids starting at position 309 at the N-terminal part of the cytoplasmic tail (RER to GAG) as well as Ala substitution of either Asp71 in the second transmembrane domain, or Arg123 in the conserved DRC (Asp-Arg-Cys) motif at the N terminus of the second intracellular loop, yielded three interesting mutant receptors with poor coupling to Gαi proteins and deficient signaling capacity. The Asp71 mutant receptor is not activated or phosphorylated in response to fMLF, whereas the Arg123Gly mutant does not bind G protein but is phosphorylated and able to bind β-arrestins (Gripentrog and Miettinen, 2008).
V. Regulation of Formyl Peptide Receptors
The activation of FPRs is regulated at the levels of receptor to G protein activation (R→G), transduction and amplification of signals from activated G proteins to effectors (G→E) including kinases and small GTPases, and integration of effector signals leading to phagocyte functions (E→F) such as chemotaxis, degranulation, and superoxide generation. This section is focused on major regulation of FPRs at the receptor level.
A. Desensitization of FPR1
After stimulation with fMLF, the cellular responses rapidly decline in intensity and the cells become refractory to subsequent stimulations with the same agonist. This loss of responsiveness, known as homologous desensitization, is a feature of G protein-coupled receptors that results from phosphorylation of the agonist-occupied receptor by G protein-coupled receptor kinases (GRKs) (Pitcher et al., 1998). A related phenomenon is that stimulation of one receptor can desensitize the cells to subsequent stimulations by ligands for another receptor. This is referred to as heterologous desensitization and is thought to result from the modification and/or inactivation of unliganded receptors after phosphorylation by second messenger-triggered kinases, such as protein kinase A and protein kinase C (PKC) (Dohlman et al., 1991).
Through the expression of different chemoattractant receptors in HEK293 cells and measurement of calcium mobilization induced by different agonists, Didsbury et al. (1991) introduced the concept of receptor class desensitization. For instance, C5aR and FPR1 were found to desensitize each other, but they cannot desensitize, or be desensitized by, receptors such as the α1 adrenergic receptor, which is not closely related to the chemoattractant receptors and has different signaling properties (Didsbury et al., 1991). Furthermore, comparison of the ability of fMLF, C5a, and IL-8 to desensitize one another in calcium mobilization assays has led to the observation of hierarchy in receptor desensitization between these chemoattractant receptors. The strength of peptide chemoattractants to desensitize calcium responses to one another (in the order of fMLF > C5a > IL-8) reflects the ability to generate desensitizing signals for both phosphorylation of an unliganded receptor by PKC and inactivation of a distal component in the calcium mobilization pathway. It is noteworthy that all three peptide chemoattractants can desensitize the responses mediated by lipid mediators such as leukotriene B4 (LTB4) and PAF. However, none of these lipids was able to desensitize the calcium response mediated by fMLF, C5a, or IL-8. Thus, this type of receptor cross-desensitization consists of at least two distinct processes. The first process possibly involves the phosphorylation of the receptor by PKC. FPR1 is resistant to this process because of the lack of PKC phosphorylation sites, yet the fMLF-mediated calcium response can be desensitized. The second process occurs downstream of the receptor-G protein complex and most likely results from the inactivation of a shared effector that is required for the calcium mobilization response mediated by peptide chemoattractants. The hierarchy of desensitization among these agonists may be important for leukocyte signal processing when multiple chemoattractants are present (e.g., in tissues with bacterial infection). Under these conditions, the “end-target chemoattractants,” such as fMLF, that are more capable of activating terminal functions of neutrophils, seem to prevail over “intermediary chemoattractants” such as IL-8 and other chemokines present at the site of inflammation (Heit et al., 2002). It has been shown that the “end-target chemoattractants” use both p38 MAPK and the PI3K pathways to maintain polarity and steer a migrating neutrophil, whereas the “intermediary chemoattractants” only activate the PI3K pathway. Through an as-yet-unknown mechanism, the chemoattractants that activate p38 MAPK can maintain PI3K phosphatase and tensin homolog (PTEN) localization to the rear and sides of the migrating cell, which helps to amplify the intracellular PtdIns(3,4,5)P3 gradient and resist any PI3K activity initiated by “intermediary chemoattractants” from other directions (Heit et al., 2008).
Studies of FPR1 have revealed the dose-dependent characteristics of receptor desensitization, such that FPR1 exposed to a given concentration of fMLF can no longer respond to fMLF at the same concentration but it can still respond to a higher concentration of fMLF. In a study of Mac-1-dependent neutrophil adhesion, Hughes et al. (1992) demonstrated that sequential, stepwise increases in fMLF concentrations could overcome desensitization and augment Mac-1 mobilization from intracellular stores to the cell's surface as well as Mac-1-mediated cell adhesion. This and other results from calcium mobilization assays indicate that full desensitization of chemoattractant receptors requires sustained exposure to saturating concentrations of agonists.
B. Phosphorylation of Formyl Peptide Receptors
It has been shown that FPR1 is rapidly phosphorylated in an agonist concentration- and time-dependent manner (Ali et al., 1993; Tardif et al., 1993). PKC is not involved in the phosphorylation of FPR1 based on several observations. First, the C-terminal region of FPR1 does not contain PKC phosphorylation sites. Second, the agonist-dependent phosphorylation of FPR1 is resistant to the PKC inhibitor staurosporin. Third, phorbol 12-myristate 13-acetate (PMA), an activator of protein kinase C, is unable to induce the phosphorylation of FPR1. The phosphorylation sites are restricted to the carboxyl terminal region, and phosphoamino acid analysis has revealed that only serines and threonines are phosphorylated (Prossnitz et al., 1995a). The carboxyl tail of FPR1 contains a total of 11 serine and threonine residues. Eight of these residues, located between and including Ser328 and Thr339, are found to be critical to FPR1 internalization and desensitization (Maestes et al., 1999). These residues are arranged in two domains, and the sequences D328STQTS332 and D333TATNST339 have the characteristics of GRK phosphorylation sites (Onorato et al., 1991). GRK2 has been identified as the primary kinase that phosphorylates FPR1, using as substrate a fusion protein consisting of the carboxyl-terminal 47 amino acids of FPR1 and glutathione transferase (Prossnitz et al., 1995a). Other GRKs are either significantly less effective (GRK3) or have no detectable activity (GRK5 and GRK6). Site-specific mutagenesis of the FPR1 carboxyl terminus has shown that acidic residues (Glu326, Asp327, and Asp333) are critical to FPR1 phosphorylation (Prossnitz et al., 1995a). Amino acid substitutions indicate that FPR1 phosphorylation uses a hierarchical mechanism in which the phosphorylation of two clusters of adjacent serine and threonine residues (Ser328/Thr329 or Thr331/Ser332) is required for subsequent phosphorylation of other serine and threonine residues. The phosphorylation of Ser328, Ser332, and Ser338 is critical to FPR1 internalization and desensitization as well as β-arrestin 2 binding (Potter et al., 2006). The phosphorylation status of these two clusters of serines and threonines modulates the affinity of FPR1 for β-arrestins and agonists (Key et al., 2003, 2005). The affinity of FPR1 for β-arrestins is controlled by the phosphorylation of serines and threonines in the sequence D328STQTS332, whereas the affinity of the β-arrestin-bound receptor for the agonist is regulated by the phosphorylation of the serines and threonines in the sequence D333TATNST339. Phosphorylation of these residues renders the β-arrestin-bound FPR1 able to form a high-affinity ternary complex with the ligand. Phosphorylation of carboxyl terminal serine and threonine residues may produce a localized concentration of negative charges, which facilitates ionic interactions with the positively charged receptor recognition domain of β-arrestin. This interaction may induce a β-arrestin intramolecular conformational change that then stabilizes a firm interaction between the two proteins and/or exposes a secondary high-affinity binding site (Han et al., 2001; Milano et al., 2002).
Like FPR1, FPR2/ALX is phosphorylated in an agonist-dependent manner, but little is known about the nature of the kinase(s) involved in the process. The determinants responsible for its internalization have not yet been identified. In contrast to FPR1 and FPR2/ALX, FPR3 displays a marked level of phosphorylation in the absence of stimulation (Christophe et al., 2001). Because phosphorylation of these receptors is known to affect their internalization, it will be interesting to know whether constitutive phosphorylation of FPR3 is related to its cell surface expression pattern.
C. Interaction between Formyl Peptide Receptors and β-Arrestins
1. Uncoupling of G Proteins.
In the current model, liganded GPCRs are phosphorylated and form a high-affinity complex with β-arrestin 1 or β-arrestin 2. The β-arrestins are thought to sterically interfere with G protein coupling and thereby inhibit the signaling capacity of the receptor. However, several studies have suggested that the desensitization of FPR1 may be unique. Using a fluorimetric assay in conjunction with solubilized receptors, Bennett et al. demonstrated that fMLF-induced phosphorylation of FPR1 can block FPR1 interaction with G proteins independently of β-arrestin (Bennett et al., 2001). Based on these findings, it is proposed that phosphorylation alone may be sufficient for uncoupling of the receptors from G proteins in vivo and this may not be the primary function of β-arrestins.
In the case of GPCRs that are coupled to the Gq or Gs class of G proteins, such as the angiotensin II type I A receptor and the V2 vasopressin receptor, β-arrestins play the role of adaptors and scaffolding molecules that have recently emerged as signal transducers for the activation of ERK in response to agonist stimulation (Shenoy and Lefkowitz, 2003; Lefkowitz and Whalen, 2004). Two studies have examined whether the binding of β-arrestins to FPR1 and FPR2/ALX, which are coupled to the PTX-sensitive Gi class of G proteins, plays a role as signal transducer to ERK activation. The first study compared the kinetics of ERK activation in both wild-type and β-arrestin-deficient mouse embryonic fibroblasts that stably expressed FPR2/ALX or in HEK293 cells that coexpressed FPR2/ALX and β-arrestin 1 or 2 (Huet et al., 2007). The second study took advantage of FPR1 mutants, such as the Arg123 mutant with a partial defect in Gαi coupling and the Asn297 mutant with a defect in β-arrestin recruitment (Gripentrog and Miettinen, 2008). Both studies have led to the conclusion that ERK activation occurs through Gαi and is not affected by β-arrestins. The lack of involvement of β-arrestins is thought to be a common characteristic among Gαi-coupled chemoattractant receptors, but additional investigations will be necessary to prove this is the case.
2. Internalization of Formyl Peptide Receptors.
After prolonged stimulation with fMLF, FPR1 was found to colocalize with β-arrestin 1 and β-arrestin 2 in endocytic vesicles in transfected monocytic U937 cells. It is well accepted that the β-arrestins serve to link phosphorylated receptors to the components of the endocytic machinery, including clathrin and the clathrin adaptor AP2 (Goodman et al., 1996; Laporte et al., 2002). This endocytic pathway seems to be used by the majority of GPCRs (for review, see Marchese et al., 2003; Lefkowitz and Whalen, 2004). However, several studies indicate that the internalization of FPR1 diverges from this common model in certain aspects (Gilbert et al., 2001; Xue et al., 2007). Flow cytometric analyses of HEK293 cells coexpressing FPR1 and dominant-negative constructs of β-arrestin 1, dynamin, and clathrin provided the first evidence that FPR1 is not internalized through clathrin-coated pits (Gilbert et al., 2001). Studies with transfected mouse embryonic fibroblasts (Vines et al., 2003) that are derived from β-arrestin 1 and β-arrestin 2 knockout mice (Kohout et al., 2001) further established that FPR1 can interact with different components of the endocytic machinery in a β-arrestin-independent manner. However, phosphorylation of liganded FPR1 is a prerequisite for the internalization whether the two β-arrestins are present or not. The ability of FPR1 to be internalized via a β-arrestin-independent pathway in mouse embryonic fibroblasts does not exclude the possibility that the classic arrestin- and clathrin-dependent pathway is involved when arrestins associate with the phosphorylated receptor (Gilbert et al., 2001; Vines et al., 2003).
In contrast to FPR1, several studies indicate that liganded FPR2/ALX undergoes clathrin-mediated and dynamin-dependent endocytosis and that it recycles in a slow pathway involving perinuclear recycling endosomes. First, FPR2/ALX internalization can be inhibited by either siRNA-mediated depletion of cellular clathrin or the expression of a dominant-negative mutant of dynamin in HeLa cells (Ernst et al., 2004b). Second, in HEK293 cells cotransfected with FPR2/ALX and a β-arrestin-enhanced green fluorescent protein, the receptor was rapidly internalized after the addition of the synthetic agonist peptide WKYMVM. It colocalized within a few minutes with β-arrestin-enhanced green fluorescent protein vesicles and gathered in the perinuclear region of the cytoplasm (Huet et al., 2007). Third, the internalization of FPR2/ALX, expressed in mouse embryonic fibroblasts lacking both β-arrestins, was highly compromised (Huet et al., 2007). In comparison, FPR2/ALX was normally internalized when only one of the two β-arrestins was expressed.
VI. Formyl Peptide Receptor Signal Transduction and Activation of Cell Functions
FPR1, along with the receptor for C5a, were the first identified phagocyte chemoattractant receptors. In comparison with chemokines and other “classic chemoattractants” such as PAF and LTB4, fMLF and C5a have several properties that qualify them as “end-target chemoattractants” (Heit et al., 2002). These two chemoattractants are predominant in cross-desensitization of other receptors (Ali et al., 1999), and they are potent agonists for stimulating bactericidal functions such as superoxide generation. This section will focus on major phagocyte activities induced by fMLF and the relevant signaling mechanisms. A more detailed description of signaling through FPR1 and similar chemoattractant receptors is provided in a recent review (Rabiet et al., 2007). It is notable that, although there are many similarities between FPR1 and FPR2/ALX (and possibly also FPR3), major differences exist between these receptors in signaling. These include for example the sensitivity to membrane cholesterol depletion (Tuluc et al., 2003), and to the intracellularly acting peptide (PBP10) derived from a PIP2 binding domains of the cytoskeletal protein gelsolin (Fu et al., 2004a). Evidence also suggests that the basic mechanisms by which these receptors trigger a transient increase in intracellular Ca2+ may be different (Partida-Sánchez et al., 2004).
A. Phagocyte Activation for Bacterial Clearance
1. Chemotaxis.
N-Formyl peptides have been extensively studied for their abilities to induce directional migration of neutrophils since their initial discoveries (Schiffmann et al., 1975a; Zigmond, 1977). Human neutrophils are able to detect a chemotactic gradient of subnanomolar concentrations of fMLF (Showell et al., 1976; Freer et al., 1980). In dose-response experiments, fMLF induces a typical bell-shaped curve seen with other leukocyte chemoattractants, indicating that increasing chemoattractant concentrations beyond a certain level actually reduces chemotaxis. At least two mechanisms can contribute to the descending curve past an optimal chemotactic concentration. First, saturation of these receptors with a high concentration of chemoattractant eliminates any meaningful gradient, thereby reducing chemotaxis. Second, chemoattractant receptors are desensitized by exposure to high concentrations of agonists, causing reduced responsiveness to subsequent agonist stimulation. It is unclear what proportion that desensitization contributes to the reduced chemotaxis when cells are exposed to a chemoattractant at high concentrations, but studies have shown that mechanisms other than heterologous desensitization are involved in prioritizing migrating cells exposed to multiple chemoattractants such as fMLF and IL-8 (Heit et al., 2008). Chemotaxis of neutrophils does not require chemoattractant receptor internalization or redistribution to the leading edge (Hsu et al., 1997; Neptune and Bourne, 1997). It requires Gβγ released from the activated Gαi, but not Gαi itself or activation of Gαq or Gαs (Neptune et al., 1999). Signaling downstream of Gβγ is important for chemotaxis because it determines the orientation of the cells. This was shown in elegant studies using video microscopy of fluorescent proteins as sensors for the detection of clustered signaling molecules at the leading edge of moving neutrophils (Servant et al., 1999). The PI3K family of lipid kinases has been found to play important roles in signaling for chemotaxis downstream of chemoattractant receptors (Niggli and Keller, 1997; Hirsch et al., 2000; Li et al., 2000; Sasaki et al., 2000). In neutrophils, the small GTPase Rac2 is also necessary for optimal cell migration (Roberts et al., 1999). Therefore, exposing neutrophils to a chemotactic gradient creates an intracellular gradient of signaling molecules that defines the “frontness” or leading edge as well as the direction of cell migration. The leading edge is characterized with increased accumulation of PtdIns(3,4,5)P3 and actin polymerization. The “backness” or uropod of a migrating cell is characterized by concentrations of molecules such as PTEN, RhoA, and the G12/13 family of G proteins (Funamoto et al., 2002; Xu et al., 2003). Whereas FPR1 is known to activate p38 MAPK, which helps to localize PTEN in the circumference other than the leading edge, exactly how FPR1 couples and activates Gα12 or Gα13 remains unclear.
Migrating neutrophils in tissues are exposed to multiple chemoattractants and must sort out different and sometimes conflicting signals. How these chemoattractants influence the migration of neutrophils has been studied in vitro using the “under agarose” chemotaxis assay (Nelson et al., 1975). Campbell et al. (1997) and Foxman et al. (1997, 1999) found that navigation of neutrophils is a multistep process, and a migrating cell not only detects multiple chemoattractants but also integrates these different signals. Therefore, the net result of cell migration is determined by the type of orienting signals, the strength of the signals, and the time sequence in which they appear. Heit et al. (2002) identified a hierarchy in chemotactic signaling that determines the direction of migrating cells in opposing chemotactic gradients. They found that “end-target chemoattractants” such as fMLF and C5a dominate over the “intermediary chemoattractants” such as IL-8 (CXCL8) and LTB4 (Heit et al., 2002). The ability of neutrophils to distinguish between these chemoattractants is crucial for their optimal migration toward bacteria with minimal interference by the “intermediary chemoattractants” that are also present in the inflammatory site. In a recent study, the same group of investigators provided new insights into the molecular mechanisms that allow neutrophils to prioritize chemotactic signals (Heit et al., 2008). As mentioned above, PI3K plays an important role in cell migration through generation of PtdIns(3,4,5)P3, which is found in the leading edge of a cell undergoing chemotaxis (Dormann et al., 2002; Wang et al., 2002). PTEN converts PtdIns(3,4,5)P3 back to phosphatidylinositol (4,5)-bisphosphate, thereby counteracting the effect of PI3K. Contrary to PtdIns(3,4,5)P3, PTEN is localized in the rear (uropod) as well as sides of a migrating cell, which helps to amplify the intracellular PtdIns(3,4,5)P3 gradient (Funamoto et al., 2002). In addition to the PI3K/PTEN pathway, “end-target chemoattractants” such as fMLF and C5a activate p38 MAPK, which is important for neutrophil chemotaxis toward these chemoattractants. Inhibition of p38 MAPK prevents neutrophil migration toward fMLF and C5a but not IL-8 (CXCL8) or LTB4, which do not stimulate p38 MAPK activation in neutrophils (Heit et al., 2002; Khan et al., 2005). Pten(-/-) neutrophils migrate normally to a single gradient of fMLF but randomly to a single gradient of an “intermediary chemoattractant” such as MIP-2 (CXCL2) (Heit et al., 2008). These cells exhibit two or more pseudopods and often lack an identifiable uropod when stimulated with MIP-2. In the presence of opposing chemotactic gradients such as fMLF and MIP-2, the Pten(-/-) neutrophils were unable to prioritize chemoattractants to migrate toward fMLF (Heit et al., 2008). The results from this study suggest an important function of p38 MAPK in maintaining proper localization of PTEN in cells migrating toward an “end-target chemoattractant,” although how p38 MAPK regulates PTEN localization is still unclear. PTEN that is present in the sides and rear of a moving cell helps the cell to resist any PI3K activity initiated by “intermediary chemoattractants” from other directions. This study raises an intriguing question of how two types of chemoattractant receptors that converge to the same PTX-sensitive Gαi protein can trigger differential activation of downstream pathways such as the p38 MAPK pathway. Further studies will be necessary to determine whether the two types of receptors are localized in different plasma membrane microdomains that may recruit different components of intracellular signaling pathways.
2. Superoxide Generation.
Stimulation of neutrophils with fMLF at concentrations higher than those required for chemotaxis leads to generation of superoxide (Boxer et al., 1979; Lehmeyer et al., 1979). In most published studies, fMLF concentrations of 50 to 100 nM are required for the induction of superoxide production by human neutrophils in suspension. In studies using mouse neutrophils, an even higher fMLF concentration (5–10 μM) is necessary for superoxide production. The response is ameliorated when mouse neutrophils are stimulated with formyl peptides of different sequences such as fMIVIL and fMIFL, which are able to activate this function in mouse neutrophils at concentrations as low as 10 nM (Southgate et al., 2008). The different concentration requirements for chemotaxis and superoxide generation effectively prevent oxidant-mediated tissue injury that may be caused by migrating neutrophils. It is not entirely clear why neutrophils require a 50- to 100-fold higher concentration of fMLF for superoxide production than chemotaxis, but a high signaling strength is probably necessary for simultaneous activation of multiple pathways leading to NADPH oxidase activation in phagocytes.
Superoxide production in neutrophils results from membrane assembly and activation of NADPH oxidase, which is a multicomponent enzyme complex for electron transfer leading to one-electron reduction of molecular oxygen (Babior et al., 2002). In the resting state, the membrane components (cytochrome b558, consisting of gp91phox and p22phox) are physically separate from the cytosolic components (p67phox, p47phox, p40phox, and the small GTPase Rac) (Fig. 6). Phosphorylation of the cytosolic factor p47phox is necessary to change its conformation and unmask the N-terminal PX domain for interaction with the membrane components p22phox and phosphatidylinositol (3,4)-bisphosphate (DeLeo and Quinn, 1996). Several studies have demonstrated that fMLF can activate kinases known to phosphorylate p47phox, including several PKC isoforms (Dang et al., 2001), Akt (Chen et al., 2003), and the MAPKs ERK and p38 (El Benna et al., 1996; Dewas et al., 2000). In reconstituted COSphox cells engineered to express all membrane and cytosolic phox proteins of phagocyte NADPH oxidase (Price et al., 2002), fMLF-induced superoxide generation requires a novel PKC isoform, PKCδ (He et al., 2004). It has been shown that fMLF stimulates rapid phosphorylation of PKCδ in its activation loop, and mutating the activation loop Thr507 to an alanine abolishes fMLF-induced, PKCδ-dependent superoxide production (Cheng et al., 2007). It is noteworthy that PAF, which is a weak agonist for activating NADPH oxidase, also fails to stimulate phosphorylation of PKCδ at Thr507. Therefore, rapid phosphorylation of PKCδ may be a mechanism for the superoxide production induced by fMLF. In addition to PKCδ, several other isoforms of PKC, including PKCα, PKCβII, and PKCζ, are likely to contribute to fMLF-induced superoxide generation in neutrophils. Genetic deletion of PLCβ2, which affects activation of the conventional and novel PKCs, abrogates fMLF-induced superoxide production in mouse neutrophils (Jiang et al., 1997).

The fMLF-induced signaling events leading to the activation of phagocyte NADPH oxidase (Nox2). Depicted schematically are major signaling pathways activated by fMLF that results in NADPH oxidase activation in neutrophils. Upon activation of the Gαi proteins, the released Gβγ subunits trigger PI3K activation, resulting in the production PIP3 and its degradation products. The Gβγ and PIP3-mediated p-Rex1 activation is key to the conversion of GDP-bound Rac small GTPase to GTP-bound Rac, which translocates to membrane and associates with gp91phox.Gβγ is also responsible for PLCβ2 activation, leading to the production of the second mesengers diacyl glycerol and 1,4,5-inositol trisphosphate (IP3), which stimulate PKC activation. Isoforms of PKC (PKCδ, PKCζ, PKCβII, and PKCα), MAPK (p38, ERK), and Akt are known to catalyze the phosphorylation of p47phox in fMLF-stimulated neutrophils, prompting membrane translocation of the cytosolic factors. The PX domain in p47phox also facilitates its membrane association. Assembly of a membrane complex of NADPH oxidase is key to its conversion of molecular oxygen to superoxide. Omitted in this drawing is the phospholipase D (PLD) activation pathway, which is reported to contribute to fMLF-induced superoxide generation.
The small GTPase Rac is an essential cytosolic component for NADPH oxidase activation (Abo et al., 1991; Knaus et al., 1991). In mouse neutrophils, deletion of Rac abrogates fMLF- and PMA-induced superoxide generation (Roberts et al., 1999). One of the mechanisms for chemoattractant-induced Rac activation involves p-Rex1, a guanine nucleotide exchange factor for Rac that is activated by PtdIns(3,4,5)P3 and Gβγ from activated heterotrimeric Gi proteins (Welch et al., 2002). The fMLF-induced Rac activation and superoxide generation is significantly reduced in mouse neutrophils lacking p-Rex1 (Dong et al., 2005; Welch et al., 2005). In addition to p-Rex1, other Rac guanine nucleotide exchange factors, such as Vav1, also play a role in fMLF-induced NADPH oxidase activation (Kim et al., 2003).
Studies of the individual kinases and guanine nucleotide exchange factors have shown a prominent role for PI3K in fMLF-induced functions, including superoxide generation. PI3K catalyzes the conversion of phosphatidylinositol (4,5)-bisphosphate to PtdIns(3,4,5)P3.Inaddition to activating p-Rex1, PtdIns(3,4,5)P3 is responsible for Akt and PKCδ activation, either directly or through the action of phosphoinositide-dependent kinase 1 (Dutil et al., 1998). The degradation products of PtdIns(3,4,5)P3, including phosphatidylinositol (3,4)-bisphosphate and phosphatidylinositol phosphate, serve as membrane binding sites for the PX domain of p47phox and p40phox, respectively (Kanai et al., 2001). Genetic deletion of PI3Kγ significantly reduces chemoattractant-stimulated neutrophil superoxide production (Hirsch et al., 2000; Li et al., 2000; Sasaki et al., 2000). The roles of other type I PI3Ks have also been implicated in fMLF-induced phagocyte NADPH oxidase activation (Condliffe et al., 2005; Boulven et al., 2006).
Human neutrophils in suspension that are pretreated with priming agents such as granulocyte macrophage–colony-stimulating factor, TNFα, PAF, LPS, and certain chemokines produce significantly more superoxide when stimulated with fMLF. These agents do not induce significant superoxide production when applied to the neutrophils in concentrations sufficient for priming. There is a large body of literature on the priming mechanisms that range from partial phosphorylation of p47phox (Dewas et al., 2003) and p67phox (Brown et al., 2004) to enhancement in PI3K activity (Kodama et al., 1999), up-regulation of NADPH oxidase assembly (DeLeo et al., 1998), and mobilization of receptors from intracellular stores (Almkvist et al., 2001; Almkvist et al., 2004), suggesting that there are multiple mechanisms for the potentiation of phagocyte NADPH oxidase activation. It is noteworthy that when used at low concentrations (e.g., 5 nM), fMLF is unable to stimulate neutrophil superoxide production but can prime the cells for a more robust response to PMA (Karnad et al., 1989). Conversely, PMA can prime the response to fMLF, as illustrated by the study performed by Tardif et al. (1998) with a variant clone of the myeloid HL60 cell line. When added to the dibutyryl cyclic AMP-differentiated HL-60 variant, PMA per se is unable to stimulate superoxide production, but it primes the cells for a robust and sustained response to fMLF at concentrations 1000-fold lower than those required for superoxide production when used alone. These findings indicate that the fMLF- and PMA-activated signaling pathways synergize and complement one another. The different studies that deal with the priming issue suggest that the ability of fMLF to stimulate neutrophil superoxide production is a function of its potency (the amount required to produce an effect of given intensity). For most priming agents, their inability to directly stimulate neutrophil superoxide production is an intrinsic property of the ligand and cannot be overcome simply by increasing ligand concentrations. Of particular interest is that IL-8 (CXCL8), known as a priming agent in some studies (e.g., Guichard et al., 2005), has been shown to directly activate neutrophil NADPH oxidase in other studies (Thelen et al., 1988; Fu et al., 2004b). Pretreatment of neutrophils with IL-8 can prime the cells for a more robust response to fMLF, whereas pretreatment of cells with fMLF reduces the IL-8-stimulated superoxide production (Fu et al., 2004b). This pattern is similar to the order of desensitization by these agonists. It remains to be determined whether experimental conditions (e.g., pretreatment of neutrophils with cytochalasin B) or properties of the reagents (e.g., lengths of the chemokine) are contributing factors for the discrepancy in IL-8 priming and activation studies carried out in different laboratories.
3. Degranulation.
In addition to the induction of superoxide generation, formyl peptides at higher concentrations (usually 10–50 times higher than the optimal concentration for chemotaxis) stimulate release of granule constituents from neutrophils (Showell et al., 1976; Bentwood and Henson, 1980). The fMLF concentrations required for the release of secretory vesicles and gelatinase granules are lower than that required for the release of other granules (Borregaard et al., 1993). The fMLF-induced mobilization of granules produces several effects, including proteolytic cleavage of membrane-localized adhesion molecules such as l-selectin, cell surface expression of new adhesion molecules, and release of proinflammatory matrix proteins such as the aforementioned FPR2/ALX agonist LL-37, as well as enzymes that can cause tissue degradation and killing of bacteria. For instance, neutrophil myeloperoxidase released from the azurophil granules helps to convert hydrogen peroxide to hypochlorous acid (Nauseef, 2007), a metabolite of importance both for killing invading microbes and for the resolution of inflammation. Most of the membrane-associated NADPH oxidase components, gp91phox (Nox2) and p22phox, are localized to mobilizable granules and these become more available for the assembly of NADPH oxidase when the granules fuse with the plasma membrane or with the membranes that surround engulfed microorganisms. Last but not the least, there are intracellular pools of FPR1 as well as of FPR2/ALX that are up-regulated to the cell surface when cells are stimulated with inflammatory mediators that mobilize the granules (Sengeløv et al., 1994; Bylund et al., 2002).
Degranulation in fMLF-stimulated neutrophils involves the second messenger diacylglycerol and PKCs that are activated by diacylglycerol and Ca2+ (Smith et al., 1988). However, fMLF is still able to induce secretory granule release when extracellular and intracellular Ca2+ is chelated, suggesting the presence of Ca2+ independent pathways for degranulation (Sengeløv et al., 1993). It has been reported that adhesion-dependent and fMLF-induced degranulation requires the Src family protein kinase Fgr and Hck, which also involves p38 MAPK (Mócsai et al., 1999, 2000). In addition, the activation of PI3K has been indicated as an important step leading to neutrophil granule release (Arcaro and Wymann, 1993; Thelen et al., 1994). The cGMP-dependent kinase PKG-1α has been implicated in fMLF-induced degranulation in transfected RBL-2H3 cells expressing FPR1 and in primary neutrophils (Nanamori et al., 2007). Further investigations will be necessary to determine the cross-talk between these kinases and their roles in regulating soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) that mediate vesicular fusion during degranulation.
B. Other Cellular Functions
1. Transcriptional Regulation and Anti-Inflammatory Functions.
Although neutrophils are terminally differentiated myeloid cells with special bactericidal functions, these cells retain the ability to synthesize selected proteins, including certain cytokines (Lloyd and Oppenheim, 1992). fMLF has been found to stimulate neutrophil transcriptional regulation and cytokine production (Cassatella et al., 1992). fMLF-induced IL-8 secretion is accompanied by the activation of NF-κB, a nuclear factor for transcription of a large number of proinflammatory genes (McDonald et al., 1997). NF-κB activation is also mediated by FPR2/ALX, in response to SAA stimulation, which leads to IL-8 secretion (He et al., 2003). Other studies have shown that agonists for FPR2/ALX, such as SAA and WKYMVm, can induce the expression of matrix metalloproteinase in fibroblast-like synoviocytes and monocytes (O'Hara et al., 2004; Sodin-Semrl et al., 2004; Lee et al., 2005). These results are consistent with the ability of certain GPCRs to regulate transcriptional activation that contributes to the proinflammatory activities of the respective ligands (Ye, 2001).
In contrast, FPR2/ALX ligands such as LXA4 and ANXA1 exhibit anti-inflammatory activities (Perretti et al., 2002; Chiang et al., 2006). ANXA1 has been shown to cause detachment of leukocytes and prevent transendothelial migration (diapedesis) (Perretti, 2003). In comparison, the anti-inflammatory effect of LXA4 is shown to involve suppression of proinflammatory gene expression. József et al. (2002) reported that LXA4 analogs attenuated nuclear accumulation of activator protein 1 and NF-κB in both polymorphonuclear and mononuclear leukocytes, resulting in the inhibition of LPS-induced IL-8 mRNA expression and IL-8 release by 50 to 65%. It is not entirely clear whether this action of LXA4 is mediated through FPR2/ALX-dependent negative signaling or blockade of FPR2/ALX binding of and activation by an endogenous, proinflammatory agonist for this receptor. The first possibility was suggested in a study that demonstrated LXA4 induction of NAB1, a transcriptional corepressor induced by glucocorticoids (Qiu et al., 2001). More recently, it was also shown that LXA4 and its analogs induce SOCS-2 expression (Machado et al., 2006) and SOCS-2-dependent proteosomal degradation of TRAF6, a molecule involved in Toll-like receptor signaling (Machado et al., 2008). The utilization of FPR2/ALX in these studies was suggested by the inhibition of LXA4 effects with pertussis toxin treatment (Qiu et al., 2001) and with Boc2 (Machado et al., 2006), although neither is specific for FPR2/ALX. In the latter study, the AhR, which is a receptor for LXA4 (Schaldach et al., 1999), has been shown to also mediate SOCS-2 expression induced by LXA4. The possibility that LXA4 blocks the access of proinflammatory FPR2/ALX ligands is suggested by a more recent study showing that LXA4 can compete off the binding of iodinated WKYMVm in a GPCR heterodimer consisting of BLT1 and a chimeric BLT1 receptor encompassing the third extracellular loop and the seventh transmembrane domain of FPR2/ALX (Damian et al., 2008). The chimeric BLT1 was previously shown to bind LXA4 and share some properties with wild-type FPR2/ALX (Chiang et al., 2000). Therefore, the possibility remains that the anti-inflammatory effect of LXA4 is mediated in part by its blocking effect (i.e., acting as an antagonist of the receptor). This notion is supported by experimental data showing that LXA4 does not have the property of a typical agonist for FPR2/ALX, despite its ability to stimulate GTPase activity in transfected Chinese hamster ovary cells (Fiore et al., 1994; Takano et al., 1997). In functional assays, most agonists for FPR2/ALX stimulate signaling pathways that lead to calcium mobilization, ERK phosphorylation, and terminal functions of neutrophils. However, LXA4 stimulation does not lead to calcium mobilization in several types of transfected cells or induce neutrophil degranulation and superoxide generation (Su et al., 1999b; Bae et al., 2003; Prat et al., 2006). In contrast, LXA4 has been shown to activate monocytes and the monocytic cell line THP-1 (Maddox et al., 1997). The discrepancy suggests that signaling molecules essential for certain LXA4-induced functions might be missing in neutrophils and epithelial cells. It also reflects that LXA4 lacks full agonistic activities at FPR2/ALX.
2. Neutrophil Apoptosis.
Neutrophils released to blood circulation have a half-life of 8 to 10 h. If not activated, these cells are destined for apoptosis (Savill, 1997). Stimulation of neutrophils with proinflammatory cytokines such as granulocyte macrophage–colony-stimulating factor, G-CSF, and IL-1β, but not with fMLF, C5a, or IL-8, prolongs the lifespan of neutrophils (Colotta et al., 1992). Other reports have shown that stimulation of neutrophils with fMLF can induce apoptosis, and this process requires superoxide generation (Kettritz et al., 1997). Neutrophil apoptosis and phagocytosis of apoptotic neutrophils are related to resolution of inflammation.
Several ligands for the formyl peptide receptors are found to play different roles in neutrophil apoptosis. SAA suppresses neutrophil apoptosis induced by anti-Fas antibody (Christenson et al., 2008). However, this effect is thought to involve the nucleotide receptor P2X7 and not FPR2/ALX. In comparison, ANXA1 has been shown to accelerate neutrophil apoptosis (Solito et al., 2003), although glucocorticoids that regulate ANXA1 production have antiapoptotic effects (Liles et al., 1995). El Kebir et al. (2007) reported that SAA could stimulate concurrent activation of the ERK and PI3K/Akt signaling pathways, resulting in the phosphorylation of BCL-XL/BCL2-associated death promoter (BAD) protein at Ser112 and Ser136, respectively, thereby preventing collapse of mitochondrial transmembrane potential, cytochrome c release, and caspase-3 activation. It is noteworthy that LXA4 treatment could reverse the antiapoptotic effect of SAA. The underlying mechanism has not been fully identified, although ERK and Akt can work together to delay neutrophil apoptosis. These results suggest a potential role of FPR2/ALX in the regulation of neutrophil apoptosis, but other receptors may be involved as well. It will be important to determine how a single class of receptors mediates different functions in cell survival and apoptosis when stimulated with different ligands.
VII. A New IUPHAR-Approved Nomenclature for the Formyl Peptide Receptor Family
Research conducted in the past 3 decades has shown that members of the human FPR gene family are G protein-coupled receptors expressed primarily in differentiated myeloid cells. All three receptors bind N-formylated peptides and respond to these peptides with similar cellular functions. The sequence identity between these receptors (56–69%) is high among G protein-coupled receptors detecting immunomodulatory peptides such as chemokines (Murphy, 1993). Therefore, these receptors are structurally similar and functionally related proteins of one gene family. Recent discovery of multiple agonists for the FPR family of receptors has raised new questions regarding their nomenclature, their physiological functions, and the mechanisms by which one receptor mediates both proinflammatory and anti-inflammatory activities. The pharmacological principle for receptor nomenclature has been based on the specific agonists for the receptor and not sequence homology between the receptor and another protein, as in the case of FPRL1 and FPRL2. This and other considerations for the nomenclature of the FPR family are discussed below.
A. Nomenclature of FPR1
Most mammalian receptors are known for their abilities to specifically recognize and respond to agonists produced by the host. Although receptor interactions with endogenous agonists are critical for homeostasis and numerous physiological functions, a fundamental basis for innate and acquired immunity is the ability of host cells to recognize exogenous ligands such as bacterial and viral products, known as nonself or foreign antigens. The structural basis for the host to recognize a large number of foreign antigens has been illustrated in both humoral and cytotoxic cell-mediated immune responses. As a first line of host defense against invading microorganisms, cells of the innate immune system serve to detect invading pathogens, and activate their antimicrobial mechanisms, such as phagocytosis, degranulation, and superoxide production. The discovery of FPR1, the first mammalian receptor known to bind N-formylated peptides, represents a major milestone in the understanding of how innate immune cells are mobilized when the host is infected with bacteria. The N-formylmethionine, characteristic of bacterial protein synthesis, can be considered a PAMP akin to bacterial cell wall peptidoglycans, CpG DNA, and lipopolysaccharides (Medzhitov and Janeway, 2000). Unlike these bacterial products, N-formylated peptides may be derived from both bacterial proteins and, in mammalian cells, mitochondrial proteins. Therefore, FPR1 may also serve homeostasis functions. Despite the presence of nonformyl peptides (e.g., acetylated peptides and peptides with no N-terminal modification) as high-affinity ligands for FPR1, there is overwhelming evidence for a key role of the N-formylmethionine in promoting ligand interaction with the receptor. Studies have shown that addition of an N-formylmethionine potentiates binding affinity as well as potency of the peptides tested on FPR1 (Freer et al., 1980, 1982; Chen et al., 1995). A physiological function of FPR1 in host defense is demonstrated in a study using mice lacking Fpr1, which displayed increased susceptibility to L. monocytogenes infection (Gao et al., 1999). Taken together, numerous studies have shown that FPR1 is a cognate receptor for N-formylated peptides of bacterial and mammalian origins.
B. Nomenclature of FPR2/ALX
The second receptor of the FPR family was initially identified through molecular cloning based on sequence homology to FPR1. It has been referred in the literature as FPR2, FPRL1 (the most frequently used name), FPRH1, HM63, LXA4R, and ALX. Of these, the name FPR2 reflects the ability of this receptor to bind N-formyl peptides, albeit at low affinity for fMLF (Ye et al., 1992). The name FPRL1 refers to the sequence homology shared between this receptor and FPR1, which in pharmacological terms does not reflect its ligand binding property. This receptor has been shown to bind a large number of endogenous and exogenous ligands, all peptides except LXA4 and ATLs. Among the characterized endogenous peptides, humanin and CCL23β are highly potent agonists (Elagoz et al., 2004; Harada et al., 2004; Ying et al., 2004; Miao et al., 2007), and SAA is an acute-phase protein with well defined physiological functions in amyloid and inflammatory diseases (Su et al., 1999b; He et al., 2003; O'Hara et al., 2004; Sodin-Semrl et al., 2004; Lee et al., 2006a,b; El Kebir et al., 2007). The primary structures of these peptides have very little in common, and these peptides come from different sources. Therefore, it is difficult to name this receptor after any one of the peptide agonists. Whereas the ALX nomenclature ratified by IUPHAR in 2003 (Brink et al., 2003) precisely indicates that LXA4 and ATLs are endogenous agonists for this receptor, it does not reflect the fact that the majority of the agonists for this receptor are peptides. One prominent feature of this receptor, shared with other members of the FPR family, is the ability to preferentially bind and respond to N-formylated peptides compared with nonformylated peptides of the same sequence (Murphy et al., 1992; Ye et al., 1992; Harada et al., 2004; Rabiet et al., 2005). For instance, an N-formylated humanin is nearly 300-fold more potent than nonformylated humanin (Harada et al., 2004). Studies conducted by Rabiet et al. (2005) showed that the receptor preferentially binds endogenous N-formyl peptides derived from mitochondrial proteins, compared with exogenous N-formyl peptides from bacterial sources. This property of the receptor is reflected in the name FPR2, indicating the receptor is the second member of the FPR family that interacts with N-formylated peptides. Based on these considerations as well as the ability of the receptor to bind and mediated the anti-inflammatory effects of LXA4 and ATLs, we recommend for this receptor the nomenclature FPR2/ALX (ALX/FPR2 when the lipoxin-binding property is of primary concern).
C. Nomenclature of FPR3
The third member of the FPR family often appears in the literature as FPRL2, a name describing its structural homology to other members of the same family. Compared with FPR1 and FPR2/ALX, fewer ligands have been identified for this receptor. One of its endogenous agonists is F2L, an acetylated N-terminal cleavage product of the mammalian heme-binding protein that is chemotactic to dendritic cells and monocytes (Migeotte et al., 2005). Like FPR2/ALX, this receptor also binds and responds to the mitochondrial peptide fMMY-ALF (Rabiet et al., 2005). Therefore, we recommend the nomenclature of FPR3 for this receptor.
In summary, the presence of an extraordinarily numerous and structurally diverse group of agonistic ligands for the FPR family of receptors has been a challenge to their nomenclature. We recommend the names FPR1, FPR2/ALX, and FPR3 primarily based on a shared property of these receptors to bind N-formylated peptides and, for the second member of this family, its ability to bind LXA4 and ATLs. Despite the diverse ligand recognition properties, these receptors are genetically clustered and functionally related proteins that play important roles in host defense and inflammation.
VIII. Unmet Challenges and Future Perspetives
Research on the FPRs conducted in the past 3 decades has generated important information for our understanding of chemoattractant receptors as GPCRs that transduce chemotactic signals in innate immune cells. Despite tremendous progress made in this area of research, a number of issues and questions remain to be addressed in the future.
A. The Functions of the Formyl Peptide Receptor Family of Receptors in Other Mammals
Most functional studies of the FPRs have been conducted in human neutrophils and, in early studies, rabbit neutrophils. In comparison, much less is known about the functions of FPRs in other species. In mice, deletion of the Fpr1 gene leads to increased susceptibility to L. monocytogenes infection (Gao et al., 1999). This and other studies indicate that mFpr1 is the ortholog of human FPR1 that serves to detect bacterially derived N-formylated peptides (Gao and Murphy, 1993; Southgate et al., 2008). However, the number of receptors in the mouse Fpr gene family (8) far exceeds the number of receptors in humans (3). The three mouse receptors found in leukocytes (mFpr1, mFpr2, and mFpr-rs1), which have been studied to some extent, have overlapping structural and functional properties with the three human receptors (Gao and Murphy, 1993; Takano et al., 1997; Gao et al., 1998, 2007; Liang et al., 2000; Vaughn et al., 2002; Wang and Ye, 2002; Southgate et al., 2008). Therefore, although mFpr1 is the ortholog of human FPR1, its putative ligand binding domain contains structural features resembling those of FPR2/ALX. The absence of high-affinity binding for fMLF and the ability to effectively recognize several other N-formylated peptides are also features shared between mFpr1 and FPR2/ALX (Rabiet et al., 2005; Southgate et al., 2008). Both mFpr2 and mFpr-rs1 share significant sequence homology with FPR2/ALX and FPR3. It is presently difficult to determine whether the gene product of Fpr-rs1 or Fpr-rs2 is the ortholog of FPR2/ALX, because both of the mouse receptors are able to detect and respond to LXA4 (Takano et al., 1997; Vaughn et al., 2002), although LXA4 activation through mFpr2 requires the presence of Gα16 (Vaughn et al., 2002). Further complicating the nomenclature of the mouse receptors is the recent finding that mFpr2 serves as a receptor for F2L, a highly potent and specific agonist for human FPR3 (Gao et al., 2007). These observations suggest that the human FPRs, especially FPR1 and FPR3, have better defined and more specialized ligand-binding properties than the mouse receptors. Clearly, a great deal of work has yet to be performed to understand the biological functions of these and other distantly related members of the mouse Fpr gene family.
B. Structural Basis for the Diversity of the Formyl Peptide Receptor Family in Ligand Recognition
The human FPRs are able to detect an incredibly diverse group of agonists. More than a dozen ligands of different structures and origins have been found for FPR2/ALX alone, making it one of the most promiscuous GPCRs characterized to date. It is presently unclear how FPR2/ALX is able to bind such a diverse group of agonists that include peptides and the eicosanoid LXA4. Studies employing chimeric receptors between FPR2/ALX and structurally similar receptors have produced useful information suggesting that this receptor has structural properties different from those of the FPR1 (Quehenberger et al., 1993; Chiang et al., 2000). With respect to binding of both peptides and LXA4 by FPR2/ALX, computer-assisted 3-D modeling of the dimensions and spatial volumes for retinal, fMLF, and LXA4 have shown that they are remarkably similar in the three-dimensional spatial volumes (Mills et al., 1999). However, this does not explain why structurally similar receptors such as FPR1 do not interact with LXA4. It is hypothesized that certain features of the FPR2/ALX binding pocket are required for its interaction with a multitude of agonists with different sequence composition and chemical nature. First, the binding pocket must be sufficiently large to accommodate ligands of various sizes, including longer peptides such as SAA (104 amino acids). Second, the receptor structure involved in ligand binding must be sufficiently flexible for proper contact with a variety of ligands. In this regard, charge-charge interactions such as those found in FPR1 (Mills et al., 1998) may actually restrict ligand access, whereas hydrophobic interactions can facilitate the binding of these ligands. In fact, most agonists for FPR2/ALX are hydrophobic or contain a significant number of hydrophobic residues. Finally, there may be multiple ligand binding sites on FPR2/ALX, thereby permitting its interaction with two or more different classes of agonists. This notion is supported by the diverse structures of the FPR2/ALX ligands, including recently identified synthetic ligands with relatively rigid structures. Moreover, binding studies have shown that the different ligands for FPR2/ALX can only partially compete off radiolabeled WKYMVm or LXA4 (Chiang et al., 2000; Nanamori et al., 2004a). More detailed structural studies will be required to test these and other possibilities and to identify the structural basis for the diverse recognition by FPR2/ALX.
C. Mechanisms Used by FPR2/ALX to Respond to Two Different Classes of Ligands for Proinflammatory and Anti-Inflammatory Effects
LXA4 and its stable analogs have been found to exert potent anti-inflammatory effects in vivo (for review, see Chiang et al., 2006). In comparison, the majority of the peptide agonists for FPR2/ALX stimulate proinflammatory activities in leukocytes through the activation of signaling pathways that are well defined for the G protein-coupled chemoattractant receptors as a class. Studies have shown that LXA4 and its stable analogs induce small G protein-dependent actin reorganization in monocytes but not in neutrophils (Maderna et al., 2002), even though FPR2/ALX is abundant in both types of cells. The mechanism underlying this difference remains undefined. Early characterization of the LXA4 receptor identified it as a high-affinity binding site for the eicosanoid and, when expressed in transfected Chinese hamster ovary cells, could mediate LXA4-stimulated activation of Gαi proteins (Fiore et al., 1994). However, unlike most peptide agonists for FPR2/ALX, high-affinity binding of LXA4 is not translated into the activation of signaling pathways from the G proteins that couples to the receptor. For instance, LXA4 does not stimulate phosphorylation of ERK1 and ERK2 even when used at micromolar concentrations (Bae et al., 2003). In comparison, several partial agonists for FPR2/ALX such as Quin-C1 are able to induce ERK phosphorylation (Nanamori et al., 2004a). In another study, it was shown that LXA4 could stimulate ERK2 phosphorylation, but through a mechanism independent of FPR2/ALX and FPR3 (Christophe et al., 2002). The same study also demonstrated that LXA4 did not affect neutrophil activation by WKYMVM, a selective agonist for FPR2/ALX. Apparently, the agonistic activity of LXA4 is not typical compared with most other agonists for FPR2/ALX. This difference may account for the anti-inflammatory effect produced by LXA4 and ATLs. Currently, there is no clear mechanism for the bifurcation of signaling pathways downstream of FPR2/ALX that may lead to proinflammatory and anti-inflammatory functions. It is possible that the anti-inflammatory effects of LXA4 require more than FPR2/ALX. Indeed, published reports indicate that, in addition to ALX, AhR is a receptor for LXA4 (Schaldach et al., 1999). AhR has been shown to be partially responsible for the anti-inflammatory functions of LXA4 in dendritic cells (Machado et al., 2006). Although AhR is structurally different from FPR2/ALX, it has been suggested that both receptors can activate SOCS-2, which is a mechanism for the anti-inflammatory actions (Machado et al., 2006, 2008). The observation that LXB4 does not compete for LXA4 binding in neutrophils but exhibits anti-inflammatory properties similar to that of LXA4 (Fiore et al., 1992; Maddox and Serhan, 1996) suggests the presence of a LXB4 receptor that differs from FPR2/ALX.
The potent anti-inflammatory effects of LXA4 and ATLs have been demonstrated in numerous in vivo studies (for review, see Serhan, 2005; O'Meara et al., 2008). Therefore, it is of great interest to understand how LXA4 and ATLs exert their anti-inflammatory functions through receptor-mediated mechanisms. An ongoing effort is focused on the identification and characterization of peptides and small molecules that serve as ligands for FPR2/ALX and that exhibit anti-inflammatory properties (Bürli et al., 2006; Hecht et al., 2009). Comparative studies of these molecules along with pharmacologically characterized FPR2/ALX ligands, such as WKYMVm, SAA, and LXA4, will likely uncover the different structural and functional changes that the ligands bring to the receptor and the receptor-mediated signaling pathways. It is our hope that future research, including the development of mice genetically deficient in Fpr-rs1 and Ahr, the two reported mouse LXA4 receptors, will lead to a better understanding of the mechanisms whereby GPCRs such as FPR2/ALX transduce multiple signals for different biological functions.
Acknowledgments
This work was supported in part by the National Institutes of Health National Institute of Allergy and Infectious Diseases [Grant AI033503]; the National Institutes of Health National Institute of General Medical Sciences [Grant GM066182] (to R.D.Y.); by the Commissariat à l'Energie Atomique, the Centre National de la Recherche Scientifique, and the University Joseph Fourier, France (to F.B.); by the Swedish Medical Research Council [Grant 298-20-3] (to C.D.); and by the Intramural Research Programs of the National Institutes of Health National Cancer Institute (to J.M.W.) and the National Institutes of Health National Institute of Allergy and Infectious Diseases (to P.M.M.). We thank Jing Deng for drawing chemical structures and Benjamin Gantner for critical reading of the manuscript.
Footnotes
-
↵1 Abbreviations: 7TM, 7-transmembrane; Aβ, β amyloid; Aβ42, 42-amino acid form of β amyloid peptide; AG-14, 1,3-benzodioxolane-5-carboxylic acid 4′-benzyloxy-3′-methoxybenzylidene-hydrazide; AhR, aryl hydrocarbon receptor; ALX, receptor for lipoxin A4 and aspirin-triggered lipoxins; ANXA1, annexin A1; ATLs, aspirin-triggered lipoxins; CDCA, chenodeoxycholic acid; CGEN-855A, TIPMFVPESTSKLQKFTSWFM-amide; CHIPS, chemotaxis inhibitory protein of S. aureus; CsA, cyclosporin A; CsH, cyclosporin H; c-suPAR, cleaved soluble uPAR; DC, dendritic cell; DCA, deoxycholic acid; ERK, extracellular signal-regulated kinase; fMLF, N-formyl-methionyl-leucyl-phenylalanine; FPR, formyl peptide receptor; FPRH1, formyl peptide receptor-homolog 1; FPRL1, formyl peptide receptor-like 1; G-CSF, granulocyte-colony-stimulating factor; GPCR, G protein-coupled receptor; GRK, G protein-coupled receptor kinase; HEK, human embryonic kidney; HSC, hematopoietic stem cells; IL, interleukin; LTB4, leukotriene B4; LXA4, lipoxin A4 (5S,6R,15S-trihydroxy-7,9,13-trans-11-eicosatetraenoic acid); MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor κB; PAF, platelet-activating factor; PAMP, pathogen-associated molecular pattern; PI3K, phosphatidylinositol 3-kinase; PI-PLC, phosphatidylinositol-specific phospholipase C; PKC, protein kinase C; PMA, phorbol 12-myristate 13-acetate; PtdIns(3,4,5)P3, phosphatidylinositol (3,4,5)-trisphosphate; PTEN, phosphatase and tensin homolog; PTX, pertussis toxin; Quin-C1, 4-butoxy-N-[2-(4-methoxy-phenyl)-4-oxo-1,4-dihydro-2H-quinazolin-3-yl]-benzamide; RBL, rat basophilic leukemia; SAA, serum amyloid A; SNP, single-nucleotide polymorphism; SOCS, suppressor of cytokine signaling; SR-BI, scavenger receptor class B type I; TA, temporin A; t-Boc, t-butyloxycarbonyl group; TLR, Toll-like receptor; TM, transmembrane; TNF, tumor necrosis factor; uPA, urokinase-type plasminogen activator; uPAR, urokinase-type plasminogen activator receptor.
-
↵2 In early reports and some recent publications, the abbreviation “fMLP” is used for fMet-Leu-Phe. This abbreviation is obsolete and can be confusing, because the letter “P” stands for proline in the now widely used one-letter amino acid code, not phenylalanine, as in fMLF. Therefore, fMLF should be the correct abbreviation for fMet-Leu-Phe.
-
This article is available online at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.109.001578.
- U.S. Government work not protected by U.S. copyright
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




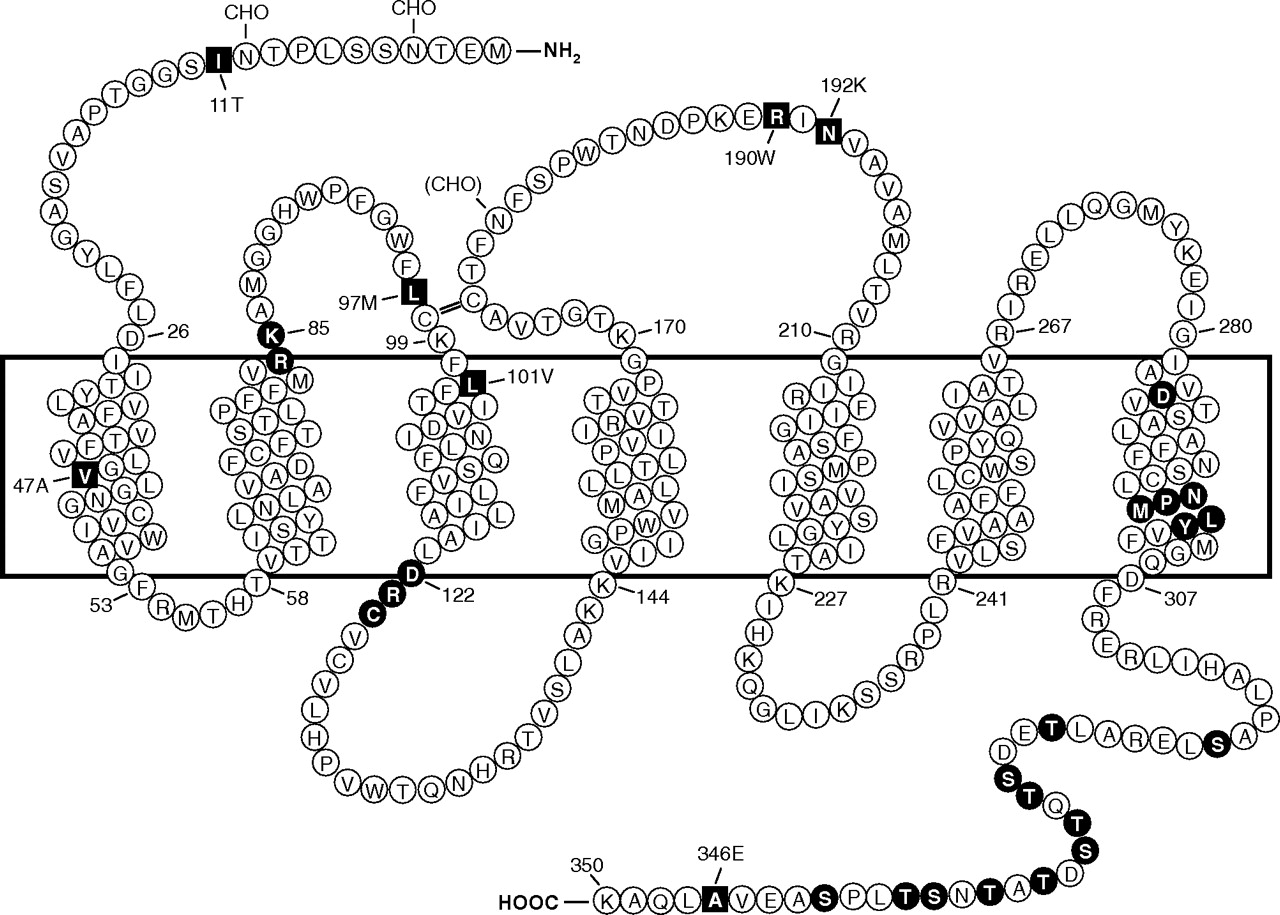{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction and Historical Overview
- II. The Expanded Family of Formyl Peptide Receptors
- III. Ligands for the Formyl Peptide Receptor Family of Receptors
- IV. Structure-Function Relationship of the Formyl Peptide Receptor Family of Receptors
- V. Regulation of Formyl Peptide Receptors
- VI. Formyl Peptide Receptor Signal Transduction and Activation of Cell Functions
- VII. A New IUPHAR-Approved Nomenclature for the Formyl Peptide Receptor Family
- VIII. Unmet Challenges and Future Perspetives
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters