


Abstract
The leukotrienes and lipoxins are biologically active metabolites derived from arachidonic acid. Their diverse and potent actions are associated with specific receptors. Recent molecular techniques have established the nucleotide and amino acid sequences and confirmed the evidence that suggested the existence of different G-protein-coupled receptors for these lipid mediators. The nomenclature for these receptors has now been established for the leukotrienes. BLT receptors are activated by leukotriene B4 and related hydroxyacids and this class of receptors can be subdivided into BLT1 and BLT2. The cysteinyl-leukotrienes (LT) activate another group called CysLT receptors, which are referred to as CysLT1 and CysLT2. A provisional nomenclature for the lipoxin receptor has also been proposed. LXA4 and LXB4 activate the ALX receptor and LXB4 may also activate another putative receptor. However this latter receptor has not been cloned. The aim of this review is to provide the molecular evidence as well as the properties and significance of the leukotriene and lipoxin receptors, which has lead to the present nomenclature.
I. Introduction
Feldberg, Kellaway, and coworkers (Feldberg and Kellaway, 1938;Feldberg et al., 1938; Kellaway and Trethewie, 1940) observed that perfusion of guinea pig lungs with antigen induced the release of a material named “slow reaction smooth muscle-stimulating substance (SRS1)” that caused a contraction of the isolated guinea pig ileum bioassay tissue. These observations were confirmed by several workers (Schild et al., 1951; Brocklehurst, 1960) who demonstrated that SRS (renamed slow-reacting substance of anaphylaxis or SRS-A) was also released from the human lung following antigen challenge. Sweatman and Collier (1968)reported that SRS-A constricted human airways and the compound FPL 55712 (Augstein et al., 1973) was shown to inhibit SRS-A-induced contractions in the guinea pig ileum assay. These observations provoked an intense interest in elucidating the biochemical nature of this entity. Initial attempts to characterize this substance revealed that the factor was a low-molecular weight derivative of arachidonic acid (Orange et al., 1973; Bach et al., 1977; Jakschik et al., 1977) containing sulfur (Orange et al., 1973, Parker et al., 1979). SRS-A was identified subsequently to be a family of lipid mediators known as leukotrienes, a name derived from their cell source (leukocytes) and their conjugated double bonds (triene) structure (Borgeat et al., 1976;Borgeat and Samuelsson, 1979a,b,c; Murphy et al., 1979; Corey et al., 1980; Lewis et al., 1980; Morris et al., 1980; Rokach et al., 1980). Leukotriene B4 (LTB4) was the first of the leukotrienes to be isolated (Borgeat et al., 1976).
The elucidation of the structures and synthetic pathways for the leukotrienes lead to a considerable amount of research on these arachidonic acid metabolites (Fig. 1). This work involved comprehensive assessments of the biological profiles of both the cysteinyl-leukotrienes (cys-LTs: LTC4, LTD4, and LTE4) as well as dihydroxy-leukotriene (LTB4) and, more recently, the lipoxins. Lipoxins (LX), an acronym for eicosanoids, which are often generated during the transcellular metabolism of arachidonic acid via the sequential actions of the 15- and 5- or 5- and 12-lipoxygenase enzymatic pathways (Serhan et al., 1984; Samuelsson et al., 1987). When the synthetic ligands were made available many studies documented a myriad of actions for these lipid mediators (Table 1) providing pertinent evidence for their possible patho-physiological roles in inflammatory diseases, in particular asthma. During the last 20 years significant efforts involving diverse chemical strategies have been directed toward the identification and development of receptor antagonists. These compounds have facilitated the identification and characterization of distinct receptors, which are activated by either the dihydroxy- or cysteinyl-leukotrienes.

Major pathways for leukotriene and lipoxin formation. The leukotrienes and lipoxins are lipid mediators derived from arachidonic acid, which is released from cell membrane phospholipids by the action of phospholipase A2. Leukotriene formation is initiated by 5-lipoxygenase, which catalyzes the dioxygenation of arachidonic acid to 5-HPETE and the subsequent conversion to LTA4. This latter unstable epoxide is transformed either to LTB4 or LTC4, and LTC4 is further catalyzed to LTD4 and LTE4. The tetraene epoxide intermediate can be formed either from LTA4 or 15-HPETE depending upon the interactions of the different lipoxygenases, and this metabolite is enzymatically hydrolyzed to the lipoxins (LXA4 and LXB4).
Major biological actions of leukotrienes and lipoxins
II. General Considerations
A. Biochemical Pathways
The leukotrienes are formed via activation of the 5-lipoxygenase enzyme (5-LO) in collaboration with the “5-lipoxygenase activating protein” (FLAP). A prerequisite for this enzymatic reaction is the hydrolyzation of arachidonic acid from membrane phospholipids by phospholipase A2. The principal 5-LO products of arachidonic acid metabolism are LTC4 and LTB4 as well as 5-hydroxyeicosatetranoic acid (5-HETE). In addition, eicosanoids that are formed by pathways that involve the dual lipoxygenation of arachidonic acid by either 15- and 5-LO or 5- and 12-LO are referred to as lipoxins (Serhan et al., 1984;Samuelsson et al., 1987). The transcellular metabolism of intermediates such as LTA4 and 15(S)-HETE is associated with LX formation (Serhan, 1994). LX and their carbon 15-epimer-LXs (aspirin-triggered lipoxins; ASA-15-epi-LX) are bioactive and structurally distinct from other eicosanoids in that they carry a conjugated tetraene system and are present in biological matrix in two main forms that are positional isomers, namely, lipoxin A4(5S,6R,15S-trihydroxyeicosa-7E,9E,11Z,13E-tetraenoic acid) and lipoxin B4(5S,14R,15S-trihydroxyeicosa-6E,8Z,10E,12E-tetraenoic acid; Serhan, 1997; Fig. 1). The aspirin-triggered form carry their C15 alcohol in the R configuration, which is inserted by COX-2 following aspirin treatment (denoted ASA-15-epi-LX). These metabolites are often produced during cell-to-cell interactions, and the principal targets appear to be platelets and leukocytes. During these cellular interactions, platelets convert neutrophil derived LTA4 to 5,6,-epoxytetraene through the action of platelet 12-LO. However, under these conditions the term 12-LO is a misnomer since this enzymatic activity was originally based on an interaction with arachidonic acid. This enzyme functions as a 15-LO (LX synthase) when the substrate is LTA4. Thus in an inflammatory condition LTA4 serves as a pivotal intermediate for both leukotriene and lipoxin formation.
B. Cellular Origins
The leukotrienes are formed in different cell types as well as via transcellular metabolism involving multiple cells such as neutrophil and platelets and vascular cells (Feinmark and Cannon, 1986;Maclouf and Murphy, 1988; Sala et al., 1993). Human eosinophils and neutrophils synthesize both LTC4 and LTB4, respectively (Bray et al., 1980;Ford-Hutchinson et al., 1980). Monocytes and macrophages also synthesize both LTB4 and the cys-LTs (Samuelsson, 1983). LTC4 is metabolized to LTD4 and LTE4 by the cells in which this mediator is formed. In addition, the cys-LTs can be transformed into 6-trans-LTB4 by hypochlorous acid, which is generated during the respiratory burst in leukocytes (Henderson et al., 1982; Lee et al., 1983). LTB4 is also metabolized in the cells which produce this metabolite, by a unique membrane bound cytochrome P450 enzyme. LTB4 is metabolized to 20-hydroxy-LTB4 (Hansson et al., 1981; Shak and Goldstein, 1985; Soberman et al., 1985). There is also evidence for a reductase dehydrogenase in polymorphonuclear leukocytes (PMN) that appears to be specific for LTB4 (Powell et al., 1989).
C. Nomenclature for Leukotriene Receptors
The previous IUPHAR publication (Coleman et al., 1995) introduced two main classes of leukotriene receptors. One based on the biological activities of leukotriene B4 and related hydroxyacids, referred to as BLT receptors, and a second class identified by the cysteinyl-leukotrienes (cys-LTs). The different profiles of biological activity for these two classes of metabolites were the initial basis for these categories and were supported by structure-activity data obtained in studies with a variety of compounds that selectively antagonized the different ligands. Activation of the BLT receptors initially was shown to produce potent chemotactic activities on leukocytes whereas the latter class (CysLT receptors) stimulated smooth muscle as well as other cells. However, the structures of the leukotriene receptors have recently been deduced from the nucleotide sequences of the cDNAs and the encoding proteins are now known for human, mouse, and rat. These data have permitted the IUPHAR committee to establish the nomenclature for the leukotriene receptors, and this is presented in Table 2. The phylogenic tree for the different eicosanoid and bioactive lipid G-protein-coupled receptors (GPCR) is illustrated in Fig.2 and shows the molecular families with the relationship between leukotrienes and lipoxins as well as other proteins with seven transmembrane helices.
Human cloned leukotriene receptors2-a

Dendogram of several GPCRs. The major receptor families for lipid mediators are indicated in this phylogenic tree. There are four known cell surface GPCRs for the leukotrienes, which are classified as either chemoattractants (BLT1 and BLT2) or nucleotide receptors (CysLT1 and CysLT2). The lipoxin receptor (ALX) is also included in the chemoattractant receptor class along with formyl peptide receptors (FPL). This evolutionary tree was constructed using the sequences from the receptors. Construction was performed by using the “All All Program” at the Computational Biochemistry Server at ETHZ (http://cbrg.inf.ethz.ch/ServerBooklet/chapter2-3.html).
The lipoxins, are chemically and functionally different from the leukotrienes (Fig. 1). Although LXA4 and LXB4 are similar in structure, these mediators display biological activities that are quite distinct. LXA4 interactions with neutrophils involves binding sites that are not recognized by LXB4(Nigam et al., 1990; Fiore et al., 1992). LXB4 is a potent agonist for stimulating proliferation and differentiation of granulocyte-monocyte colonies from human mononuclear cells (Popov et al., 1989), increasing the S-phase in the cell cycle and enhancing nuclear protein kinase C activity (Beckman et al., 1992) actions, which have not been reported for LXA4. However, LXB4 has also been shown to share actions with LXA4, such as, both selectively stimulate human peripheral blood monocytes (Maddox and Serhan, 1996) and enhance growth of myeloid progenitor cells (Stenke et al., 1991). Furthermore, LXA4 does not activate BLT (Fiore et al., 1992) but activates FPRL-1 receptors (Chiang et al., 2000; Resnati et al., 2002; Perretti et al., 2002). These investigators have shown that ALX and FPRL-1 are the same receptor and that LXA4 is the natural and most potent ligand. In addition, Takano et al. (1997)have identified the amino acid sequence for the receptor associated with the LXA4 responses. In line with the IUPHAR nomenclature directives, this committee recommends that ALX be used to designate the receptor that has been cloned and is activated by the native ligand LXA4 (Table 2). LXB4-induced responses, although different from those of LXA4, have not to date provided sufficient evidence to specify another receptor. Since this receptor has not been cloned, the LXB4 response is associated with activation of a putative receptor.
The aim of this review is to present the evidence that led to the leukotriene nomenclature. To this end, information not only from the molecular database but also derived from the properties and significance of leukotriene receptors will be presented. Furthermore, the above nomenclature for the LX receptors is recommended as the framework for this evolving area of receptor research.
III. Molecular Database for Leukotriene Receptors
A. Molecular and Structural Aspects of Dihydro-Leukotriene Receptors
1. BLT1.
The cloning and characterization of the BLT1 receptor was achieved by cDNA subtraction using human leukemic cells HL-60, which were differentiated into granulocyte-like cells (Yokomizo et al., 1997). The BLT1 receptor was identified as a putative seven transmembrane domain receptor with 352 amino acids. This receptor had been initially misidentified as a purinergic receptor, P2Y7 (Akbar et al., 1996). BLT1 shares low homology to P2Y receptors and belongs to a family of receptors for chemoattractants including complement receptors and a recently identified novel prostaglandin D2 receptor, CRTH2 (Hirai et al., 2001). The homology between the BLT1 receptor for mouse and humans is presented in Fig. 3.

The sequence alignment of BLT1 and BLT2 from human and mouse receptors. The amino acid sequences were aligned using ClustalW and converted using Boxshade 3.21. The putative transmembrane domains of hBLT1 predicted by Kyte-Doolittle hydrophobicity analysis are overlined and labeled as I–VII. Consensus matches are boxed and shaded with darker shading for identities and light shading for conservative substitutions. The amino acid identity between human and mouse BLT1 was 78.6% whereas BLT2 was 92.7%. The mouse sequence data are available from Swiss-Prot under accession numbers (mBLT1: no entry presently available) and (mBLT2: Q9JJL9).
Kato et al. (2000) reported that the BLT1 gene consists of three exons located in a region of 5 kbp on chromosome 14. Whereas the open reading frame (ORF) for BLT1 is in the last exon (exon 3), the basal promoter activity is found 100-bp upstream from the transcriptional initiation site. This report and the data of Yokomizo et al. (2000) demonstrated that there was no TATA or CAT element near the transcription site. However, they did observe the presence of an Sp-1 site at −50 bp, which played a major role in the basal transcription of BLT1. Since the promoter region of BLT1 is rich in GC sequences and methylated in nonleukocyte cells but nonmethylated in leukocyte cells expressing BLT1, Kato et al. (2000) have suggested that the methylation state may, in part, be responsible for transcription of the BLT1 gene.
COS-7 membranes transfected with human BLT1exhibited [3H]LTB4binding with a K d of 0.15 nM, comparable to the K d of 0.14 nM for the differentiated HL-60 cells (Fiore et al., 1993; Yokomizo et al., 1997; Table 3). Various eicosanoids also competed for the [3H]LTB4binding to COS-7 membranes expressing human BLT1and the potency ranking (K i values) were: LTB4 (0.38 nM) > 20-hydroxy-LTB4 (7.6 nM) = 12-oxo-LTB4 (7.6 nM) > 12R-HETE (30 nM) > 20-COOH-LTB4 (190 nM). This profile agrees with the previously characterized LTB4 binding sites in human granulocytes. The structures and potencies of several antagonist for these receptors in Chinese hamster ovary (CHO) cells expressing human BLT (CHO-BLT) are presented in Table 4.
Recombinant BLT receptors in transfected cells and [3H]LTB4 binding
Several BLT antagonists and structures
BLT1 mRNA is predominantly expressed in leukocytes, granulocytes, macrophages, and eosinophils with high amounts of BLT1 in human and mouse cells (Yokomizo et al., 1997; Huang et al., 1998). In addition, BLT1 expression is inducible in activated macrophages (Toda et al., 1999) and eosinophils treated with IL-5 (Huang et al., 1998), suggesting an association of BLT1 with various inflammatory diseases. BLT1-transgenic mice showed enhanced PMN responses in the lung following ischemia-reperfusion and increased expression of 5-lipoxygenase in peritonitis (Chiang et al., 1999). Recently, two lines of BLT1-deficient mice have been reported, and a reduction in leukocytes migration was demonstrated (Haribabu et al., 2000; Tager et al., 2000).
2. BLT2.
During the analysis of transcriptional regulation of human BLT1 gene (Kato et al., 2000), a putative ORF for a novel GPCR with structural similarity to BLT1 was identified (Yokomizo et al., 2000). This novel receptor was also found in a human genome sequence database, reported to act as a low-affinity receptor activated by LTB4 (Kamohara et al., 2000; Tryselius et al., 2000; Wang et al., 2000) and subsequently referred to as the BLT2 receptor (Yokomizo et al., 2001b). The gene structure for BLT2 has also been established (Yokomizo et al., 2001b). Of considerable interest is that the promoter region (Fig. 4) of human BLT1 overlaps BLT2 ORF (Kato et al., 2000). This represents the “promoter in ORF”, as has been reported in prokaryotes but the biological significance of this rare gene structure is presently not clear. However, there is sufficient evidence that BLT1 and BLT2 form a gene cluster both in human (chromosome 14 q11.2-q12;) and mouse (Yokomizo et al., 2000) chromosomes suggesting that these receptors may be generated by gene duplication.

Structure of the human genomic DNA containing BLT1 and BLT2 located on human chromosome 14q11.2-q12. Chromosome 14 is indicated by the line; the transcribed segments are indicated by open boxes; putative ORFs are shown as filled boxes. Note that the promotor region for BLT1 is located in the ORF of BLT2. This is the first mammalian example of “promotor in ORF” (Yokomizo et al., 2001b).
The K d values for LTB4 derived from transfection assays were 23 nM in human embryonic kidney (HEK)-293 cells (Yokomizo et al., 2000), and a K d value of 0.17 nM in COS-7 cells (Wang et al., 2000). Although there are differences in the reportedK d values (Table 3 and Table 4), generally BLT2 is considered as a low-affinity receptor since the pEC50 value of LTB4 required for adenylyl cyclase inhibition and the concentrations required for LTB4-dependent chemotaxis were higher than those for BLT1receptor activaton (Kamohara et al., 2000; Yokomizo et al., 2000). The tissue distribution of BLT2 assessed by different groups using Northern blots are varied. Using the ORF probe of BLT2 (Kamohara et al., 2000; Yokomizo et al., 2000) the highest expression was found in spleen, followed by leukocytes and ovary. Other groups reported that BLT2 is expressed highest in liver and intestine (Tryselius et al., 2000; Wang et al., 2000) using the 5′-UTR probes. The abundance of low-affinity binding sites for LTB4 in spleen membrane supports the data showing BLT2 expression in spleen (Showell et al., 1998). Of interest, the rank order of potency of several eicosanoids for BLT2 activation was LTB4 > 12-epi-LTB4 > 12S-HETE > 12S-HPETE > 12R-HETE > 20-hydroxy-LTB4, which is quite different from BLT1 (Yokomizo et al., 2001c). Currently, there are no data on specific in vivo functions for BLT2 receptors, however, lymphocytes have been suggested to be the target, since BLT2 is highly expressed in splenic T-cells (Yokomizo et al., 2001a).
3. Phenotypes Involving BLT Receptors.
Investigations with transgenic mice expressing the human BLT1receptor on leukocytes (Chiang et al., 1999) as well as targeted gene disruption of the BLT1 receptor in knockout mice (BLT−/−) indicate that an apparent phenotypic difference (Haribabu et al., 2000; Tager et al., 2000) from wild type littermates is not observed unless the animals are subject to experimental disease or injury, which are known to stress the effector immune system (vide infra).
In BLT1−/− mice (Haribabu et al., 2000; Tager et al., 2000) leukocytes exhibited a normal response to C5a and platelet-activating factor (PAF), however, there was a selective loss of responsiveness to exogenous LTB4. Tager et al. (2000) also reported that adhesion to the endothelium in response to LTB4 was diminished in leukocytes from BLT1 −/− animals. These findings are in line with earlier observations with LTB4 in the hamster cheek pouch (Raud et al., 1991) and suggest that the BLT receptor is pivotal for the LTB4-induced leukocyte activation.
Unfortunately, there are only a limited number of investigations examining the relative input of LTB4 in chemotaxis during inflammation in vivo in genetically modified animals. Two groups (Haribabu et al., 2000; Tager et al., 2000) have provided evidence that peritoneal inflammation is suppressed in these deficient mice. Interestingly, Haribabu et al. (2000) described a loss of this protection at 72 h between −/− and +/+ animals, whereas Tager and colleagues (2000) observed an increase in protection between −/− and +/+ with time intervals greater than 50 h. These latter investigators also reported a marked diminution in the number of eosinophils, which accounted for virtually all of the changes in cellular influx. In contrast, Haribabu et al. (2000) noted reductions in both PMNs and macrophage populations. The basis for these differences is presently not known, but may be related to the different stimuli (zymosan versus thioglycollate) used to induce the inflammatory cellular infiltration in vivo.
Expression of the human BLT1 receptor in leukocytes has also been carried out using the CD11b promoter (Chiang et al., 1999). BLT receptor-transgenic mice displayed enhanced leukocyte responsiveness in acute dermal inflammation, with leukocyte trafficking to remote organs (as in secondary organ reperfusion injury), or in leukocyte recruitment following a peritoneal challenge. These results are consistent with the interpretation that LTB4 is associated with an excessive activation of leukocytes and the 5-LO pathway provoking leukocyte-mediated injury. Expression of the human BLT1 receptor in vivo lead to an up-regulation of 5-LO expression and leukotriene biosynthesis (Chiang et al., 1999). These results suggest that receptor expression may amplify proinflammatory circuits in vivo.
B. Molecular and Structural Aspects of Cysteinyl-Leukotriene Receptors
1. CysLT1.
The cloning and characterization of the human CysLT1 receptor (hCysLT1) was achieved by two groups under the general program of identifying cognate ligands for orphan GPCRs, a process which has been termed “ligand fishing” (Lynch et al., 1999;Sarau et al., 1999). The hCysLT1 receptor was identified as a 337-amino acid putative seven transmembrane domain receptor, termed either HG55 (Lynch et al., 1999) or HMTMF81 (Sarau et al., 1999) (Fig. 5). The former investigators demonstrated that LTD4 produced activation of a calcium-activated chloride channel in Xenopus laevis oocytes expressing the cRNA for HG55 but not in control cells or oocytes expressing other GPCRs. This LTD4-induced stimulation of oocytes was blocked by the selective CysLT1 receptor antagonist MK-571 (Lynch et al., 1999) (Table 5). Similar results were obtained using the X. laevismelanophore signaling assay and in mammalian monkey kidney COS-7 cells expressing the HG55 (hCysLT1) receptor (Lynch et al., 1999).

Comparison of amino acid sequences of the human CysLT1 and CysLT2 receptors. A G-protein-coupled receptor snake diagram depiction of the amino acid sequences of the human CysLT1 and CysLT2receptors. The amino acid identities between the hCysLT1and hCysLT2 receptors is 37.3%.
CysLT1-selective and nonselective antagonists and structures
Researchers at SmithKline Beecham identified a GPCR (HMTMF81) that, when expressed in HEK-293 cells, responded selectivity with calcium mobilization to LTC4, LTD4, or LTE4 (0.1–10 μM) but not to more than 900 other ligands, including greater than 200 ligands for GPCRs.
The genomic organization of the hCysLT1 receptor has not been reported but consists of at least three exons with all of the open reading frame of the receptor in one exon (M. Metzker, Merck unpublished results). Three single nucleotide polymorphisms have been identified in the hCysLT1 receptor but none relate to the asthmatic phenotype (J. Drazen, unpublished data). The hCysLT1 receptor is located to chromosome Xq13-q21 (Lynch et al., 1999) and has 31% amino acid identity to the P2Y purinoceptor (Sarau et al., 1999).
Full Scatchard analyses of [3H]LTD4 binding to the HG55 (hCysLT1) COS-7 membranes demonstrated aK d of 0.3 nM andB max of 50 fmol/mg of protein (Lynch et al., 1999). In the structure-affinity relationships, the IC50 for the agonists were LTD4 (0.9 nM), LTC4 (350 nM), and LTE4 (200 nM). The affinities of antagonists (Table 5) were similar to that demonstrated functionally in other systems for the CysLT1 receptor as well as in binding experiments on human lung or U937 cells (Frey et al., 1993;Lynch et al., 1999). Likewise the HMTMF81 (hCysLT1) receptor expressed in HEK-293 cells responded selectively to the cys-LTs with rank order of potency (pEC50 value) were LTD4(2.5 nM) > LTC4 (24 nM) > LTE4 (240 nM) (Sarau et al., 1999). In these functional studies LTE4 was shown to be a partial agonist of the HMTMF18 (hCysLT1) receptor expressed in HEK-293 cells (Sarau et al., 1999). This is similar to evidence derived from the human bronchus (Labat et al., 1992), sheep trachea (Mong et al., 1988), and U937 cells (Saussy et al., 1989). [3H]LTD4 binding and LTD4-induced calcium mobilization in HEK-293 cells expressing the HMTMF18 (hCysLT1) receptor were potently inhibited by the structurally distinct CysLT receptor antagonists and the rank order of potency in this system was pranlukast = zafirlukast > montelukast > pobilukast (Sarau et al., 1999). LTD4-induced calcium mobilization in HEK-293 cells expressing the HMTMF18 (hCysLT1) receptor was not affected by pertussis toxin and appeared to result from the release of intracellular calcium stores (Sarau et al., 1999). In X. laevis melanophore signaling system, the HG55 (hCysLT1) receptor was shown not to couple via Gαi (Lynch et al., 1999).
Northern analyses of human tissues showed that the hCysLT1 mRNA was detected as an approximately 2.8-kb species and was expressed in spleen, peripheral blood leukocytes, less strongly in lung, bronchus small intestine, colon, skeletal muscle, pancreas, placenta, and weakly in the prostate, heart, brain, liver, kidney, and adipose tissues (Lynch et al., 1999; Sarau et al., 1999). Further work is necessary to confirm whether expression in some of these tissues may be attributable to expression of the receptor on interstitial myeloid cells. The receptor was also detected in U937 cells and HL-60 cells, with an increase observed in dimethyl sulfoxide-differentiated HL-60 cells (Sarau et al., 1999). In HL-60 cells differentiated with sodium butyrate to eosinophilic-like cells, interleukin-5 treatment resulted in a 5-fold increase in the expression of the hCysLT1 (Thivierge et al., 2000). Northern analysis in normal and asthmatic lungs suggested no total differences in hCysLT1 mRNA expression, although in situ studies in asthmatic tissues have not been reported (Sarau et al., 1999). Using in situ hybridization, the hCysLT1 mRNA was shown in lung smooth muscle cells and interstitial macrophages, with little or no expression in epithelial cells (Lynch et al., 1999). These in situ findings were confirmed with immunohistochemical localization of the receptor protein using a specific hCysLT1 antiserum (Figueroa et al., 2001). In addition, the hCysLT1 mRNA and protein have been detected recently in normal peripheral blood eosinophils, subsets of monocytes and macrophages and in pregranulocytic CD34+ cells (Figueroa et al., 2001). The hCysLT1 protein was not observed on CD4+ or CD8+ peripheral T cell populations (Figueroa et al., 2001). In situ and immunohistochemical studies of the hCysLT1 in diseased lung and other tissues are needed to determine whether the expression of the hCysLT1 changes in asthma, allergy, or other pulmonary and inflammatory diseases.
Two isoforms of the mouse CysLT1 receptor (mCysLT1) have been cloned from a mouse lung cDNA library (Maekawa et al., 2001; Martin et al., 2001; Mollerup et al., 2001). A short isoform cDNA containing two exons encodes a polypeptide of 339 amino acids with 87.3% amino acid identity to the hCysLT1 (Lynch et al., 1999; Sarau et al., 1999,Maekawa et al., 2001). A long isoform has two additional exons and an in-frame upstream start codon resulting in a 13-amino acid extension at the N terminus of the receptor (Maekawa et al., 2001). Northern blot analysis of the mCysLT1 detecting both isoforms of the receptor showed expression of a 3.5-kb transcript in the lung and skin, whereas reverse transcriptase polymerase chain reaction showed wide expression of the long isoform, with strongest expression in the lung and skin. Unlike the hCysLT1, neither form of the mouse receptor is expressed in the spleen. In addition, the long rather than the short isoform of the mCysLT1receptor is the closer counterpart to the human receptor, and that is expressed more abundantly in the mouse tissues. The mCysLT1 receptor maps to the X chromosome at band XD. LTD4-induced intracellular calcium mobilization in CHO cells stably expressing either isoform of mCysLT1 was blocked by the CysLT1 receptor antagonist MK-571 (Maekawa et al., 2001). The rank order agonist functional potency for calcium mobilization or the potency for competition of LTD4 binding to the recombinant mouse receptor was similar to that described above for the human receptor, except that LTC4 was relatively less potent for the mouse than for hCysLT1 (Lynch et al., 1999; Sarau et al., 1999; Maekawa et al., 2001).
2. CysLT2.
The cloning and characterization of the CysLT2 receptor (hCysLT2) was initially reported by Heise et al. (2000) (Fig. 5). This publication confirmed the previous pharmacological characterization of a human CysLT2 receptor in different tissues, based upon the relative potencies of the cys-LT agonists and the lack of sensitivity of the responses to classical CysLT1receptor antagonists, and the antagonist activity of the partial agonist BAY u9773 (Labat et al., 1992; Tudhope et al., 1994; Heise et al., 2000). Subsequent to this initial publication, the Takeda group published an article confirming the identification of the hCysLT2 (Takasaki et al., 2000), and then a third report by the Nothacker et al. (2000), on the characteristics of the hCysLT2 was published, which revealed similar distribution and functional data to the previous publications but with more details on the partial agonist activity of BAY u9773 (Nothacker et al., 2000). Recently, the cloned mCysLT2 has also been reported (Hui et al., 2001).
The initial CysLT2 receptor identification was made through homology to a rat CysLT receptor homolog, namely, RSPBT32, followed by cloning of a human homolog of RSPBT32 (Heise et al., 2000). Using FASTA, the hCysLT2 was shown to have 37.5% amino acid identity with the hCysLT1 over a 315-amino acid overlap and the extreme carboxyl termini of these proteins have little homology (Heise et al., 2000). The hCysLT2 receptor was also identified from the Helix Research Institute database (Takasaki et al., 2000). The human chromosomal localization of the hCysLT2 in 13q14, a region that has been identified as a polygenic atopic linkage (Kimura et al., 1999).
The cRNA for the hCysLT2 was expressed inX. laevis oocytes and either LTD4 or LTC4 produced a calcium-dependent chloride flux in these cells that was not blocked by the selective CysLT1 receptor antagonist, MK-571, but was inhibited by the dual CysLT1/CysLT2 receptor antagonist BAY u9773 (Heise et al., 2000). In addition, LTD4 and LTC4 elicited a concentration-dependent activation of calcium flux in HEK-293 cells coexpressing the aequorin bioluminescent protein and the recombinant hCysLT2 (Heise et al., 2000). This activation was blocked by BAY u9773 but not significantly by the CysLT1 antagonists MK-571, montelukast, zafirlukast, or pranlukast (Heise et al., 2000). Experiments involving radiolabeled LTD4 binding to COS-7 cell membranes expressing the hCysLT2 demonstrated high-affinity competition by LTD4 and LTC4, with weaker competition by LTE4 and no effect by LTB4(Heise et al., 2000). Similar studies using radiolabeled LTD4 binding to COS-7 cell membranes expressing the hCysLT2, showed that the dual antagonist BAY u9773 fully competed for the binding, whereas CysLT1 antagonists showed no or minimal competition (Heise et al., 2000). Furthermore, Kamohara et al. (2001)have shown by in situ hybridization the presence of hCysLT2 mRNA in human atrium, ventricle, and at intermediate coronary arteries with little detection of hCysLT1. These investigators also reported that human coronary smooth muscle cells stimulated with LTC4 caused an increase in calcium mobilization. Thus the existence of a CysLT2 functional receptor in the human heart has been documented.
Northern blot analyses of hCysLT2 expression showed high expression in the human heart, adrenals, peripheral blood leukocytes, placenta, spleen, and lymph nodes with weaker expression in the brain (Heise et al., 2000; Nothacker et al., 2000; Takasaki et al., 2000). A reverse transcription polymerase chain reaction comparison of the CysLT receptors showed negligible hCysLT1expression but high expression of hCysLT2, in the heart and eosinophils (Mita et al., 2001), whereas only hCysLT1 was expressed in tracheal tissue (Takasaki et al., 2000). Both receptors were highly expressed in spleen (Heise et al., 2000; Nothacker et al., 2000; Takasaki et al., 2000). In situ hybridization analyses in the human lung demonstrated hCysLT2 mRNA strongly expressed on interstitial macrophages and weak expression on smooth muscle cells (Heise et al., 2000). In human peripheral blood monocytes, about 30% of cells expressed the hCysLT2, whereas greater than 90% of purified human eosinophils expressed the receptor (Heise et al., 2000). In the adrenal gland, in situ hybridization for the hCysLT2 demonstrated localization to the chromaffin-containing adrenal medulla cells (Heise et al., 2000). In the heart, the hCysLT2 was expressed in Purkinje fiber conducting cells (Heise et al., 2000).
The recent molecular cloning, expression and characterization of the hCysLT1 and hCysLT2receptors (Table 2) represents a significant milestone in the history of CysLT research and is predicted to lead to a renaissance in interest in this area of biology and therapeutics. The findings have confirmed much of the earlier pharmacological characterization of the two receptors, which were based solely on the functional profiling of the effects of agonists and antagonists in different cellular and tissue systems. However, there are some unexpected findings with regard to genomic structure and tissue expression, as well as the relatively low homology (38%) between the two receptors. The cloning of the receptors allows the generation of tools to investigate in more detail the regulation of CysLT1 and CysLT2 receptor expression, the roles of these CysLT receptors in normal and diseased states, and their potential as new therapeutic targets. The ultimate goals will be the identification of potent and selective CysLT2 receptor antagonists and combined CysLT1/CysLT2 receptor antagonists and their clinical evaluation in the diverse diseases in which the cys-LTs have been implicated as significant pathophysiological mediators.
C. Lipoxin Receptors
Of the nonprostanoid eicosanoid GPCRs, the LXA4 receptor (ALX) was the first recognized at the molecular level (Fiore et al., 1993, 1994). In addition, ALX was initially identified as the only inhibitory or anti-inflammatory receptor that acts via an agonist role as a “stop signal” (Fiore et al., 1994; Serhan, 1994, 1997; Takano et al., 1997). This action appears to be a unique flexibility of GPCR that functions within the immune system. Since LXA4 shares some structural features with LTC4 and LTD4as well as prostaglandins, LXA4 competed for CysLT1 receptors identified on isolated human vascular endothelial cells (Gronert et al., 2001) and mesangial cells (McMahon et al., 2000) and antagonized either LTC4- or LTD4-induced bronchoconstriction in humans (Christie et al., 1992) and animals (Badr et al., 1989; Gronert et al., 2001). In addition, lipoxin B4 has also been reported to activate another receptor. The present nomenclature for the lipoxin receptors is therefore based on the cloned receptor sequence as well as the observation that LXA4 is the natural and most potent ligand. In contrast, the putative receptor activated by LXB4 has not been cloned. ALX activation has been reported to generate intracellular stop signals (Serhan et al., 1994; Levy et al., 1997, 1999) and thereby promote resolution of inflammation.
1. Molecular and Structural Aspects of Lipoxin Receptors.
Based on the finding that functional ALX are inducible in promyelocytic lineages (HL-60 cells) (Fiore et al., 1993), several putative receptor cDNAs cloned earlier from myeloid lineages and designated orphans were screened for their ability to bind and signal in response to LXA4 (Fiore et al., 1994). When transfected into CHO cells, one of the orphans (previously denoted as pINF114 or a formyl peptide receptor-like-1 (FPRL-1), displayed both specific [3H]LXA4 binding with high affinity (K d of 1.7 nM) and demonstrated ligand selectivity when compared with LXB4, LTB4, LTD4, and prostaglandin E2(Fiore et al., 1994). LXB4 did not act via the ALX receptor and interacted with a specific receptor present on human leukocytes (Maddox and Serhan, 1996). In transfected CHO cells, LXA4 activated both GTPase and released arachidonic acid from membrane phospholipids, indicating that this cDNA encodes a functional receptor for ALX in myeloid cells. A mouse ALX receptor cDNA was also identified and cloned from a spleen cDNA library. This receptor expressed in CHO cells displayed specific [3H]LXA4 binding, and LXA4 initiated GTPase activity (Takano et al., 1997).
The ALX cDNA was originally cloned by several groups who were evaluating fMLP receptor (FPR) using the FPR cDNA as a probe under low-stringency hybridization conditions. ALX has high sequence homology (∼70%) to FPR. In light of this homology, the term FPRL1 (FPR-like-1) (Murphy et al., 1992) or FPRH1 (Bao et al., 1992) was designated by different research groups. Similarly this receptor was also known as FPR2 (Ye et al., 1992) or RFP (receptor related to FPR) (Perez et al., 1992). ALX was also cloned by Nomura et al. (1993) from human monocyte cDNA library and was designated as an orphan receptor (HM63). Both human and mouse ALX cDNA contain an open reading frame of 1051 nucleotides that encode a protein of 351 amino acids. Northern blot analysis demonstrated that ALX mRNA is ∼1.4 kb in both human and mouse (Takano et al., 1997). Deduced amino acid sequences indicated that ALX belongs to the GPCR superfamily characterized by seven putative transmembrane segments with N termini on the extracellular side of the membrane and C terminus on the intracellular side.
The overall homology between human and mouse ALX is 76% in nucleotide sequence and 73% in deduced amino acid (Takano et al., 1997). A high homology was present in the sixth transmembrane segment and the second intracellular loop, a finding that suggests an essential role for these regions in ligand recognition and signaling. The molecular evolution, as assessed from computer-based sequence analyses indicates that ALX (Fig. 2) is not related to prostanoid receptors and belongs to a rapidly growing cluster of chemoattractive peptide and chemokine receptors, exemplified by fMLP, C5a, and IL-8 receptors (Toh et al., 1995). The BLT1 receptor cloned from human HL-60 cells (Yokomizo et al., 1997) and mouse eosinophils (Huang et al., 1998) shares an overall homology of ∼30% with ALX in deduced amino acid sequences.
Human and mouse ALX cDNA, when transfected into CHO cells, displayed specific binding to [3H]LXA4, with aK d of 1.7 nM (Fiore et al., 1994) and 1.5 nM (Takano et al., 1997), respectively. Human ALX-transfected CHO cells were also tested for binding with other eicosanoids, including LXB4, LTD4, LTB4, and prostaglandin E2. Only LTD4 shows competition with [3H]LXA4 binding, with aK i value of 80 nM (Fiore et al., 1994). Moreover, LXA4 does not compete with radiolabeled LTB4 binding to either recombinant human BLT1 or isolated human peripheral blood neutrophils (Fiore et al., 1992; Chiang et al., 2000). Hence, the ability of LXA4 and 15-epi-LXA4 to block LTB4-induced responses in vivo and in isolated PMNs may not be via direct interactions at either BLT1 or BLT2. Although ALX shares ∼70% homology with FPR, ALX binds [3H]fMLP with only low affinity (K d ∼5 μM) and therefore is selective for LXA4 by 3 orders of magnitude (Fiore and Serhan, 1995).
Preparation of radiolabeled [11,12-3H]LXA4 (Brezinski and Serhan, 1991) enabled the direct characterization of specific LXA4 binding sites present on PMNs (Fiore et al., 1992). Intact PMN demonstrated specific and reversible [11,12-3H]LXA4 binding (K d ∼0.5 nM andB max ∼1,830 sites/PMN) that are modulated by guanosine stable analogs. These LXA4-specific binding sites are inducible in HL-60 cells exposed to differentiating agents (e.g., retinoic acid, dimethyl sulfoxide, and phorbol 12-myristate 13-acetate) and confer LXA4-induced phospholipase D activation in these cells (Fiore et al., 1993). The various ligands that activate the ALX receptor are presented in Table 6. These results provided further evidence that LXA4interacts with specific membrane-associated receptors on human leukocytes. These binding data have corroborated with the functional results for LXA4. In contrast, although functional studies (Maddox et al., 1998) have indicated the existence of a receptor activated by LXB4, this receptor has not been cloned and is presently referred to as a putative receptor. In addition, the receptor binding investigations with LXB4 must await the synthesis of a suitable LXB4 radiolabel with high specificity. Such an analog has proven to be quite difficult to achieve.
Ligand specificity for human ALX
Northern blot analysis of multiple murine tissues demonstrated that, in the absence of challenge to the host, ALX mRNA is most abundant in neutrophils, spleen, and lung with lesser amounts in heart and liver (Takano et al., 1997). In humans, ALX mRNA is also abundant in PMNs, as well as in spleen, lung, placenta, and liver (Fiore et al., 1994;Takano et al., 1997). In human enterocytes, ALX is inducible by IL-13 and interferon present in crypt and brush-border colonic epithelial cells (Gronert et al., 1998). The location of this receptor provides additional evidence that ALX is not associated with bacterial chemotaxis as observed for N-formyl peptide FPR-signaling since the gastrointestinal tract always has a very high level of bacteria present.
D. Receptors and Cellular Signals
Whereas the cascades of cellular events subsequent to GPCR activation have been the subject of many investigations, the exact signal transduction mechanisms for either the leukotrienes or the lipoxins have not been completely elucidated. Generally, agonist interactions with GPCRs involve activation of heterotrimeric G-proteins associated with a group of conventional cellular events. However, effectors for GPCRs that are independent of G-proteins are also known to exist (Hall et al., 1999). G-proteins, composed of α-, β-, and γ-subunits each encoded by a different gene, appear often to be cell specific. Upon ligand-receptor activation, the Gα- and Gβγ-subunits stimulate a variety of intracellular molecular systems. Furthermore, G-protein activation leads to increases in intracellular Ca2+ and modifications in a number of membrane ion channels.
The cellular responses to ligand activation of GPCRs can also be up-regulated through priming of cells and down-regulated by desensitization. Two types of desensitization have been described, one that results from phosphorylation of the agonist-occupied receptor by G-protein-coupled receptor kinases. These phosphorylated receptors are associated with the arrestin family of proteins. A second type of rapid desensitization (loss of response) following phosphorylation by either second messenger-activated kinases (protein kinase A, protein kinase C) or inhibition of phospholipase C, which are activated by different receptors or signaling processes. Generally, this second type of desensitization does not require agonist-receptor occupancy. In addition, Didsbury et al. (1991) also demonstrated “cross-receptor desensitization”, a phenomenon that has been reported for the chemoattractant family receptors. Presently, an exploration of these latter mechanisms associated with the actions of leukotrienes and lipoxins at the molecular level has received little attention.
1. BLT.
Investigations involving the intracellular signaling of BLT receptor activation have been performed in peripheral leukocytes specifically granulocytes. One of the problems involved in such studies is that these cells have a limited life span (24 h) making drug and transfection studies difficult. These limitations have caused several investigators to use either CHO cells expressing human BLT receptors (Yokomizo et al., 1997) or to perform reconstitutional studies with the heterotrimeric GTP-binding proteins (Miki et al., 1990; Igarashi et al., 1999). Although high-affinity binding of LTB4 (BLT1 receptor) is found essentially in leukocytes and macrophages, the G-proteins associated with the functions in these cells has not been clearly established. Furthermore, the intracellular signaling pathways for BLT may depend on the G-proteins expressed in the different cells. For example, most of the LTB4-dependent signals in granulocytes appear to be mediated by Gi-like G-proteins, (granulocytes express abundant Gαiproteins, mainly Gαi2), whereas in the nervous system Gi1 and Go are mainly present (Simon et al., 1991). In several cell types, LTB4 signals via Gi-proteins are inhibited by pretreatment of pertussis toxin (PTX). However, LTB4-induced calcium mobilization in CHO-BLT1 was not affected by PTX, suggesting the coupling with Gq-like molecules in these latter cells. Chemotaxis and inhibition of adenylyl cyclase by LTB4 were completely PTX-sensitive in CHO-BLT1 cells. The coupling of BLT1 with various Gα-subunits was examined by cotransfection studies using COS-7 cells, and BLT1-mediated phospholipase C activation was shown to be mediated by Gαi6- and Gβγ-subunits released from Gαi (Gaudreau et al., 1998). When expressed heterologously in CHO, HeLa, and COS-7 cells, BLT2 activation led to the inhibition of adenylyl cyclase and an increase in calcium. However, BLT2 activation was less potent in mobilizing calcium than BLT1 receptor activation (Yokomizo et al., 2000). BLT2 was also shown to mediate LTB4-dependent chemotaxis through Gi-like G-proteins (Kamohara et al., 2000;Yokomizo et al., 2000). Recently, Woo et al. (2002) have suggested that LTB4 stimulation of the Rac-extracellular signal-regulated kinase cascade associated with the generation of reactive oxygen species-mediated chemotaxis in Rat-2 cells was via activation of the BLT2 receptor. This suggestion, although not conclusive, was supported by the observations that BLT1 expression has not been detected in Rat-2 fibroblasts whereas BLT2 was expressed. Furthermore, the LTB4 stimulation of reactive oxygen species was observed at high concentrations (0.3–1 μM), which are within the range for BLT2 activation and are 2 orders of magnitude higher than that observed for activation of BLT1. In addition, this LTB4 stimulation was blocked by ZK 158252. In an attempt to understand the mechanisms involved in BLT receptor desensitization, Gaudreau et al. (2002) have reported some initial molecular evidence. These investigators showed that the cytoplasmic tail of BLT1 receptor was intimately involved in the regulation of desensitization and that the amino acid threonine (Thr308) was implicated in the GPCR-specific kinase phosphorylation associated with this phenomenon. This study therefore provides pertinent leads for understanding those structural elements associated with BLT1 receptor regulation.
The exact role of calcium activation subsequent to the interaction of LTB4 with a specific receptor exhibits similar complexity. Chemotaxis of human PMN or monocytes to LTB4 does not absolutely require the mobilization of intracellular Ca2+ (Romano et al., 1996; Kuhns et al., 1998). While activation of the LTB4receptor does stimulate intracellular Ca2+mobilization (Powell et al., 1996), this Ca2+appears not to be essential for chemotaxis and plays a role only in degranulation and other responses of human neutrophils (Serhan et al., 1982; Luscinskas et al., 1990; Rola-Pleszczynski et al., 1993). Such studies indicate that the unraveling of the intracellular events associated with the BLT receptor activation with the second messenger cascades are only beginning to be understood.
2. CysLT.
Unfortunately, most studies concerning the CysLT receptors have involved only LTD4 activation of CysLT1 receptors. There is little information available concerning G-protein and Ca2+mobilization when the CysLT2 receptor is activated. Initial studies (Kuehl et al., 1984; Crooke et al., 1989,1990; Watanabe et al., 1990) demonstrated that LTD4 activation of the CysLT1 receptor lead to G-protein activation and the release of several second intracellular messengers, namely, diacylglycerol, inositol phosphates, and Ca2+, events which were followed by activation of protein kinase C (PKC) and accompanied by the mobilization of Ca2+ derived from both intracellular and extracellular stores. Clark et al. (1985)demonstrated that LTD4 activation of CysLT1 receptors also led to the release of arachidonic acid via stimulation of phospholipase A2, which was associated with an enhanced transciption of phospholipase A2 activating protein. Expression of this latter protein was controlled by activation of topoisomerase I, which in turn was regulated by PKC (Mattern et al., 1991).
LTD4 is known to induce either Ca2+ influx through the plasma membrane without any Ca2+ release from intracellular stores (Baud et al., 1987a; Oliva et al., 1994), or Ca2+ release without influx (Bouchelouche et al., 1990) and in many cell types can induce both Ca2+influx and release (Mong et al., 1984b, 1987a, 1988; Sjolander et al., 1990). In human airways, Gorenne et al. (1998) demonstrated that LTD4-induced contractions may mobilize Ca2+ via a receptor-operated channel. Recently,Accomazzo et al. (2001) provided evidence that LTD4 contractions in human airways did not elicit a marked elevation in [Ca2+]i but rather activated Ca2+ via a Ca2+-independent isoform of protein kinase C (PKCε). These observations supported an initial report that PKC activation may be associated with increased force development at constant [Ca2+]i (Masuo et al., 1994). In addition, PKCε was shown to be necessary for the generation of the LTD4-induced Ca2+ signal in intestinal epithelial cells. Together these results suggest that the Ca2+signaling for LTD4 contractions in human airways may involve several intracellular pathways. Unfortunately, the other ligands (LTC4 and LTE4) have not been examined in these studies. Interestingly, Sjolander et al. (1990) proposed that LTD4 and LTE4 activated different receptors associated with different intracellular signals. However, these observations have not been pursued. The initial observations of Gronroos et al. (1995,1996, 1998) have recently received support from several investigators (Hoshino et al., 1998; Thodeti et al., 2000; Massoumi and Sjolander, 2001; Paruchuri et al., 2002) and provided evidence that LTD4 activation of an epithelial cell line (THP-1) led to an association of a protein other than the heterotrimeric G-proteins and this mobilization was necessary for the mobilization of calcium. Although these investigators observed that LTD4 activation of THP-1 cells altered mitogen-activated protein kinase via protein kinase C and Raf-1, an effect which was PTX-insensitive, they also reported that the chemotactic response of these cells was PTX-sensitive. These observations suggest that diverse pathways at the G-protein level may also exist when LTD4 interacts with the receptor. However, these investigators did not evaluate different receptor antagonists to determine which receptor was associated with these different pathways.
3. Lipoxins.
The cytoplasmic signaling cascade of the ALX receptor is also highly specific and selective for different cell types. In human PMNs, LXA4 stimulates rapid lipid remodeling and release of arachidonic acid via a PTX-sensitive G-protein (Nigam et al., 1990) and blocked intracellular generation of inositol 1,4,5-trisphosphate (Grandordy et al., 1990) as well as Ca2+ mobilization (Lee et al., 1989). In contrast, in human monocytes and THP-1 cells, LXA4 triggers intracellular calcium release (Romano et al., 1996; Maddox et al., 1997), suggesting a different intracellular signaling pathway than in PMNs despite identical receptor sequences. In addition, distinct signaling in monocyte and PMNs was further supported by different responses to LXA4in these cell types. LXA4 modulates mitogen-activated protein kinase activities in mesangial cells in a PTX-insensitive manner (McMahon et al., 2000), suggesting the presence of an additional ALX receptor subtype and/or signaling pathway for ALX. Since the ALX receptor has been shown to switch recognition and function with certain chemotactic peptides, the G-proteins and intracellular pathways involved may prove to be a difficult but fascinating area to explore. One of the problems presently confronting investigators in this area of research is the availability of the ligands. Studies on G-protein and intracellular messengers are presently limited (Kang et al., 2000), since stable analogs for LXA4 and LXB4 have only recently become available.
E. Summary
Within the last few years, a considerable effort at the molecular level has been undertaken to identify the leukotriene receptors. However, data involving chimeric constructs of the leukotriene receptors have only recently been reported (Gaudreau et al., 2002). In contrast, there are several observations that warrant further investigation. For example, the mouse CysLT1cloned receptor is activated by all three native ligands and antagonized by MK-571. However, the ligand profile for the mCysLT1 is quite different from that observed in the human CysLT1 receptor, since the mouse CysLT1 receptor exhibited little response to LTC4. An explanation for this difference is not readily apparent. In addition, MK-571 potentiated Ca2+ mobilization in CHO cells transfected with mCysLT1 long isoform cDNA (Maekawa et al., 2001). The exact reason for this specific effect has not been explored. Recently, Ogasawara et al. (2002) reported different pharmacological properties of the CysLT2 receptor between human and mouse, and they also showed that mCysLT1 and mCysLT2 tissue expression was different in some strains of mice. These data suggest that receptor distributions may be different within and between species.
IV. Properties and Significance of Leukotriene Receptors
Well before the cloning of the leukotriene receptors, a considerable amount of indirect evidence had already suggested their existence. To date, the molecular database has confirmed the pharmacological database. However, a more thorough appreciation of ligand-receptor interactions under both physiological and pathophysiological conditions can only be achieved by an assessment of both these databases. Therefore, an effort has been made in this section to provide the evidence that reported the properties and significance of leukotriene receptors and to highlight those observations that suggested other receptor subtypes may exist.
A. BLT Functional and Radioligand Binding Studies
As early as 1974, certain monohydroxy eicosanoids, including the platelet arachidonate lipoxygenase product (12-HETE), were shown to be neutrophil chemoattractants (Turner et al., 1975) as well as other monohydroxy products (5-HETE, 12-HETE) (Goetzl and Pickett, 1980;Powell et al., 1996). In addition, certain derivatives of LTB4, namely, 12-oxo-LTB4and 20-hydroxy-LTB4 were reported to induce chemotaxis (Yokomizo et al., 1993, 1996). However, each required higher concentrations than LTB4 to induce chemotaxis in experiments when cells were placed in isolated Boyden-type chambers (Ford-Hutchinson et al., 1980). Furthermore, radioligand binding studies (Goldman and Goetzl, 1982; Snyder et al., 1984; Goldman et al., 1985; Gorman et al., 1985; Falcone and Aharony, 1990; Maghni et al., 1991) provided more support for the existence and distribution of specific receptors for LTB4. The differences in relative potencies between LTB4 and the mono-HETEs (LTB4 ≫ HETE) also provided evidence and facilitated the identification and classification of the cognate receptor. However, the leukocyte surface receptors on neutrophils involved in chemotaxis by which other mono-HETEs activate these cells (Powell et al., 1996; Falgueyret and Riendeau, 2000) have not been identified.
B. Cysteinyl-Leukotriene Functional Studies
1. Airway Smooth Muscle Contraction.
Drazen et al. (1980) reported that LTC4 and LTD4 were equipotent in guinea pig tracheal smooth muscle preparations whereas LTD4 was a more selective peripheral airway agonist, since LTD4 exhibited a 100-fold greater potency than LTC4 in the lung parenchymal strip suggesting the presence of two CysLT receptors. The heterogeneity of leukotriene receptors received further support from Krell et al. (1983) who showed that the potency of LTD4 in the guinea pig lung parenchymal tissues (pEC50, 9.2) was significantly different from that observed in the tracheal preparations (pEC50, 7.8). In addition, Fleisch et al. (1982)demonstrated that the potency of LTD4 was different in tissue preparations derived from various organs. These investigators reported that in the guinea pig ileum LTD4 had a pEC50 of 9.2, whereas in the trachea from this species the pEC50 was 8.5. However, when contractile studies were performed on human airways, results showed that LTC4, LTD4, and LTE4 were equipotent as contractile agonists (Buckner et al., 1990; Labat et al., 1992) with pEC50 values of approximately 10 nM, that is, about one-thousandth of the pEC50 values for histamine in the same tissues (Dahlén et al., 1980). However, LTE4 was markedly less active than LTC4 and LTD4 in these tissues (Buckner et al., 1986). A comparison of the cys-LT agonist potencies between different tissues illustrates that considerable variation exists in functional assays (pEC50values 11 to 7) (Jones et al., 1984; Hand and Schwalm, 1987; Gardiner et al., 1990; Gieske et al., 1990; Norman et al., 1990; DeLegge et al., 1993). Based solely upon agonist potencies, one is unable to surmise whether either a single or several receptors are present in these preparations. This requires additional information from structural, operational, and molecular biological studies. However, the data suggest a marked difference between LTE4 and the other cys-LTs. Interestingly, human vascular preparations exhibited exquisite sensitivity to these mediators (Allen et al., 1994).
Results using the SRS-A receptor antagonist, FPL 55712, further supported the hypothesis for the existence of distinct receptors for the cys-LTs. Drazen et al. (1980) reported a pK B value for FPL 55712 of 6.9 in the lung parenchymal tissues when LTD4 was used as the agonist with a Schild plot slope of 0.72; similar results were demonstrated by Krell et al. (1981). Furthermore, these latter investigators showed that the potency of FPL 55712 (pK B) for antagonism of LTD4-induced contraction in the parenchyma decreased with increasing concentrations of this antagonist, suggesting that in this tissue either factors may be released which were not blocked by FPL 55712 or the profile of antagonism of this compound was not competitive. In the guinea pig trachea, Krell et al. (1981) calculated a pK B (6.4) for FPL 55712 for inhibition of LTD4 contraction. When these preparations were contracted with LTC4, the pK B for FPL 55712 was similar to that obtained against LTD4. In contrast, when tracheal tissues were treated with indomethacin the pK B obtained against LTC4 was 5.7. Thus, in the trachea, while FPL 55712 antagonized both LTD4-induced and LTC4-induced contractions, the pK B values differed (6.4 and 5.7, respectively). In parenchymal tissues, contractions to LTD4, but not those to LTC4, were inhibited. Fleisch et al. (1982)reported their results with FPL 55712 against LTD4 contractions in different tissues and demonstrated pK B values of 6.1 and 6.3 in parenchyma and trachea whereas in the ileum the value was 7.1; the pA 2 values were calculated to be 7.3 for the ileum, 6.5 for the trachea, and 6.0 for the parenchyma. These data also indicated that the dissociation constants for FPL 55712 derived from the trachea and parenchyma were markedly different from that obtained in the ileum. Additional experiments with FPL 55712 supported the notion that different leukotriene receptors existed, since FPL 55712 failed to antagonize the LTC4contractions in the lung parenchymal strips and exhibited a pK B of 9.2 against LTD4-induced contractions in the guinea pig trachea. Since FPL 55712 was reported to have effects other than leukotriene antagonism, including PDE inhibition (Welton et al., 1981), the development of other specific compounds was required to establish more definitive evidence for the presence of at least two receptors. Many compounds with much greater potency (K i values of about 0.1–10 nM; pA 2 values or pK B values of 9.5–7.5) and markedly improved selectivity compared with FPL 55712 have been pharmacologically identified and comprehensively characterized (Jones et al., 1983; Buckner et al., 1986; Tucker and Weichman, 1986; Hay et al., 1987; Krell et al., 1987, 1990; Muccitelli et al., 1987; Jones et al., 1989, 1995; Torphy et al., 1989; Labat et al., 1992; Gardiner et al., 1994). Collectively, the results with these chemically distinct antagonists (Table 5) provided pertinent pharmacological support for the presence of two receptors in the various tissue preparations.
2. Vascular Smooth Muscle Contraction.
Hanna et al. (1981)reported that cys-LTs contracted not only isolated human airways but also human pulmonary veins and noted that the maximum responses on human pulmonary arteries were small (Schellenberg and Foster, 1984;Bourdillat et al., 1987). Berkowitz et al. (1984) studied vascular preparations from several species (rat, rabbit, and guinea pig) and observed small contractions in guinea pig pulmonary veins, the inferior vena cava and jugular vein; the effects of LTC4were not examined. In the rabbit renal vein, LTC4- and LTD4-induced contractions (Kito et al., 1981), although only small or no contractile responses, were observed in pulmonary and portal veins. Gleason et al. (1983) also reported that LTD4-induced contractions in the guinea pig-isolated pulmonary artery were small. Together these observations suggested that a number of vascular preparations exhibited little or no contractile response to the cys-LTs. Whether this lack of contraction was due to the absence of receptors or to factors that are released and mask the response has not been systematically studied (Bäck et., 2002). However, in human vascular preparations, Allen et al. (1992) reported a pronounced constriction by LTC4 and LTD4 in human-isolated saphenous veins and the agonists were equipotent.
Nishiye et al. (1988) showed that FPL 55712 and ONO-RS-411 blocked the LTD4 contractions in the guinea pig basilar artery demonstrating a single specific receptor. However, in human pulmonary veins, unlike those in human airways, the contractions induced by cys-LTs were not affected by the CysLT1 receptor-selective antagonists MK-571 and ICI 198615 (Labat et al., 1992; Gorenne et al., 1996). These results suggested the presence of distinct CysLT receptors on the human pulmonary veins compared with either human bronchus or trachea (Jones et al., 1982; Buckner et al., 1986). In contrast, Rinkema et al. (1993)showed that the LTD4 contractions in the guinea pig inferior vena cava were blocked by tomelukast and WY 48252. However, the contractions induced by LTC4 were blocked in a biphasic fashion by these two antagonists, that is, the low concentrations of LTC4 were not affected by the antagonists suggesting two CysLT receptors. Therefore, in some species, such as the guinea pig, vascular smooth muscle may contain either one or two CysLT receptors associated with the contractions. Whether the CysLT receptors in the human pulmonary veins, which are resistant to these CysLT1 receptor antagonists, are the same as the receptors in the guinea pig vena cava, which are activated by LTC4, remains to be established. Recently, Bäck et al. (2000b) have reported that contractions induced by LTC4 in porcine pulmonary arteries were resistant to MK-571 (CysLT1antagonist) as well as BAY u9773 (dual antagonist, CysLT1/CysLT2), suggesting that another receptor may be associated with this tissue response.
3. Vascular Smooth Muscle Relaxation.
Secrest et al. (1985)reported that, in canine renal arteries where tone had been induced by a contractile agonist, LTD4 provoked relaxations. Similar results were obtained with LTC4 and LTD4 in guinea pig pulmonary artery (Sakuma et al., 1987; Sakuma and Levi, 1988) and isolated human saphenous veins (Allen et al., 1992), and LTD4 in isolated human pulmonary arteries and veins (Ortiz et al., 1995). LTD4 relaxed canine renal blood vessels and was approximately 100-fold more potent on veins than arteries. Whether or not this latter difference is due to receptor density or receptor-effector coupling is presently unknown. LTC4 was not examined in these tissues. In canine splanchnic venous capacitance vessels similar relaxations for LTC4 and LTD4 have been observed. Furthermore, in canine coronary arteries both LTC4 and LTD4 relaxed the tissues with the latter agonist being 10 times more potent. The data reported indicate that the agonist potencies for relaxation of human vascular preparations (pEC50 values, approximately, 11–10) (Allen et al., 1992; Ortiz et al., 1995) are markedly different from the potencies required to produce contractions in the same tissue (pEC50 values, approximately, 9–7) (Schellenberg and Foster, 1984; Bourdillat et al., 1987; Allen et al., 1992; Labat et al., 1992; Ortiz et al., 1995; Stanke-Labesque et al., 2000). Such data suggest that either the receptors on the endothelium associated with the relaxation are different from the receptors responsible for the contraction or the G-protein second messengers may be coupled more efficiently. In addition, CysLT1 receptor antagonism of contractions in preparations from the human lung (Table7) suggest that the receptor present in vascular smooth muscle may be different from that in the human airways. Interestingly, the LTD4-induced relaxation of isolated human pulmonary arteries and veins has also been reported to be resistant to CysLT1 antagonists, MK-571 and ICI 198,615 (Ortiz et al., 1995). The question remains as to whether or not the receptor present on the endothelium and associated with the relaxation is the same as that present on the vascular smooth muscle and responsible for the contraction.
CysLT1 antagonists: effects in human lung preparations
Secrest et al. (Secrest et al., 1985; Secrest and Chapnick, 1988) reported that the endothelium-dependent relaxations were produced by activation of a specific receptor since the response was attenuated by FPL 55712. Sakuma and Levi (1988) observed that the relaxations induced by LTC4 and LTD4 in the isolated guinea pig thoracic aorta were antagonized with similar potency by the CysLT1 receptor antagonist, ICI 198,615, suggesting that both agonists activate a single receptor (CysLT1). This antagonism was also observed in the guinea pig pulmonary artery. Therefore, on the endothelium of guinea pig arterial preparations, a single receptor was present (Sakuma and Levi, 1988), and activation was associated with relaxation. In contrast, this does not appear to be the case in the canine renal arteries and veins (Pawloski and Chapwick, 1993a; Pawloski and Chapwick, 1993b) or in the human pulmonary arteries and veins (Ortiz et al., 1995). In the latter study, the endothelium present on the human pulmonary veins contains two CysLT receptors: a CysLT2-type responsible for relaxation and a CysLT1 receptor associated with the release of a contractile factor (Ortiz et al., 1995).
4. Cardiovascular Effects.
Subsequent to the early investigations (Levi and Burke, 1980; Levi et al., 1980), which reported that partially purified SRS markedly reduced coronary blood flow, a number of other investigators have explored the cardiovascular effects of the cys-LTs in a variety of different animal models. The early studies have shown that the cys-LTs are potent vasoconstrictors of the coronary vasculature in both isolated perfused hearts (Woodman and Dusting, 1982; Letts and Piper, 1983; Letts et al., 1983) and in a number of species in vivo (Michelassi et al., 1982; Panzenbeck and Kaley, 1983; Fiedler et al., 1984, 1987; Leffler et al., 1984; Fiedler and Abram, 1987; Kopia et al., 1987; Michelassi et al., 1987) including human (Marone et al., 1988). In sheep and pigs, cys-LTs induced not only coronary vasoconstriction but also ischemia and impaired left ventriclar function (Michelassi et al., 1982; Ezra et al., 1983;Fiedler et al., 1985). In isolated perfused guinea pig heart preparations (Terashita et al., 1982; Letts and Piper, 1983; Roth et al., 1985), LTC4 and LTD4caused a reduction in myocardial contractility concommitant with the vasoconstriction. In perfused rat hearts (Bittl et al., 1985), LTD4 caused not only a reduction in flow but also reduced the spontaneous heart rate to a greater extent than contractility, suggesting an action on conductivity in this species (Feuerstein et al., 1981; Zukowska-Grojec et al., 1982; Zukowska-Grojec et al., 1984; Tomoike et al., 1987).
Burke et al. (1982) also showed that FPL 55712 antagonized the cardiodepressant effects of the cys-LTs suggesting that these effects were via activation of a specific receptor and noted that the potency of FPL 55712 was greater against ventricular contraction induced by LTD4 compared with those of LTC4, suggesting that there may be two receptors present in the guinea pig heart. These investigators also reported that in the human heart, the negative inotropic effect of cys-LTs was similar to that of the guinea pig and the rank order potency (LTD4 > LTC4>LTE4) was the same for both species. In other studies using isolated guinea pig cardiac muscle preparations, the data have been contradictory. Letts and Piper (1983) reported no myocardial effects whereas other investigators (Falcone et al., 1991a,b) have demonstrated positive inotropic affects.
The use of potent and selective CysLT1 receptor antagonists provided an alternate approach for establishing the effects of cys-LTs in the perfused heart preparations. McLeod and Piper (1991)reported a differential antagonistic effect against LTC4 and LTD4. ICI 198,615 selectively antagonized LTD4-induced increases in coronary vascular resistance with no significant effect against LTC4. Pobilukast antagonized both LTC4 and LTD4 responses whereas MK-571 was without effect on cardiac vascular resistance. However, ICI 198,615 antagonized both LTC4- and LTD4-induced ventricular contractility. These data suggested that the reduction in contractility by LTC4 may be more sensitive to the actions of the antagonists when compared with their action against the vasoconstrictory effect of LTC4. The failure of MK-571 to antagonize the coronary vasoconstriction was also observed against ventricular contractility induced by LTC4and LTD4. In contrast, Falcone et al. (1990)showed specific binding of [3H]ICI 198,615 to guinea pig cardiac ventricular membranes, suggesting the existence of CysLT1 receptors in this tissue, whereas Egan et al. (1989) demonstrated that selective antagonism of the leukotriene responses did not reduce myocardial effects. Although the data from animal studies remain controversial, the results derived from the guinea pig indicate that the antagonist profile in the guinea pig heart and lung preparations were not similar and suggested that the receptors in cardiac vessels may be different from those in the airways of this species.
Marone et al. (1988) reported the effects of cys-LTs following intracoronary injection in man. These investigators showed that low doses of exogenous LTD4 and LTC4 induced immediate changes of cardiovascular function consisting of a fall in blood pressure, a rise in heart rate, and sympathoadrenergic activation. These initial alterations were followed by a marked vasoconstriction of small resistance coronary vessels. In another study, Albazzaz et al. (1989) assessed the cardiopulmonary effects of leukotriene C4inhalation in nonasthmatic and asthmatic patients. Although the mean cardiac output decreased without changes in blood pressure and heart rate in both subjects, there was difficulty in separating the cardiac output modifications from the relative contribution of the pulmonary vasoconstriction, bronchoconstriction, and reduced cardiac output, which followed the challenge. The use of the potent and selective CysLT1 receptor antagonists for such in vivo studies in man may provide an alternative for understanding the role of these mediators in cardiovascular disease.
5. Diverse Effects of Cysteinyl-Leukotrienes.
Human umbilical vein endothelial cells (HUVECs) have been reported to produce (Sjostrom et al., 2001) as well as respond to all cys-LTs (Datta et al., 1995) by releasing von Willebrand Factor as well as an increased expression of P-selectin The effects were blocked by the CysLT1receptor antagonist, pobilukast. These investigators demonstrated a concomitant surface expression of P-selectin and release of high-molecular weight von Willebrand Factor by the cys-LTs due to stimulation of exocytosis of the Weibel-Palade body pools rather than promoting the constitutive release from endothelial cells. In contrast,Pedersen et al. (1997) showed that the P-selectin secretion induced by LTD4 and LTC4 was not inhibited by three different CysLT1 receptor antagonists (zafirlukast, SB 205,312 and pobilukast). However, in this latter study the pool of P-selectin monitored by their assay was not described. The difference between the effects of the CysLT1 antagonists may depend on which pool of P-selectin was assayed rather than a subset of receptors.
There are several reports that the cys-LTs increase microvascular permeability in guinea pig airways (Woodward et al., 1983a,b; Evans et al., 1989; Obata et al., 1992; Bochnowicz and Underwood, 1995); the mechanism may involve contraction of endothelial cells resulting in gaps in the endothelium of venules (Joris et al., 1987). This influence of the cys-LTs, which appeared to involve both direct and indirect pathways, was inhibited by either FPL 55712 (Woodward et al., 1983a) or pranlukast (Nakagawa et al., 1992; Bochnowicz and Underwood, 1995) indicating that a CysLT1 receptor was involved.
There is increasing evidence that the cys-LTs may also influence inflammatory cell function since they enhance the recruitment of eosinophils. For example, inhaled LTE4 (Laitinen et al., 1993) or LTD4 selectively increased airway eosinophil numbers when assessed in bronchoalveolar lavage from humans (Smith et al., 1993). Similar results were obtained with LTC4 or LTD4 in guinea pigs (Foster and Chan, 1991; Underwood et al., 1996), with single administration of inhaled LTD4 elicited a marked increase in eosinophils, which was maintained for at least 4 weeks (Underwood et al., 1996). The receptor responsible for this phenomenon of the cys-LTs appears to be CysLT1 receptor (Foster and Chan, 1991; Underwood et al., 1996) and, at least in guinea pigs, appeared to involve IL-5 (Underwood et al., 1996), a key cytokine in eosinophil proliferation, activation, and survival. Pobilukast inhibited LTD4-induced chemotaxis of peripheral blood eosinophils from nonasthmatic volunteers (Spada et al., 1994). Furthermore, the CysLT1 receptor antagonists pranlukast, MK-571, ICI 198,615 inhibited antigen-induced eosinophil influx in rats (Salmon et al., 1999), guinea pigs (Foster and Chan, 1991), and cynomolgus monkeys (Turner et al., 1994).
The effects of the cys-LTs on mucus secretion have been explored only to a limited degree. Initial studies involved the incorporation of radiolabels such as [3H]glucosamine into secretory cells (Shelhamer et al., 1980; Coles et al., 1983; Kim et al., 1989, 1993). Although the cys-LTs were shown to increase release of radiolabeled material from the secretory elements of the human bronchus, there is little information on the effects of selective antagonists (Lundgren et al., 1990). LTC4 or LTD4 induced an increase in mucus secretion from cultured human airway mucosal explants, an effect which was antagonized by FPL 55712 (Marom et al., 1982; Coles et al., 1983). Pobilukast inhibited the aerosolized LTD4-induced increase in epithelial mucus secretion in guinea pig airways in vitro (Hoffstein et al., 1990), suggesting that the effects of the cys-LTs on mucus secretion are CysLT1 receptor-mediated. Labat et al. (1999) have reported that challenge of human bronchial preparations with LTD4 failed to elicit increased amounts of the MUC5AC gene product, suggesting that there may or may not be alterations in the secretory activity of human airways depending upon which secretory product is monitored.
Panettieri et al. (1998) reported that although LTD4 alone had no significant effect on DNA synthesis in human cultured airway smooth muscle cells, there was a marked potentiation in the airway smooth muscle proliferation induced by the classical mitogens, epidermal growth factor, or thrombin. This effect of LTD4 was inhibited by either pranlukast or pobilukast, but not zafirlukast, suggesting activation of a CysLT receptor other than CysLT1 (Panettieri et al., 1998). Evidence for an influence of the cys-LTs on airway smooth muscle proliferation is derived from the results of two in vivo studies in rats following chronic antigen challenge where airway smooth muscle proliferation and hyperreactivity (Wang et al., 1993) or eosinophil infiltration (Salmon et al., 1999) were attenuated by the CysLT1 receptor antagonists, MK-571 (Wang et al., 1993) or pranlukast (Salmon et al., 1999).
There is also evidence in guinea pigs that the cys-LTs may influence lung function by modulating the afferent nervous system, specifically enhancing activity of capsaicin-sensitive sensory C fibers (Stewart et al., 1984; Undem and Weinreich, 1993; Undem et al., 1993). For example, LTD4 enhanced the release of substance P (Bloomquist and Kream, 1990) and potentiated tachykinin-mediated, nerve-induced responses (Ellis and Undem, 1991) in guinea pig trachea. Vagal nerve-induced contraction and microvascular permeability in guinea pig airways are inhibited by CysLT1receptor antagonists (Ellis and Undem, 1991), indicating that the effects of the cys-LTs on tachykinin-containing sensory C fibers are mediated via the CysLT1 receptor in guinea pig airways.
There are a number of other observations concerning the cys-LTs that require further investigation. Few studies have been performed to examine these mediators in angiogenesis (Tsopanoglou et al., 1994) where pobilukast has been reported to block responses to LTD4, albeit with less potency than those to LTC4. The effects of these mediators on liver metabolism has been overlooked but an investigation has demonstrated that both LTD4 and LTC4 are equipotent in altering glucose and lactate output, and this modification is blocked by CGP 35949, a CysLT1antagonist (Iwai and Jungermann, 1988, 1989). In addition, investigations on the actions of the cys-LTs on renal function (Chapnick, 1984; Filep et al., 1985; Piper et al., 1985; Petric and Ford-Hutchinson, 1995), the pulmonary circulation (Smedegard et al., 1982; Kadowitz and Hyman, 1984; Jones and Masson, 1985; Malik et al., 1985; Garcia et al., 1987; Ohtaka et al., 1987; Schreiber et al., 1987), systemic circulation (Pfeffer et al., 1983), and on the microcirculation (Dahlén et al., 1981; Bisgaard et al., 1982,1985, 1986; Soter et al., 1983; Chan and Ford-Hutchinson, 1985; Hua et al., 1985) to established specific CysLT receptors have not been pursued.
C. CysLT Radioligand Binding Studies
The presence of heterogeneous CysLT receptors was also supported by data from experiments using classical radioligand binding techniques. Cheng et al. (1985) provided evidence that the CysLT receptors were widely distributed in different organs, tissues, and species. Furthermore, autoradiographic studies with the labeled agonists have also demonstrated the presence of CysLT receptors in lung (Carstairs et al., 1988), endothelial cells (Chau et al., 1986), uterus (Chegini and Rao, 1988b), corpus lutea (Chegini and Rao, 1988a), as well as brain (Goffinet, 1986). In addition, a number of smooth muscle or transformed cells in culture were also shown to possess these receptors (Krilis et al., 1983; Mong et al., 1988; Frey et al., 1993). The radioligand binding studies have not only demonstrated the presence of specific sites for [3H]LTC4, [3H]LTD4, and [3H]LTE4 binding but also have shown that there is often a preferential binding for one ligand and not the other (Mong et al., 1984a, 1985; Cheng et al., 1985). These latter observations suggested that certain tissues may contain a single class of receptor responsible for the physiological actions of cys-LTs. Although a considerable amount of evidence demonstrated that membrane preparations from a number of tissues bind all the radiolabeled agonists, there are data suggesting the existence of at least two sites: one associated with [3H]LTC4 and one that binds [3H]LTD4 and [3H]LTE4 (Cheng and Townley, 1984b).
However, three major points became apparent from the radioligand binding studies that need to be highlighted. First, interpretation of the results of [3H]LTC4binding assays were complicated by the rapid metabolic transformation of LTC4 to LTD4 if this was not controlled by enzymatic inhibition. Inhibitors of this metabolism must frequently be incorporated in [3H]LTC4 binding studies. In addition, recent binding studies (Ravasi et al., 2000) have also suggested the necessity to include S-decyl-glutathione (a high-affinity ligand for nonreceptor LTC4 binding sites) in the membrane preparations. Second, there was often no correlation between the [3H]LTC4 binding studies and the functional studies (contraction) as in the case of either the guinea pig ileum (Norman et al., 1990), guinea pig lung strip (Mong et al., 1985; Norman et al., 1987), or guinea pig uterus (Weichman and Tucker, 1982; Levinson, 1984). In contrast, a correlation was observed between the antagonist effects in the [3H]LTD4 binding studies and the ability of the antagonists to block LTD4-induced contractions (Fig.6). Furthermore, in the rat lung the radioligand binding data provided evidence for the presence of two sites (Pong et al., 1983; Metters et al., 1991) whereas the functional data (contraction) suggested a single receptor was associated with the contractions with LTC4 as the full agonist (Norman et al., 1994; Gardiner et al., 1994). Third, the number of [3H]LTC4 binding sites in most tissues was in excess of 10 pmol/mg of protein whereas the number of binding sites for [3H]LTD4 was markedly lower. These observations suggested that [3H]LTC4 may bind to other cellular entities, such as glutathione S-transferase (GST) as has been reported by several investigators (Sun et al., 1987;Metters et al., 1994; Bannenberg et al., 1999). In addition, LTC4 may also bind to specific exporters in human eosinophils (Lam et al., 1992), to ATP-dependent carriers in rat liver (Ishikawa et al., 1990; Keppler et al., 1992), and to multidrug-resistant protein 1 (Qian et al., 2001). The GST data (Sun et al., 1987) supported the original observations of Mong et al. (1985)who demonstrated that the glutathione conjugates (SK&F 103,050 and SK&F 103,024), which were devoid of contractile activity in the guinea pig lung and trachea, exhibited a high affinity to the specific site labeled by [3H]LTC4. Furthermore, these glutathione conjugates did not antagonize the LTC4-induced contractions in the guinea pig trachea. These observations suggest [3H]LTC4 binding to GST may mask the LTC4 receptors present in the preparations and thereby explain the reason for the lower affinity for LTC4 binding sites when compared with the functional data. However, the evidence reported by several groups in functional studies have shown that in guinea pig tracheal preparations the LTC4-induced contractions, in the presence of inhibitors of γ-glutamyl transpeptidase (Snyder and Krell, 1984;Charette and Jones, 1987), are resistant to the classical CysLT1 antagonists suggesting a distinct receptor for LTC4.

Cysteinyl-leukotriene antagonists in binding versus functional assays. Data (pK B/pA 2 values) are derived from guinea pig tissues contracted with LTD4and compared with results (pK i values) obtained in [3H]LTD4 radioligand binding assays performed on membrane fractions from the same species. The different CysLT1 receptor antagonists are presented. There was a significant correlation (r 2 = 0.92). The values are derived and modified from Shaw and Krell (1991).
1. [3H]LTC4 Binding Sites.
Despite the [3H]LTC4 binding to nonreceptor proteins, which is now well established, [3H]LTC4 binding has been demonstrated in the central nervous system (Schalling et al., 1986), brain capillaries (Mayhan et al., 1986; Black et al., 1987), endothelial cell membranes (Muller et al., 1987), as well as human erythrocytes (Ghiglieri-Bertez et al., 1986) and leukocytes (Baud et al., 1987b). Unfortunately, the significance of this [3H]LTC4 binding is presently unknown. Cheng et al. (1985) showed that membrane preparations from the guinea pig uterus did not bind [3H]LTD4, data that were also confirmed in kinetic binding experiments (Mong et al., 1985). The [3H]LTC4 bindingK d value (10 nM) in the membrane fractions in the uterus (Levinson, 1984) was similar to that reported for binding to intact cultured smooth muscle cells from the hamster vas deferens (Krilis et al., 1983) but slightly lower than that reported for the guinea pig and rat lung membranes (Hogaboom et al., 1983: Pong et al., 1983). Similar concentrations of LTC4competed for [3H]LTC4binding in the guinea pig ileal muscle (Nicosia et al., 1984). Together these data are consistent with the notion that a specific high-affinity receptor was present in the uterus. Further studies have been performed using brain homogenates, which contain the highest activity of [3H]LTC4 binding sites, and the potency order of the cys-LTs for inhibition of the brain [3H]LTC4 binding is identical to their inhibitory order in the uterine homogenate. In addition, these studies have demonstrated that the relative ability of LTC4, LTD4, and LTE4 to inhibit [3H]LTC4 binding in the brain and uterine membrane fraction was the same as their ability to reduce binding in the smooth muscle cell and lung homogenates (Krilis et al., 1983, 1984; Pong et al., 1983; Cheng et al., 1985). In contrast, the IC50 of LTC4to inhibit brain [3H]LTC4binding differs from that value obtained in the uterine homogenate. This suggests a difference between the dissociation constant (K d) of brain and uterine [3H]LTC4 binding sites. Data from initial studies are comparable with these results and have shown that the K d of lung [3H]LTC4 binding apparently differed from that demonstrated in the smooth muscle cell (Krilis et al., 1983; Pong et al., 1983). Whether this difference is due to the existence of high- and low-affinity LTC4 receptor sites (Krilis et al., 1983) awaits further studies. Interestingly, Cheng and Townley (1984a) also demonstrated that LTC4 was more potent than LTD4 in the ability to produce uterine contractions. These results would suggest that in the guinea pig uterine preparations [3H]LTC4 binds to a receptor that may be responsible for cys-LT-induced uterine contraction in this species.
2. [3H]LTD4 Binding Sites.
The initial data from radioligand binding in a variety of membrane preparations demonstrated the presence of [3H]LTD4 binding sites. There was also a correlation between the tissue distribution and the binding sites for [3H]LTD4 and [3H]LTE4 (Cheng et al., 1985). In addition, the apparent affinity constants for agonists and antagonists (Mong et al., 1985) suggested that [3H]LTD4 and [3H]LTE4 bound to the same receptor in guinea pig lung preparations. Aharony et al. (1989)provided the pertinent evidence that this was indeed the case in the guinea pig lung. However, these investigators demonstrated that not only was the rank order inhibition potency similar when obtained against either ligand but also the absolute potency for LTC4, LTD4, and LTE4 was higher against [3H]LTE4 than that observed with [3H]LTD4.These results suggested a discriminative binding of [3H]LTE4 toward a subset of high-affinity LTD4 receptors. Although ICI 198,615 exhibited equal potency for inhibiting both [3H]LTE4 and [3H]LTD4, there were subtle differences; namely, LTD4 was significantly less potent than ICI 198,615 for inhibiting [3H]LTD4 binding whereas no such difference was observed against [3H]LTE4. Furthermore, the rank order antagonist potency against [3H]LTD4 was ICI 198,615 analog > ICI 198,615 > zafirlukast whereas against [3H]LTE4 the antagonists were equipotent. These investigators also reported that there was a higher sensitivity to inhibition by sodium ions and GTP analogs for [3H]LTE4 binding compared with [3H]LTD4. These data provided evidence for the existence of heterogeneous LTD4 receptors. This suggestion has received some support from Shirley and Cheng (1991) who demonstrated that either LTD4 (K i = 0.49 nM) or ICI 198,615 (K i = 6.89 nM) interacted with a single homogeneous [3H]LTD4 binding site, whereas the competitive binding results of either LTC4 (in the presence of AT-125) or LTE4 exhibited high- and low-affinity [3H]LTD4 binding sites.
While ligand binding studies have indicated that tissues may contain different receptors (Rovati et al., 1985, 1992), the binding sites for the same radioligand in one tissue may also be different from that observed in another preparation. In guinea pig lung membranes, [3H]LTD4 exhibits aK d of ∼0.43 nM whereas in the membranes obtained from the guinea pig myocardium theK d is 3.4 nM. In the latter study,Hogaboom et al. (1985a,b) reported that the myocardial [3H]LTD4 binding sites were neither modulated by guanine nucleotides nor divalent cations and the sulfhydryl-reducing reagents altered the maximum number of sites without any effect on binding affinity. Unfortunately the significance of these myocardium sites has not been explored.
In a study involving [3H]ICI 198,615, Aharony et al. (1988) reported that a CysLT receptor (which corresponds to the current CysLT1 receptor) was coupled to a G-protein and exhibited high- and low-affinity states. In addition,Metters and Zamboni (1993) identified the CysLT1receptor as a single polypeptide with a molecular mass of 45 kDa using direct photoaffinity labeling. These investigators demonstrated that this protein was inhibited by both agonist and antagonists with potencies (IC50) similar to those reported in radioligand binding studies. Furthermore, both cations and nucleotide analogs modulated the photolabeled protein. This latter characteristic was analogous to [3H]LTD4specific binding in the guinea pig lung membranes.
D. Evidence for Additional CysLT Receptor Subtypes
The reported functional data suggest that other CysLT receptors may exist. Initial reports have shown that the LTC4 contractions of the guinea pig trachea are resistant to the classical CysLT1 antagonists (Snyder and Krell, 1984). Furthermore, in the guinea pig lung preparation, MK-571 exhibited a low affinity for the cys-LTs (pK B 5.8) and BAY u9773 did not block the cys-LT contractions (Gardiner et al., 1993; Sakata and Bäck, 2002), data which do not fit the classical CysLT1profile. One interpretation is that there may be a CysLT1 receptor subtype. In addition, Tudhope et al. (1994) observed a residual contraction in the guinea pig lung strip subsequent to CysLT1/CysLT2antagonism by BAY u9773. Recently, Bäck et al. (2000a,b) have reported that the contractions to LTC4 in porcine and human pulmonary arteries were not antagonized by either MK-571 or BAY u9773, data which further suggest the presence of another CysLT receptor responsible for the LTC4-induced contractions. These latter observations have received support from the work of Northacker et al. (2000) who demonstrated that BAY u9773 was a selective CysLT2 agonist. Although BAY u9773 contracted human pulmonary veins (Labat et al., 1992) in porcine and human pulmonary arterial preparations, this compound did not modify basal tone. These data suggest that the CysLT receptor associated with the contractions of human pulmonary arteries may be different from those receptors characterized as CysLT1 and CysLT2. These observations have been extended byWalch et al. (2002) who suggested a “novel CysLT receptor” based on the data in the human pulmonary artery (Table 7).
Panettieri et al. (1998) demonstrated an enhanced LTD4 proliferation of human smooth muscle cells in the presence of epidermal growth factor. These investigators suggested that the CysLT1 receptor associated with proliferation may be different from the CysLT1 receptor activated in human airways responsible for contraction. This interpretation was based on the low affinity of the well known CysLT1 antagonists, pranlukast and pobilukast, which blocked the effect of LTD4 in this assay. In addition, the authors also reported that zafilukast did not work at a high concentration. Together these data do not suggest that based only on zafirlukast the receptor is different but that all of the antagonists were less active at concentrations that caused a significant shift of LTD4 contractions in the human airways. However, the contractile response at CysLT1 receptors in human airways was also induced by LTC4 and LTE4, the proliferative response should therefore also be induced by these ligands as well. Until such data are presented, the results are preliminary and not conclusive for the existence of another receptor subtype.
Using molecular techniques, Ogasawara et al. (2002) demonstrated different pharmacological characteristics of mCysLT2 and hCysLT2. These investigators reported that paranlukast, which inhibited hCysLT1, antagonized mCysLT2 responses as determined by Ca2+ mobilization and receptor-induced promoter activation. Furthermore, the distribution of mRNA expression of mCysLT1 and mCysLT2 in tissues between different strains of mice was also different These results provide further evidence that other CysLT receptor subtypes may exist as has previously been suggested (Mellor et al., 2001). Indeed the up-regulation of the CysLT1 receptor by different cytokines (Mellor et al., 2001; Thivierge et al., 2000,2001) raises the further question as to whether or not the “induced receptor” is similar to the “constitutive receptor”.
Recently, results generated from radioligand binding studies in human lung membrane preparations suggested that [3H]LTC4 binding (Capra et al., 1998) may be associated with a specific LTC4 receptor different from the classical CysLT1 and CysLT2 receptors (Ravasi et al., 2000, 2002). These latter investigators demonstrated that the LTC4 contractions of human lung parenchyma were via a classical CysLT1 receptor activation whereas the [3H]LTC4 binding (high-affinity site) was insensitive to GTP. Furthermore, the CysLT1 receptor antagonists, zafirlukast and iralukast, did not interact with this site.
Although, the above results suggest that other receptor subtypes may be present in different preparations, this hypothesis is based solely on indirect pharmacological assessment and should be interpreted cautiously. Extension of the current CysLT receptor classification requires additional investigation, namely, structural, operational, and molecular biological information.
E. Clinical Studies Involving CysLT Receptors
Most molecules that have been identified as antagonists of the CysLT1 receptor in isolated guinea pig tissue antagonize the effects of LTC4 and LTD4 to an equal extent (Buckner et al., 1986,1990) in human isolated bronchial tissues. These data suggest that in healthy human medium-sized airways CysLT1 is the only leukotriene receptor responsible for the contractile response. These data have also received some support from Civelli et al. (1987)who demonstrated that the number of [3H]LTC4 binding sites in membrane preparations derived from human segmental bronchi at different levels of the respiratory tract were similar even though there was large interindividual variation. Furthermore, these investigators reported that [3H]LTC4may actually label LTD4 sites as well, since LTD4 bound with higher affinity to the lower capacity class site. Although certainly not conclusive, these latter observations suggest that this uniform distribution of sites may reflect the functional receptors reported by other investigators (Buckner et al., 1986; Labat et al., 1992).
Inhalation of aerosols generated from solutions of cysteinyl leukotrienes results in airway obstruction manifested as decreased specific airway conductance (S Gaw) or as decreased flow rates (measured from partial or full expiratory flow-volume curves) (Drazen, 1986); LTC4 and LTD4 are nearly equipotent contractile agonists in intact humans. Nebulizer concentrations on the order of 10 μM are required to reduce the maximal expiratory flow rate (measured from a partial flow-volume curve,V 30-P) by 30% in nonasthmatic subjects. Approximately 30-fold greater concentrations of LTE4 are required for an equivalent physiological effect in nonasthmatic subjects. Comparable nebulizer concentrations are required for reduction of a similar magnitude in theS Gaw. Compared with a reference agonist in nonasthmatic subjects (withV 30-P orS Gaw as the index of airway obstruction), LTC4 and LTD4are approximately 2000 times more potent than histamine, whereas LTE4 is 30 to 100 times more potent than histamine as a bronchoconstrictor agonist (Weiss et al., 1982; Griffin et al., 1983; Barnes et al., 1984; Smith et al., 1985; Adelroth et al., 1986; Greenberger et al., 1986; Kern et al., 1986; Davidson et al., 1987). Complete dose-response curves show that the plateau response to LTD4 is greater than that to methacholine (Bel et al., 1989). Since no such difference is evident with dose-response curves for 2- to 4-mm isolated bronchi, these findings are consistent with the hypothesis that airways from physically distinct loci (i.e., small versus large airways) respond to both stimuli (LTD4 and methacholine) in normal human subjects.
Patients with asthma also exhibit bronchoconstrictor responses when they inhale aerosols generated from solutions of cys-LTs (Griffin et al., 1983; Adelroth et al., 1986; Davidson et al., 1987; Pichurko et al., 1989; Smith et al., 1993); the aerosol generator concentrations of LTC4, LTD4, and LTE4 required to decrease theV 30-P by approximately 30% are about 30- to 100-fold less than is required in normal human subjects. The bronchoconstrictor responses of asthmatic subjects to these leukotrienes are all manifest within 3 to 5 min after aerosol inhalation; the duration of the effect is related to the severity of the bronchospasm but is on the order of 20 to 30 min when the decrement in the V 30-P is 30%.
Thus the predominant biological effect of administration of leukotrienes by inhalation is broncoconstriction mediated by the CysLT1 receptor. There may be other effects when leukotrienes are administered systemically but at present there are inadequate data to make a statement.
F. Summary
The development of selective antagonist for specific CysLT cell surface receptors was undertaken to harness the detrimental aspects of the inflammatory process specifically in asthma. This research has lead to compounds with clinical benefits in asthmatic patients and the milestones in the sequence of events which have led to this therapy are presented in Table 8.
Milestones in the development of CysLT1 therapeutic antagonists
Therefore, based on the CysLT1 antagonists, certain therapeutic goals have been achieved. However, the evidence to suggest that the metabolites of the 5-lipoxygenase enzymatic pathway activate other CysLT receptors, which may have further implications in pathophysiological conditions, needs to be addressed. Unfortunately, there are no selective antagonists for the CysLT2receptor. Recent molecular studies have shown that this receptor is expressed in several human tissues (heart, brain, peripheral blood leukocytes) but an exploration of their role in these tissues remains markedly compromised until selective pharmacological compounds become available.
Interestingly, there is some evidence from vascular studies that a CysLT receptor may be induced during pathological conditions, such as atherosclerosis (Allen et al., 1993, 1998) since cys-LT-induced contractions were only observed in tissues derived from patients with this disease. In addition, the potency of the cys-LTs are markedly altered during aging (Duncan and Douglas, 1985). Whether these data reflect modification in receptor number, affinity, or second messenger pathways has never been pursued. Recently, Mellor et al. (2001) have reported that the CysLT1 receptor may function as a UDP pyrimidinergic receptor. These latter results indicate that the eicosanoid GPCRs may exhibit a dual receptor role. UDP acting with the CysLT1 receptor is analogous in some respects to the peptides that can act at the ALX. These observations provide future perspectives for studies in receptor classification.
G. Lipoxins Receptors
1. Functional and Radioligand Binding Studies.
Although LXs are ancient molecular structures conserved and produced by fish as well as frog blood cells (Rowley et al., 1994), the potential physiologic roles of LX and related compounds have not been widely tested outside of the context of inflammation and the immune system. The principal actions reported for LX focus on leukocytes, in particular neutrophils, monocytes, and eosinophils in vitro and in vivo, suggesting that these cells are the major targets. The potency of action of LX on human leukocytes is in the nanomolar to subnanomolar range. Experiments with LXA4 were initially performed on guinea pig lung strips to establish structure-activity relationships. These early reports indicated stereospecificity, that is, the 5S,6R-orientation of the two hydroxyl groups positioned immediately adjacent to the carboxylic end of the conjugated tetraene was essential for the contractile activity in the submicromolar range. These stereospecific requirements suggested the presence of specific LXA4 recognition sites. Although this evidence was essential in establishing the biological activities evoked by LXA4, the use of isolated lung strip as a target tissue model for endogenous LX actions in view of recent findings may be misleading. These compounds, in particular LXA4, act in the subnanomolar ranges to initiate protective actions. In this regard, LXA4possesses human leukocyte-selective actions that implicate lipoxins as endogenous stop signals or chalones (Serhan, 1994, 1997). In addition, LXA4 stimulates rapid lipid remodeling and release of arachidonic acid in human PMN, which is sensitive to PTX treatment (Grandordy et al., 1990; Nigam et al., 1990). These results point to the involvement of a GPCR and indicate that LXA4 stimulates selective functions of human leukocytes (PMN and monocytes) that are markedly different from the leukotrienes.
LXA4, in addition to activating ALX to regulate leukocyte motility, may also interact at CysLT receptors (Badr et al., 1989; Fiore et al., 1992; McMahon et al., 2000; Gronert et al., 2001) and block LTD4 binding to mesangial cells (Badr et al., 1989) and isolated HUVECs (Takano et al., 1997). HUVECs specifically bind [3H]LXA4 with aK d of 11 nM, which can be inhibited by LTD4 and the CysLT1receptor antagonist, pobilukast (Fiore et al., 1993). This receptor is inducible in HUVECs and identical to the CysLT1(Gronert et al., 2001). Therefore, LXA4 interacts with at least two classes of cell surface receptors: one specific for LXA4 on leukocytes, the other shared by LTD4 that is present on HUVEC and mesangial cells, namely, CysLT1.
In human PMNs, subcellular fractionation showed that [3H]LXA4 binding sites are associated with plasma membrane and endoplasmic reticulum (42.1%) and granule (34.5%) as well as nuclear-enriched fractions (23.3%), a distribution distinct from [3H]LTB4 binding (Fiore et al., 1992). LXA4 was also reported recently to bind to the Ah receptor, which is a ligand-activated transcription factor (Schaldach et al., 1999). The finding that LXA4 blocks either PAF- or fMLP-stimulated eosinophil chemotaxis (Soyombo et al., 1994) suggests that functional ALX is also present on eosinophils.
ALX activation on human PMNs evokes inhibition of LTB4- and fMLP-stimulated PMN adhesion (by down-regulating CD11/CD18), chemotaxis and transmigration as well as degranulation (Serhan, 1997). In human enterocytes (T84), LX analogs inhibit TNFα-induced IL-8 release (Gronert et al., 1998) as well as pathogen-induced IL-8 secretion at the mRNA level (Gewirtz et al., 1998). In addition, LXA4 analogs inhibit rat leukocyte rolling and adherence by attenuating P-selectin expression in mesenteric microvasculature (Scalia et al., 1997). When applied to mouse ears, LX analogs markedly reduced LTB4-induced PMN infiltration and vascular permeability (Takano et al., 1998). Moreover, LXA4 inhibits PMN recruitment to inflamed glomeruli in vivo (Papayianni et al., 1995), further supporting the anti-inflammatory actions of LX to activate “stop signaling” via the ALX receptor.
To further investigate ALX receptors and their function in inflammation, analogs based on the native structures of LXA4 and LXB4 were designed to resist rapid inactivation (Serhan et al., 1995; Takano et al., 1998). Methyl groups were placed on carbon-15 and carbon-5 of LXA4 and LXB4 structures, respectively, to block dehydrogenation (Serhan et al., 1995; Clish et al., 2000). 15(R,S)-methyl-LXA4 is a racemic stable analog of both LXA4 and the aspirin-triggered 15-epi-LXA4. Additional analogs of LXA4 were synthesized with a phenoxy group bonded to carbon-16 and replacing the ω-end of the molecule. This design permits 16-phenoxy-LXA4 to resist potential ω-oxidation and to be protected from dehydrogenation and potential ω-oxidation in vivo by the stearic hindrance of the bulky aromatic ring. The actions of LX analogs were tested in vitro and in vivo and proved to be potent LX mimetics (Clish et al., 1999, 2000). Furthermore, the topical application of LX and 15-epi-LX analogs in a mouse ear model of acute inflammation demonstrated that these analogs are potent inhibitors of LTB4- and phorbol 12-myristate 13-acetate-initiated neutrophil-recruitment as well as PMN-mediated vascular injury (Takano et al., 1997, 1998). These LX mimetics proved to be more potent as topical agents than LTB4 receptor antagonists within the range of the clinically used anti-inflammatory steroid dexamethasone (Takano et al., 1998). In addition, in a rat model of endothelial dysfunction characterized by reduced nitric oxide release and up-regulation of adhesion molecules such as P-selectin, superfusion with LX analogs dramatically inhibited leukocyte rolling (Scalia et al., 1997). Thus, these analogs of LX and 15-epi-LX serve as potent topically active agents that inhibit PMN recruitment and PMN-mediated changes in vascular permeability.
In addition to LXA4 and aspirin-triggered LXA4 analogs, ALX is activated by and competed with a series of rogue synthetic peptides that are also chemotactic (Chiang et al., 2000). CHO cells with Gq/oexpressing ALX can chemotax in response to either the synthetic peptide MMK-1 or 15-epi-LXA4 analog. CHO cell chemotaxis to 15-epi-LXA4 was not only ligand-dependent but also Gq/o-dependent in that without Gq/o neither LXA4 nor 15-epi-LXA4 or their analogs could stimulate chemotaxis. Several synthetic nonphysiological peptides interact with ALX-related receptors such as FPRL2 (Christophe et al., 2001). The bioactive LX and 15-epi-LX analogs, as well as select small peptides, each competed for specific [3H]LXA4 binding with recombinant human ALX, and the N-glycosylation of the receptor proved essential for peptide but not LXA4 recognition. Several chimeric receptors (Chiang et al., 2000) were constructed from receptors with opposing functions, namely ALX and BLT1. Results with these chimeric receptors also showed that the seven-transmembrane segment and adjacent regions of ALX are essential for LXA4 recognition, and additional regions of ALX are required for high-affinity binding of the peptide ligands. These initial findings indicated that a single seven-transmembrane receptor can switch recognition as well as function with certain chemotactic peptides from stimulatory to inhibitory with aspirin-triggered lipoxins (ATL) and LX (lipid ligands). This evidence also raises the possibility that ALX activation by LX or ATL can protect the host from potentially deleterious PMN-induced responses associated with innate immunity and direct effector responses to sites of tissue injury by recognition of mitochondrial-derived peptide protein fragments. Hence, activation of ALX by LXA4, aspirin-triggered 15-epi-LXA4 or their analogs can dampen inflammation by evoking a stop signal as well as blocking either LTC4 or LTD4 responses at CysLT1 receptors (Gronert et al., 2001). The in vivo combination of these sites of LXA4 action (ALX and CysLT1) favor resolution of inflammation and protection from acute PMN-mediated tissue injury.
2. Summary.
The ability to inhibit the expression of pro-inflammatory messengers, regulate trafficking, and sequestration of leukocytes as well as antagonize the CysLT1receptor, illustrate that LX and ATL by activating specific receptors have clearly established roles in a variety of vascular-related inflammatory phenomenon. Of considerable interest is the ability of the stable aspirin-triggered lipoxin analogs (ATLa) to antagonize the LTD4 activation of a CysLT1receptor in the mouse ear model (Gronert et al., 2001). Previous investigators (Ortiz et al., 1995) have reported that LTD4, via CysLT1 receptor activation, released a contractile factor from endothelial cells in human pulmonary vessels. This factor masked the functional relaxations associated with nitric oxide. Whether or not the CysLT1 receptor antagonized by ATLa in the murine vascular model is identical to that present on the endothelium of the human pulmonary vessels remains to be determined. However, lipoxin activation of an ALX receptor also inhibits LTD4-induced mesangial cell proliferation (McMahon et al., 2000). These investigators demonstrated that two receptor subtypes were present in these cells, namely, an ALX receptor and an ALX/CysLT shared receptor. Recently, McMahon et al. (2002) have extended these observations to include further cross-talk between receptors, since leukotrienes and lipoxins also interact with PDGF receptor β in mesangial cell proliferation. These data provide evidence of the complex mechanisms involved in mediator-receptor activation associated with cellular proliferation.
V. General Conclusions
The classification of the receptors that are activated by leukotrienes and lipoxins was initially derived from data obtained in functional investigations. The pharmacological studies provided substantial evidence for the existence of specific and distinct receptors that were activated by these lipid mediators and are now well supported by the recombinant systems. However, the molecular cellular mechanisms for the contractions of vascular smooth muscle by LTC4 and LTD4, which are resistant to the classical CysLT1 antagonists has yet to be elucidated. Since the observations of Labat et al. (1992), no known selective antagonists has been reported. Without such compounds the identification of CysLT receptors is severely compromised and our nomenclature for the receptors associated with vascular inflammation will be difficult to postulate. In fact, the CysLT receptor on the human pulmonary endothelium is also resistant to the classical CysLT antagonists (Ortiz et al., 1995). Presently, there is no way of identifying the receptor associated with the relaxations induced by LTD4 in human pulmonary vessels from the receptor responsible for the contractions induced by this same ligand.
In addition to these areas of research, future work in splice variants of the different receptors may lead to a better understanding of the cellular signal transduction pathways subsequent to activation by the ligand. Presently, little information is available based on this approach.
The use of transgenic animals have clearly demonstrated that BLT1 plays a role in the recruitment of eosinophils to the inflamed site as well as a gender-associated effect related to mouse survival in an anaphylactic model. These results strongly suggest that BLT1 receptors are intimately linked with acute inflammation in vivo. One of the most promising aspects of knockout mice is the uncovering of new receptors or the possible roles for receptor subtypes. The role of CysLT receptors in these knockout animals remains to be elucidated
The recent evidence provided by Ogasawara et al. (2002) concerning the pharmacological differences between hCysLT2 and mCysLT2 together with CysLT receptor distribution in different mouse strains suggests that the choice of the animal model and the extrapolation to human inflammation warrants considerable reflection. Furthermore, in human airways activation of the CysLT1 receptor is responsible for the bronchoconstriction both in normal and asthmatic subjects. However, there is no evidence for airway constriction in mice (Martin et al., 1988; Richter and Sirois, 2000), and the mCysLT1receptor has only been implicated in constriction of the microvascular smooth muscles leading to increased permeability and extravasation. Although the genomic organization of the mCysLT1has been reported by Maekawa et al. (2001), there is a need for a similar exploration of the human CysLT1 receptor.
The relative potencies of LTC4, LTD4, and LTE4 in a number of functional studies demonstrated that LTE4 is generally less potent and markedly less efficient in activating the CysLT receptors. The receptor studies involving cells transfected with specific CysLT receptors have confirmed this observation. However, there are now very few investigations that are based on the use of LTE4. This is unfortunate, since there are several reports suggesting that this ligand may activate another receptor (Snyder and Krell, 1984; Mong et al., 1985; Sjolander et al., 1990; Sakata and Bäck, 2002) or may not activate the known classical CysLT receptors (Walch et al., 2002). Although these observations are only suggestive, care should be taken so as not to overlook these pertinent published data, which may offer interesting leads to our understanding the leukotriene receptors.
Young (1989) postulated that a hydrophobic pocket existed at the receptor for the tetraene chain (the unstable triene moiety found in LTC4 or LTD4). An earlier report (Snyder and Bernstein, 1987) had demonstrated that several leukotriene analogs, where the heptyl (C7H15) was positioned either para, meta, or ortho on the phenyl ring altered the pharmacological potency profile of the analog. Whether or not the use of stable but pharmacologically active analogs of the cys-LTs would provide clearer results for the presence of two separate CysLT receptors in functional studies based on agonist potency has received little attention. Such tools may be of some benefit in stereochemical investigations for the identification of receptorial proteins. Interestingly, the functional identification and classification of the ALX receptor has been systematically evaluated using the LX stable analogs.
The identification and classification of the CysLT receptors has been based essentially on the ability of antagonists to block or not to block a physiological response. Although this classification has proven to be reliable at the functional and more recently at the molecular levels, future studies need to evaluate not only several antagonists but also the different ligands, especially when the functional profiles of the antagonists do not appear to match what has previously been reported.
Based on investigations involving ALX and BLT receptors, there are several phenomenon that need to be highlighted. “Receptor induction” as seen in animal models of vascular inflammation (mouse ear model) (Gronert et al., 2001) has now been widely accepted. These data demonstrated that receptor message is markedly increased in tissues or cells where a pro-inflammatory agent has been used as a primer. In addition, other receptor subtypes may be induced, as in the case of the human mast cell, where IL-4 induced a receptor with a pharmacological profile different from the classical CysLT receptors (Mellor et al., 2001). These data warrant further attention since inducible receptors may not be identical to constitutive receptors (receptors that are present under normal conditions). In addition, the G-protein second messenger systems must equally be explored. Along similar lines, there is an intriguing observation that at least one receptor, namely, CysLT1 may act as a pyrimidinergic (UDP) receptor (Mellor et al., 2001) suggesting that GPCRs may be quite versatile at the ligand-G-protein interface. Although the significance of these latter findings remains to be established, both observations provide fruitful areas for future work. Furthermore, the observations that LTB4 is associated with an excessive activation of leukocytes as well as the 5-LO pathway suggests a role for BLT receptor “tone” and indicates that receptor expression amplifies pro-inflammatory circuits in vivo.
In the last few years there has been some controversy as to whether or not the BLT receptor may act as a coreceptor for macrophage-trophic human immunodeficiency virus (HIV) of the type 1 strains. Owman et al. (1998) reported that this receptor mediated entry of primary HIV isolates into CD14 cells. However, results from transfected cells apparently raise some question as to such a role for the BLT1 receptor (Martin et al., 1999). Whether or not BLT1 antagonists can be used to prevent HIV infection warrants perhaps further pursuit.
Although there is considerable optimism about these recent developments in lipid mediator receptor classification, only by further work in this area, as well as in vascular inflammation models, will a potential for therapeutic exploitation be realized.
Footnotes
-
↵† During the preparation of this manuscript, Professor Nicosia passed away following a long illness. Her efforts in the organization of this committee and her scientific work on the leukotriene receptors over the past years will be missed.
-
Address correspondence to: Charles Brink (Chairman), Researcher (CNRS CR-1), CNRS UMR 7131, Hôpital Broussais, Bâtiment René Leriche, 96, rue Didot, 75014 Paris, France. E-mail:charlesbrink{at}hotmail.com
-
DOI: 10.1124/pr.55.1.8
Abbreviations
- SRS
- slow reaction smooth muscle-stimulating substance
- SRS-A
- slow reacting substance of anaphylaxis
- LT
- leukotriene
- LX
- lipoxin
- LO
- lipoxygenase
- HETE
- hydroxyeicosatetraenoic acid
- PMN
- polymorphonuclear leukocyte
- cys-LT
- cysteinyl-leukotriene
- IUPHAR
- International Union of Pharmacology
- GPCR
- G-protein-coupled receptor
- kbp
- kilobase pair(s)
- ORF
- open reading frame
- bp
- base pair(s)
- CHO
- Chinese hamster ovary
- IL
- interleukin
- UTR
- untranslated region
- HPETE
- hydroperoxyeicosatetraenoic acid
- PAF
- platelet-activating factor
- HEK
- human embryonic kidney
- FPR
- formyl peptide receptor
- fMLP
- formyl-methionyl-leucyl-phenylalanine
- PTX
- pertussis toxin
- PKC
- protein kinase C
- HUVEC
- human umbilical vein endothelial cell
- GST
- glutathione S-transferase
- ATL
- aspirin-triggered lipoxin
- HIV
- human immunodeficiency virus
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵




{kind=link}
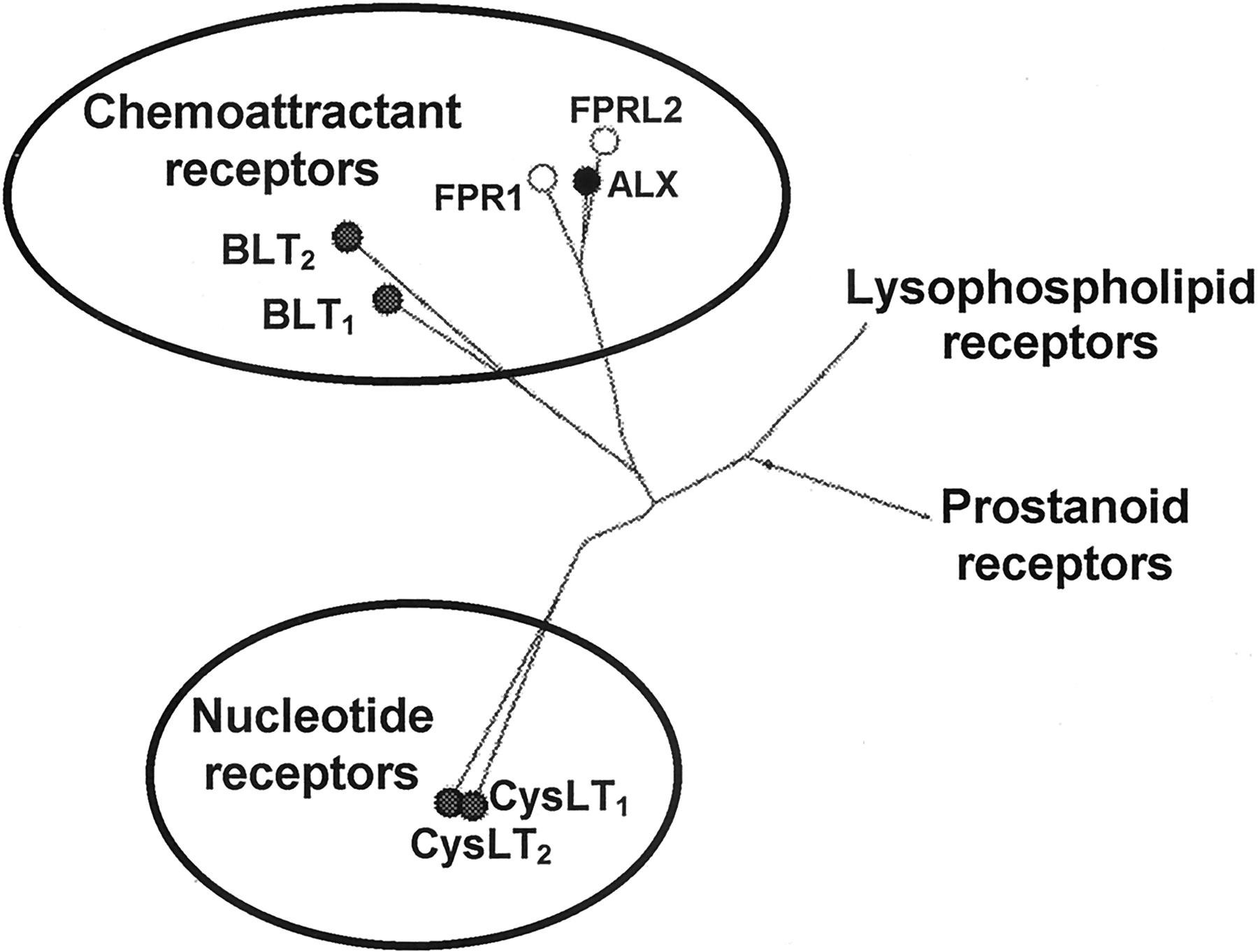{kind=link}
{kind=link}
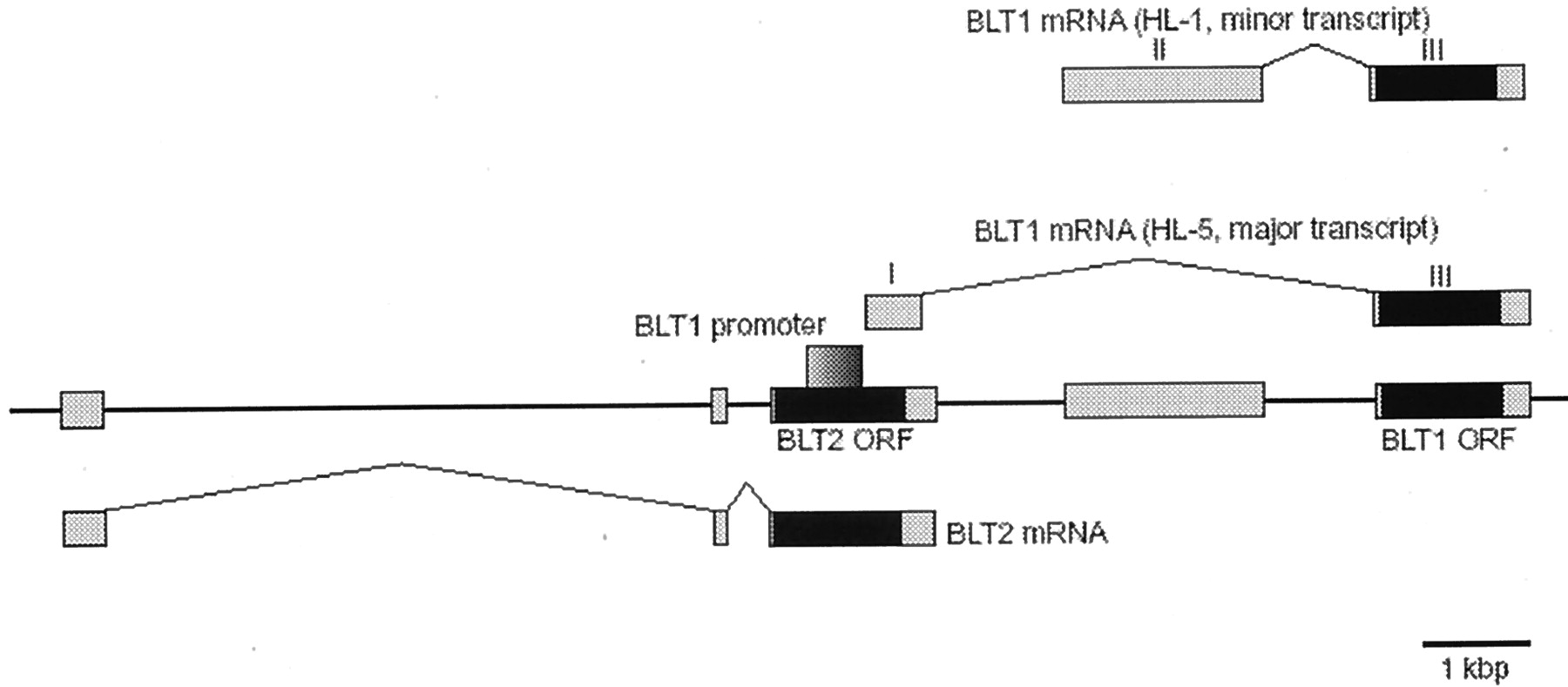{kind=link}
{kind=link}
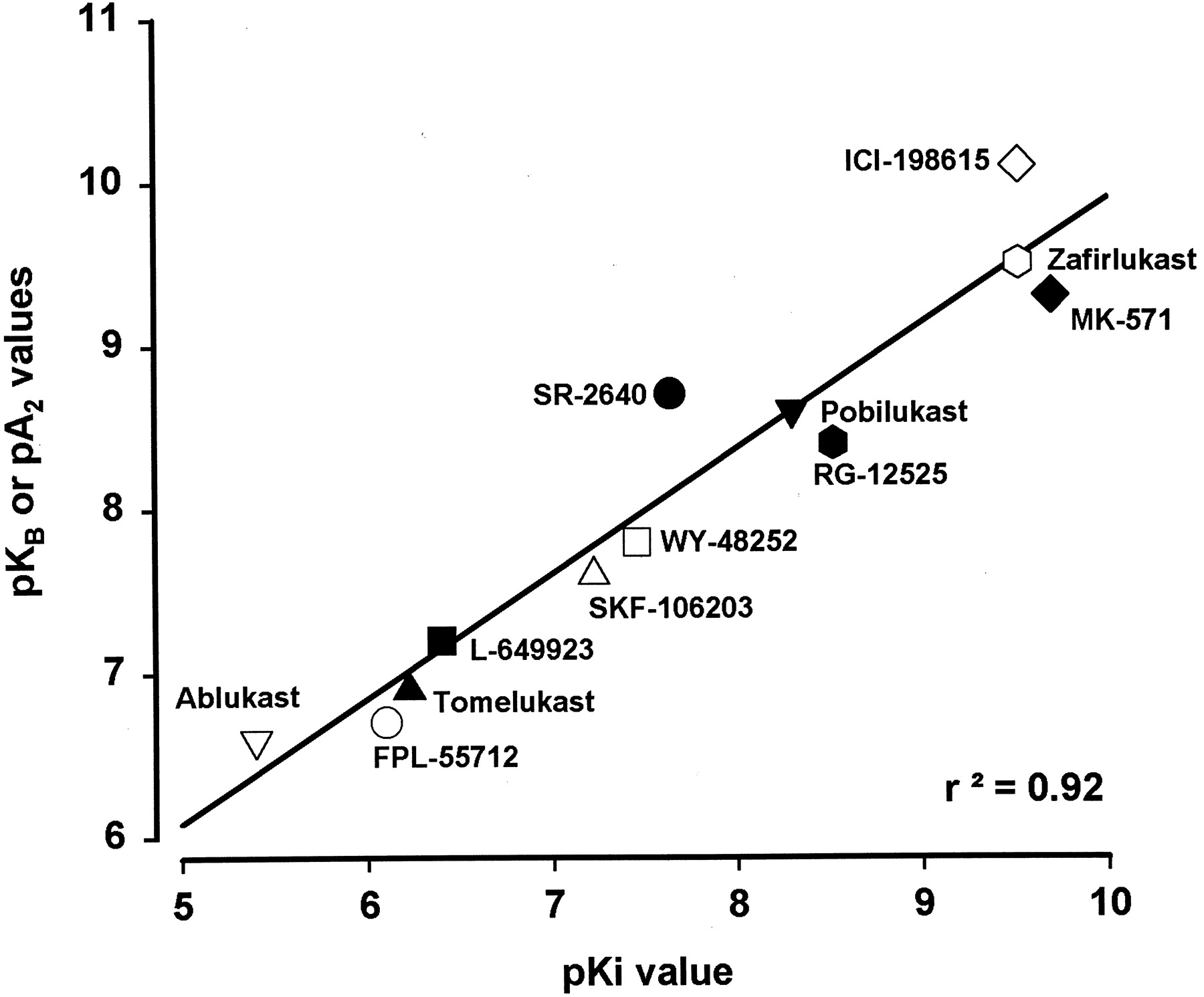{kind=link}