


Abstract
Heparin has been used extensively as an antithrombotic and anticoagulant for close to 100 years. This anticoagulant activity is attributed mainly to the pentasaccharide sequence, which potentiates the inhibitory action of antithrombin, a major inhibitor of the coagulation cascade. More recently it has been elucidated that heparin exhibits anti-inflammatory effect via interference of the formation of neutrophil extracellular traps and this may also contribute to heparin's antithrombotic activity. This illustrates that heparin interacts with a broad range of biomolecules, exerting both anticoagulant and nonanticoagulant actions. Since our previous review, there has been an increased interest in these nonanticoagulant effects of heparin, with the beneficial role in patients infected with SARS2-coronavirus a highly topical example. This article provides an update on our previous review with more recent developments and observations made for these novel uses of heparin and an overview of the development status of heparin-based drugs.
Significance Statement This state-of-the-art review covers recent developments in the use of heparin and heparin-like materials as anticoagulant, now including immunothrombosis observations, and as nonanticoagulant including a role in the treatment of SARS-coronavirus and inflammatory conditions.
I. Introduction
The recent thrombotic events related to COVID-19 infection and vaccination have highlighted the efficacy of unfractionated heparin (UFH) and low molecular weight heparin (LMWH) not only as antithrombotic/anticoagulants but potentially for the anti-inflammatory and antiviral properties of these drugs (van Haren et al., 2020). The history of the discovery of heparin and its subsequent use as an anticoagulant are covered by several detailed reviews (Barrowcliffe, 2012; Hemker, 2016). Briefly, heparin as an anticoagulant was first described by Maurice Doyon in 1910 (Doyon et al., 1911). However, the discovery of heparin has been ascribed to Jay Mclean, who copurified an anticoagulant substance while extracting procoagulant thromboplastin fractions from different tissue sources (McLean, 1916). While UFH was first used as a clinical product in the 1930s, developed by Charles Best in Canada and Erik Jorpes in Sweden, the critical antithrombin binding pentasaccharide sequence and its mechanism of action were not elucidated until the 1970s (Lindahl et al., 1979; Rosenberg and Lam, 1979; Choay et al., 1980). The 1970s also heralded the discovery of LMWH (Johnson et al., 1976). Figure 1 illustrates the important chronological milestones in the development and use of heparin and LMWH.

Timeline events in the history of heparin. Key: EP, European Pharmacopeia; IS, International Standard; IU, International Unit; NIBSC, National Institute for Biological Standards and Control; NMR, Nuclear Magnetic Resonance; USP, United States Pharmacopeia.
Heparin is a complex biologic, extracted and purified from tissues of different species. The heparins from different species and tissue types vary in their structures (Fu et al., 2013) and therefore express varying pharmacological activities (both anticoagulant and nonanticoagulant activity). Currently, the predominant source of heparin used clinically in the United States and Europe is porcine intestinal mucosa, although some countries do use bovine heparin preparations, while other non-mammalian sources are under investigation (see later discussion). As established clinical products, UFH and LMWH are under tight regulatory control, with provision of specifications in pharmacopeial monographs to ensure their safety and efficacy (US Pharmacopeial Convention, 2014; European Pharmacopeia (EP), 2015). Nonetheless, adulteration of heparin that can impact on the safety of the pharmaceutical products has periodically surfaced, sometimes resulting in mortality and morbidity of patients. The most recent example of adulteration was the contamination of UFH with over-sulfated chondroitin sulfate (OSCS) in 2008 (Kishimoto et al., 2008). While revision of pharmacopeial methods has reduced the risk of contamination with OSCS, and maybe other yet unidentified contaminants (Szajek et al., 2016), this incident has highlighted the risk of relying on a single source of raw material from porcine mucosa. Outbreaks of diseases of the pig and the fragility of the supply chain can lead to shortages of raw material for porcine heparin, which in turn led to an unfavorable impact on the availability of this important medicine. This was evident in some countries during the recent COVID-19 pandemic (McCarthy et al., 2020; Rosovsky et al., 2020; Sharma et al., 2020). Thus, the US Food and Drug Administration is encouraging the introduction of bovine heparin into the United States (Al-Hakim, 2021) to counter these supply issues with porcine heparin, but new pharmacopeial methods will be needed to safeguard the quality of bovine heparin.
This review builds on our earlier article published more than 6 years ago (Mulloy et al., 2016) and serves to provide an insight into the anticoagulant and nonanticoagulant actions of heparin, its interaction with multiple biologic targets, and its use as an anti-inflammatory and antiviral medicine in the treatment of a range of diseases beyond thrombosis, including recently COVID-19.
II. Structural Aspects of Heparin and Related Drugs
A. Structure, Biosynthesis, and Turnover of Heparin
The structure and biosynthesis of heparin have been described in considerable detail elsewhere (Mulloy, 2012; Mulloy et al., 2016).
Heparin as currently used in medicine is a complex mixture of closely related polysaccharides made up of a limited range of disaccharides in which uronic acid [β-D-glucuronic (GlcA) or α-L-iduronic (IdoA)] and N-acetyl or N-sulfamido α-D-glucosamine (GlcNAc, GlcNS) are alternately joined by 1-4 glycosidic linkages. Heparin is a member of the heparan sulfate (HS) family with an unusually high degree of sulfate substitution, found principally in the granules of mast cells (Mulloy et al., 2017). Variations in the degree and positions of sulfation confer an extra degree of complexity.
The exact geometry of sulfate substitution and hence the structure of any sequence selectively recognized by a heparin-binding protein is also dependent on the conformation of the heparin/HS polysaccharide, as discussed in the original version of this review (Mulloy et al., 2016). A major contributor to the conformational complexity of heparin is the flexibility of the six-membered pyranose ring of iduronic acid. The conformational equilibrium in solution of this monosaccharide has recently been described as involving two well-defined chair forms (1C4 and 4C1) and a somewhat less well-defined skew boat form (2S0). This study not only provides a secure experimental basis for the relationship between the ring conformational equilibrium and nuclear magnetic resonance (NMR) coupling constants generally used to determine pyranose ring conformations but also offers a detailed commentary on the relevant literature (Haasnoot et al., 2020). Furthermore, an NMR study of 15N, 13C doubly labeled heparin octasaccharide has identified thermally induced conformational changes that do not, however, affect binding to calcium (Hughes et al., 2017).
The biosynthesis of heparin is essentially identical with that of heparan sulfate but occurs exclusively in mast cells (Fig. 2). The polysaccharide backbone of alternating GlcA and GlcNAc is extended from a linker tetrasaccharide attached to the serglycin protein core by the exostosin EXT glycosyltransferases and subsequently modified by de-N-acetylation and re-N-sulfation of GlcNAc, epimerization, and 2-O-sulfation of GlcA to give IdoA2S, followed by 6-O-sulfation of GlcNS and occasionally by 3-O-sulfation of GlcNS/GlcNS6S. This results in a heparin polysaccharide consisting predominantly of IdoA2S and GlcNS6S, interspersed with less common GlcA-GlcNAc sequences and a rich selection of complex intermediate regions in which some sequences are, with more or less selectivity, recognized by protein ligands such as antithrombin (Fig. 2). The resulting heparin polysaccharide chains are then shortened by the action of heparanase, an endoglycosidase that cuts the heparin chains into shorter lengths (Lindahl and Li, 2020).

Structure and biosynthesis of HS and heparin (adapted from Weiss et al., 2017). Upper inset: Pentasaccharide AT binding sequence (where R = S). Middle section: the heparin/HS polysaccharide is built up from a serine residue in the proteoglycan protein backbone by a series of glycosyltransferases: XYTL1 and 2, xylosyltransferase isoforms 1 and 2; FAM20B, xylose kinase GALT, galactosyltransferase; GLCAT1, glucuronyltransferase; EXTL3, exostosin-like glycosyltransferase 3; EXT, GlcNAc and GlcA transferases (known as exostosins). The extended polymer is then modified by sulfotransferases and an epimerase: NDST isoforms 1-4, GlcNAc N-deacetylase/N-sulfotransferase; HSGLCE, glucuronyl C5 epimerase; HS2ST, uronic acid 2-O-sulfotransferase; HS6ST1-3, glucosamine 6-O-sulfotransferase; HS3ST1-6, glucosamine 3-O-sulfotransferase. The orange oval shape depicts the protein binding sequence for antithrombin. Lower panel: the constituent monosaccharides of heparin/HS, their structures and symbolic representations (https://www.ncbi.nlm.nih.gov/glycans/snfg.html).
Heparin is currently prepared from tissues rich in mast cells, at present principally from porcine intestinal mucosa or bovine mucosa (but see later for other current and potential sources) (van der Meer et al., 2017). Despite heparin’s heterogeneity, it is a remarkably consistent product, especially now that pharmacopeial methods and acceptance criteria have been modernized (Szajek et al., 2016). Heparin active pharmaceutical ingredient (API), with an average molecular weight (MW) of about 16,000 g/mole, can then be converted to a range of LMWHs with an average MW 4000 to 6000 g/mole by enzymatic or chemical depolymerization. UFH and LMWH products are used extensively for different clinical situations as has been previously discussed (Hao et al., 2019; Lyman et al., 2021).
The strong influence of MW on clearance of heparin from the circulation was recognized several decades ago (Johnson et al., 1976). Two major mechanisms appear to be involved: a renal route that is nonsaturable and a saturable, nonrenal route involving heparin endocytosis and lysosomal breakdown largely in the liver (Johansen and Balchen, 2013). LMWH is cleared largely by the renal mechanism, but the longer heparin chains in UFH are more rapidly bound by the scavenger receptors of endothelial cells in the liver and lymph nodes (Johansen and Balchen, 2013; Weigel, 2020), the saturable mechanism. The heparin scavenger has been identified as the hyaluronic acid receptor for endocytosis (HARE), an isoform of stabilin-2 (Harris and Cabral, 2019). Synthetic heparin oligosaccharides have been used to establish that HARE binds to chains at least 10 to 12 monomers in length and has a preference for 3-O-sulfation (Pempe et al., 2012). This is consistent with observations that heparin with high affinity for antithrombin is eliminated preferentially by the saturable, HARE-based mechanism (Johansen and Balchen, 2013).
Endocytosis of heparin to lysosomes leads to the comprehensive dismantling of molecular structure by a series of enzymes such as specific sulfatases for each type of sulfate substitution in heparin (Lübke and Damme, 2020) and other hydrolases (Filocamo et al., 2018). Deficiency in any one of these degradative enzymes leads to one or other of the lysosomal storage diseases known as the mucopolysaccharidoses (Filocamo et al., 2018). An alternative fate for heparin/HS in the gut is as a nutrient source for gut bacteria such as Bacteroides thetaiotaomicron, that expresses a variety of heparin/HS degrading enzymes (Cartmell et al., 2017).
B. Synthetic Heparin
There are many reasons to design and produce chemically synthesized heparin oligosaccharides, both as research reagents and for therapeutic purposes. Synthesis can provide single molecular species of known structure, useful in research to examine the molecular basis of interactions with proteins, and in the pharmaceutical industry for quality control, relative ease of regulatory oversight, and readily defined intellectual property. Recent reviews of synthetic and chemoenzymatic heparin analogs are recommended (Tsai et al., 2017; Baytas and Linhardt, 2020) for a more comprehensive survey of the field.
The only completely chemically synthesized heparin oligosaccharide in current medicinal use is fondaparinux, a pentasaccharide with the sequence that binds with high affinity to antithrombin (AT) (Fig. 2). It was first synthesized in the early 1980s (Choay et al., 1983) with numerous chemical steps. Novel synthesis strategies are still being developed for this compound (Ding et al., 2017; Dey et al., 2020), and some of the structurally related impurities produced in fondaparinux synthesis have been found to be as potent as, if not more so than the main product (Zhang et al., 2017).
The first single-crystal structure of fondaparinux (i.e., not in complex with a protein) has been determined (Wildt et al., 2017). The iduronic acid residue in this solid-state structure adopts a very irregular chair conformation, and the overall conformation differs from both the protein-bound crystal structures and the solution structure as determined by NMR (Langeslay et al., 2012). It is interesting to note that the glucuronic acid in the pentasaccharide (Fig. 2) may be replaced by 2-O-sulfated iduronic acid without loss of binding to antithrombin, and in this case the two internal iduronates adopt different conformations (Elli et al., 2020). On the other hand, the replacement of the original iduronate in the pentasaccharide sequence with anhydrotalose resulted in a compound with no anti-Xa activity (Demeter et al., 2018).
Synthetic heparin oligosaccharides with a defined sulfation pattern are invaluable for study of the dependence of heparin/HS biologic activities on the fine structure of highly sulfated domains. A microarray of HS-like synthetic oligosaccharides has supplied evidence of differential binding of several chemokines and growth factors to HS sequences with varied sulfation patterns, supporting the contention that changes in cell surface HS composition can modulate protein function (Zong et al., 2017). A set of synthetic, structurally defined dodecasaccharides made up of alternating N-sulfated glucosamine and 2-O-sulfated iduronate that contained no, one, or six glucosamine 6-O-sulfates (Jayson et al., 2015; Avizienyte et al., 2016) have been shown to selectively inhibit the chemokines CXCL8 or CXCL12 (Jayson et al., 2015) and fibroblast growth factor (FGF2) or vascular endothelial growth factor (Avizienyte et al., 2016). These dodecasaccharide structures are 4-O-sulfated at the nonreducing terminal and so are not naturally occurring sequences, but the point is made that there exists differential recognition of HS/heparin fine structure by proteins discussed in more detail later.
Heparin tetrasaccharide, hexasaccharide, and decasaccharides of the trisulfated disaccharide type have also been synthesized and used to establish the interaction between heparin oligosaccharides and the oligomeric form of Tau protein (see Section IV.E) (Wang et al., 2018).
C. Chemoenzymatic Synthesis of Heparin/Heparan Sulfate Structures
Chemoenzymatic synthesis is a promising strategy for the production of glycosaminoglycans (GAGs) from nonanimal sources (Zhang et al., 2020d; Gottschalk and Elling, 2021), particularly suitable for the generation of LMWH-like molecules (Wang et al., 2020b). Its application to heparin production depends on the use of recombinant biosynthetic enzymes, particularly those that affect the post-polymerization substitution and epimerization reactions that transform the precursor heparosan polysaccharide to heparin-like structures. The heparosan starting material is available as a capsular polysaccharide of bacteria, the best known of which is E. coli K5, though other bacteria such as Pasteurella multocida have also been investigated (Na et al., 2020). The bacterial source may itself also be engineered to modify the yield (Nehru et al., 2021) and/or the molecular weight of the resulting heparosan (Roy et al., 2021). It has even been possible to engineer a strain of E. coli to produce both heparosan (secreted) and N-deactylase/N-sulfotransferase (intracellular) simultaneously (Li et al., 2021c). Heparosan, recombinant sulfotransferases, and epimerase, with the necessary cofactors, have been combined to give a one-pot synthesis of heparin products (Bhaskar et al., 2015).
Not all chemoenzymatic syntheses require a prepolymerized heparosan. Homogenous heparin-like dodecamers can be synthesized from UDP-monosaccharides using recombinant glycosyltransferase steps interspersed with sulfotransferase and epimerase steps, to give gram quantities of a compound with promising anticoagulant activity, neutralizable by protamine (Xu et al., 2017).
D. Bioengineered Heparin
Recombinant heparin resulting from cells expressing high levels of heparin biosynthetic enzymes is a potential way to produce a more controllable though still heterogenous product (Glass, 2018). Recently, the production of heparin from recombinant human serglycin, expressed in human cells, has been proposed as a possible alternative to animal sources (Lord et al., 2016, 2016; Kim et al., 2017a). The serglycin so formed carries both heparin/HS and chondroitin sulfate (CS)/dermatan sulfate (DS) GAG chains and has anticoagulant (Lord et al., 2016) and growth factor (Kim et al., 2017a) activity. An alternative approach is to use CHO cells expressing enhanced quantities of the enzymes involved in heparin biosynthesis under optimized bioprocessing conditions (Glass, 2018; Thacker et al., 2022).
E. Heparin Mimetics
Compounds that have similar biologic properties to heparin have been the subject of much research (see Section IX), but there is relatively little clinical information on the use of such drugs compared with heparin itself. Heparin is a potent anticoagulant as a result of its ability to potentiate antithrombin and in addition can act as a HS mimetic, interacting with the numerous proteins that use HS to interact with some cells, and with elements of the extracellular matrix (see Section IV). One of the major aims in devising heparin mimetics is to separate out different actions of heparin, for example anti-inflammatory compounds lacking anticoagulant activity, given the broad spectrum of biologic activity exhibited by heparin itself (see Section VII). Also, when heparin is administered as an anticoagulant, it can bind to other proteins in plasma, sometimes causing adverse side-effects such as heparin-induced thrombocytopenia (HIT) (see Section VI.C).
There are multiple other reasons why both anticoagulant and nonanticoagulant heparin mimetics might be desirable. As discussed earlier, heparin is currently extracted from mammalian tissues and so could potentially be a source of disease-causing entities such as viruses or prions; this has led to some countries not allowing bovine heparin to be used clinically (though the current chemical treatments used for the manufacture of heparin reduce this risk to acceptable levels) (Andrews et al., 2020). Heparin is also a heterogenous mixture of GAG molecules, and no two heparin samples are exactly identical, even if prepared by the same protocol from the same tissue source.
The term “heparin mimetics” therefore covers a very wide range of preparations, from single molecular species with a single well defined biologic activity—such as the pentasaccharide fondaparinux, which is based on the high-affinity monosaccharide sequence for antithrombin—through to naturally occurring sulfated polysaccharides of uncertain structure that share some anti-inflammatory properties with heparin but that often have much lower anticoagulant activity.
F. Naturally Occurring Sulfated Polysaccharides
The process of heparin manufacture (van der Meer et al., 2017) separates heparin API from a crude GAG mixture; the residual GAGs can then be used to manufacture antithrombotic GAG preparations consisting of HS, DS, and CS such as danaparoid and sulodexide, for use in cases where heparin itself is not suitable (Dou et al., 2019). Though these GAG mixtures are if anything even more complex than heparin itself, the spectroscopic and mass spectrometric analytical methods recently developed for heparin can be applied to assess the consistency of these preparations (Ustün et al., 2011; Gardini et al., 2017; Veraldi et al., 2018).
Sulfated polysaccharides also occur widely in marine plants and animals, with a range of structures that have many potentially useful biologic activities in common with the most highly sulfated mammalian polysaccharide, heparin (Vasconcelos and Pomin, 2017). Some marine animals such as echinoderms and tunicates contain complex sulfated polysaccharides, in addition to the GAGs that are present throughout the animal kingdom (see later discussion). Some of the sulfated GAGs identified in invertebrates have structures that have not been seen in mammalian systems (Thomson et al., 2016; Karamanou et al., 2017) and an example has recently been reported from a snail made up entirely of the nonmammalian sequence [→4-)-α-GlcNAc (1→4)-α-IdoA2S (1→] n (Wu et al., 2020).
The literature on the sulfated polysaccharides found in macroalgae has also expanded rapidly in the past few years. Fucoidans, for example, are sulfated polysaccharides in which L-fucose is the predominant monosaccharide component, though other monosaccharides such as galactose, mannose, glucose, and uronic acids may be present (Zayed et al., 2020). Algal sulfated polysaccharides have long been known to have anticoagulant properties, but recent research has concentrated more on the nonanticoagulant potential of such molecules for therapeutic use (Zaporozhets and Besednova, 2016; Hans et al., 2021).
An alternative source of highly sulfated fucans with relatively simple structures is found in some echinoderm species (Pomin, 2009). Some details of anticoagulant activity are routinely provided in publications describing new examples of this polysaccharide class; for example, the fucan from Stichopus hermanii (Li et al., 2021b) is a homopolymer of 3-linked, 2-sulfated α-fucose with a very high molecular weight and some ability to prolong the activated partial thromboplastin time (APTT), but not the prothrombin (PT) or thrombin time. This molecule has no effect on AT-mediated anti-Xa or anti-IIa activity but does have some ability to inhibit IIa via the serpin heparin cofactor II (HCII). This is a typical anticoagulant profile for an echinoderm fucan and is shared by the polysaccharide from another sea cucumber Acaudina leucoprocta (He et al., 2020), though a regular repeating structure for this fucan has not been demonstrated. In contrast, the fucan from Holothuria albiventer (Cai et al., 2018), with a hexasaccharide repeat unit of variously sulfated 3-linked fucose residues, prolonged both the APTT and the thrombin time and also inhibits the action of the tenase complex that generates factor Xa from factor X (see Section V for details on the coagulation cascade). This activity has been described for other sulfated polysaccharides from echinoderms such as fucosylated chondroitin sulfate (Glauser et al., 2013; Cai et al., 2019). The anti-tenase activity of these complex polysaccharides may prove to be the basis for their antithrombotic activity (Li et al., 2021a), and some of these polysaccharides may even be active when administered orally (Fonseca et al., 2017).
In most of these studies, anticoagulant potential is identified as an ability to increase the APTT of a plasma sample. However, it should be noted that it is difficult to make quantitative comparisons between anticoagulant activities of different compounds as each laboratory has its own APTT protocol and presentation of results in clotting times rather than in units of activity against a recognized reference standard.
G. Chemically Sulfated Polysaccharides
Polysaccharides from any origin can be chemically sulfated to mimic heparin’s polyanionic nature. One of the most widely used mimetics of this type is pentosan polysulfate (PPS), an artificially sulfated xylan of plant origin that is licensed as a treatment of interstitial cystitis in the United States, United Kingdom, and Europe; a recent meta-analysis of clinical trials has confirmed the effectiveness of this drug in this indication (Taneja, 2021). Detailed structural analysis of PPS indicates that it is more heavily sulfated than heparin, with four sulfate substituents per disaccharide (Lin et al., 2019; Alekseeva et al., 2020). Another artificially sulfated polysaccharide with the same level of sulfation is the OSCS preparation that was found as a contaminant in certain heparin lots that were associated with severe adverse events. OSCS activates the contact system, promoting the production of kallikrein and through that route the generation of bradykinin, leading to profound hypotension in affected patients (Hogwood et al., 2018). There are no reports of PPS causing similar adverse effects, but long-term PPS use may be associated with a vision-threatening maculopathy (Lindeke-Myers et al., 2022). Both PPS and OSCS have some anticoagulant activity in vitro, mediated through heparin cofactor II rather than antithrombin (Colwell et al., 1999; Hogwood et al., 2018).
Alginates derived from seaweed may be chemically sulfated to give compounds with heparin-like properties. However, this process reduces the gel-forming ability of the native polysaccharide. The design of partially sulfated alginate hydrogels might lead to useful matrices for tissue engineering, incorporating heparin-binding proteins (see Section VIII); for a review see Arlov and Skjåk-Bræk (2017).
Dextran sulfate (DexS) is another heavily sulfated semisynthetic polysaccharide that exhibits non-serpin mediated anticoagulant activity (Drozd et al., 2017). Like heparin it can be quantified by its interaction with protamine (Gordon et al., 2021), and it is cleared from the circulation by the HARE scavenger of endothelial cells (Weigel, 2020). DexS can be used to improve the performance of anti-Xa assays of heparin in plasma, improving recovery and avoiding the impact of heparin binding to neutralizing plasma proteins (Amiral et al., 2021). As an aside, DexS-induced colitis in mice is a frequently used model of inflammatory bowel disorder (Xie et al., 2021).
H. Modified Heparins
Unfractionated heparin is isolated by fractionation of a crude GAG mixture without any deliberate structural modification, resulting in a highly sulfated and potent anticoagulant polysaccharide. Chemical or enzymatic modification of heparin alters the balance of its biologic properties. For example, the partial depolymerization of heparin to give LMWH results in an altered ratio of anti-Xa activity to anti-IIa activity (Gray et al., 2008) and decreases affinity for the stabilin-2/HARE heparin clearance receptor, resulting in delayed clearance and therefore increased bioavailability (Pempe et al., 2012; Johansen and Balchen, 2013). Reduction of anticoagulant activity does not require depolymerization but can be achieved by other chemical modifications such as partial, systematic desulfation, leaving anti-inflammatory properties relatively intact (Hogwood et al., 2020). Heparin that has been 2-O-, 3-O- desulfated is an effective neutrophil elastase inhibitor (Voynow et al., 2020) and has anti-inflammatory properties in a model of brain injury, inhibiting recruitment of leukocytes and reducing edema (Nagata et al., 2018). Periodate oxidation of heparin followed by reduction with borohydride gives the “glycol-split” heparins such as roneparstat and necuparanib, in which unsulfated uronic acid is cleaved between C2 and C3. Structurally, roneparstat is glycol-split N-acetylated heparin, and necuparanib is a glycol-split LMWH. Both are intended primarily as heparanase inhibitors in cancer therapy, to slow the progress of metastasis (Cassinelli et al., 2020) (see Section VII.E). However, there are other settings in which these HS mimetics may prove useful, such as the anti-inflammatory properties of glycol-split or N-acetylated heparin in reducing the complications of pseudomonas infection (Lorè et al., 2018). Heparin can also be modified by complexation, either to soluble molecules or to biomaterials for regenerative medicine; this field has been recently reviewed (Banik et al., 2021; and see Section VIII).
I. Heparin from Bovine and Other Nonporcine Sources
At present all heparin licensed for medicinal use in the United States and Europe is derived from porcine intestinal mucosa, most of it originating from Chinese pigs. This reliance on a single species reduces the robustness of the heparin supply worldwide, especially considering recent outbreaks of porcine disease such as African swine fever (Vilanova et al., 2019a). Such considerations have motivated the US Food and Drug Administration to encourage the reintroduction of bovine-sourced heparin to clinical use in the United States. Though heparin (of relatively modest potency) can be extracted from other species such as turkeys (Warda et al., 2003b) or camels (Warda et al., 2003a; Warda and Linhardt, 2006), the most feasible sources for large-scale production are those that have been used for heparin manufacture in the past: sheep and cattle. Bovine lung heparin (BLH) was in use until the 1990s but was discontinued as a response to the emergence of bovine spongiform encephalopathy. However, in some countries, for example in South America, bovine mucosal heparin (BMH) did not go out of use at that time and is still manufactured today (Vilanova et al., 2019b). Ovine mucosal heparin (OMH) has been found to resemble porcine mucosal heparin (PMH) more closely than does BMH and may be in the future an additional resource for heparin production (Kouta et al., 2019). Indeed, ovine LMWH is now in clinical use in Indonesia.
Concerns about potential contamination of bovine-derived heparin products with the prion protein infectious agents of BSE have been investigated. The normal processes in heparin manufacture are sufficiently severe that no special treatment should be necessary, especially with care taken to ensure that bovine material is sourced from disease-free herds (Andrews et al., 2020; Bett et al., 2020). The risk of contracting vCJD from heparin from US or Canadian cattle has been estimated to be extremely small—one in many millions (Huang et al., 2020).
There are clear differences between the structures, properties, and hemostatic effects between BMH and PMH (St Ange et al., 2016; Tovar et al., 2016). Both NMR and disaccharide analysis by heparinase digestion indicate that BMH has a lower degree of 6-O-sulfation and of 3-O-sulfation than PMH (St Ange et al., 2016; Tovar et al., 2016). Sequence differences between PMH, BMH, and OMH have also been identified by “building block” analysis using exhaustive heparinase digestion and subsequent reductive amination with sulfanilic acid. In particular, the nonreducing end sequence GlcA-GlcNS,3S,6S was recently identified as a porcine-specific marker (Mourier, 2020).
Molecular weights for BMH and PMH are not consistently different (St Ange et al., 2016; Tovar et al., 2016) and a survey of BMH samples from different manufacturers found some examples that would meet current US Pharmacopeia (USP) acceptance criteria for MW of porcine heparin (Mulloy et al., 2014), while others fell outside those limits with both higher and lower average molecular weights (Bertini et al., 2017c).
The specific anticoagulant activity of BMH is lower than that of PMH and can be as low as half that of porcine mucosal heparin (St Ange et al., 2016; Tovar et al., 2016; Kouta et al., 2019; Tovar et al., 2019). Though it has been confirmed that PMH and BMH are equivalent anticoagulants on the basis of potency in International Units (Jeske et al., 2018b), properties dependent on the mass of heparin are not equivalent, including neutralization by protamine: more protamine is required to neutralize one anticoagulant unit of BMH than for PMH (Hogwood et al., 2017; Glauser et al., 2018). However, in a primate model, 0.5 mg/kg protamine was adequate to neutralize UFH at either 0.5 mg/kg or 100 units/kg (Kouta et al., 2021). Like PMH, BMH and OMH can inhibit the extrinsic coagulation pathway and release tissue factor pathway inhibitor (TFPI) to an equivalent extent in terms of units of anticoagulant activity rather than by mass, when measured in vivo (Kouta et al., 2020). Relatively little work has been reported on the capacity of heparin of non-porcine origin to cause adverse side-effects, although a physicochemical study has shown that lower concentrations of OMH are required to form large platelet factor 4 (PF4)/heparin complexes (that are believed to cause HIT q.v.) as compared with PMH and BMH (Bertini et al., 2017a).
Taken altogether it has been accepted that the two most common heparins, BMH and PMH, are not equivalent and should be treated as distinct drugs; for example, they now have separate monographs in the Brazilian Pharmacopeia (Vilanova et al., 2019b). However, some studies have established that BMH could be used as a basis for the production of a more PMH-like heparin product either by fractionation (Tovar et al., 2019) or by chemoenzymatic enhancement of 3-O-sulfate and 6-O-sulfate (Fu et al., 2017; Baytas and Linhardt, 2020; Baytas et al., 2021), but there may be cost disadvantages to these options, at least in the near future.
As well as providing an alternative source for UFH, bovine and ovine heparin can be processed further to give LMWH. Chemical beta-elimination depolymerization of BLH can yield a product similar to enoxaparin, but the lower specific activity of the parent heparin (compared with PMH) is reflected in the product (Guan et al., 2016). Detailed structural and in vitro activity profiles of enoxaparin-like preparations from bovine lung, bovine mucosa, and porcine mucosa have indicated that LMWH with properties within or close to current US requirements for PM enoxaparin can readily be prepared (Liu et al., 2017a). This is also the case for the corresponding ovine preparations (Chen et al., 2019), which in addition have almost identical pharmacokinetics to porcine enoxaparin (Jeske et al., 2018a). Nitrous acid depolymerization of bovine and ovine heparin can also be used to make a product that meets EP specifications for dalteparin (although excluded due to porcine being the source origin requirement), though with differences in fine structure compared with the PM product (Xie et al., 2018).
Bovine mucosal heparin with enhanced 3-O- and 6-O-sulfation provides “enoxaparin” that is closer to the originator’s product in fine structure and activity (Baytas et al., 2021). There is no doubt that bovine and ovine LMWH products are both possible and desirable; the question that remains is whether they will be considered as biosimilar or generic enoxaparin/dalteparin or as completely novel therapeutics by regulatory authorities.
III. Analysis of Heparin
A. Analytical Methods for Pharmaceutical Heparin
Disaccharide compositional analysis for quality control of pharmaceutical heparin can be achieved by either 2D NMR (Mauri et al., 2017a) or chromatographic separation of disaccharides from exhaustive digestion with heparinases, detected by fluorescence and mass spectrometry (Galeotti and Volpi, 2016). NMR-based and chromatographic approaches have been compared (Spelta et al., 2019), and a combination of these approaches was found to provide accurate differentiation of species and organ sources of heparin.
The emphasis on structural similarity between generic/biosimilar and originator LMWHs has given rise to complex strategies of physicochemical analysis (Mourier et al., 2016). A combination of liquid chromatography mass spectroscopy with NMR spectrometry can be applied to such comparisons (Liu et al., 2017b), and such use can even correlate LMWH heparin samples with their parent UFH (Liu et al., 2017c).
The anticoagulant methods used to determine heparin activity are described in Section 5.9.
B. Response to Contaminated Heparin
Since the episode of contamination of pharmaceutical heparin with OSCS in 2007–2008 that led to serious adverse events associated with the clinical use of certain heparin preparations, including fatalities (Chess et al., 2012), development of new methods for the assessment of heparin continues (Devlin et al., 2019). It has now been established that the contaminant OSCS was added at an early stage of heparin manufacture, so methods applicable to the efficient screening of crude heparin samples rather than API and final product are particularly useful (Mauri et al., 2017b; Mendes et al., 2019) in ensuring such contamination does not occur in the future.
After a period of rapid evolution, pharmacopeial monographs have adopted a stable set of orthogonal methods for the determination of identity, purity and potency of heparin samples; see for example the USP (Szajek et al., 2016). In addition to these, the challenges of potential contamination and the prospect of introduction to the United States and Europe of heparin from sources other than porcine mucosa have inspired the development of novel spectroscopic methods, often using data manipulation by multivariate analysis (Rudd et al., 2019). These chemometric tools are increasingly useful in the quality assessment of pharmaceuticals (Monakhova et al., 2018b) and can be used to generate protocols that make a complex spectrum more easily interpretable for routine use. The regular analysis of heparin lots can over time generate datasets of considerable size for the “training” of such protocols (Monakhova and Diehl, 2019). In the course of collection of NMR data for heparin, the acquisition of diffusion-ordered NMR spectrometry data allows rapid estimation of average molecular weight for both UFH and LMWH samples, calibrated against GPC results using partial least squares regression (Monakhova et al., 2018a).
Screening of finished heparin product, typically an aqueous solution, can be achieved using a combination of NMR, UV-vis, Fourier transform infrared (FTIR) spectroscopy, and a potentiometric multisensory system (for chloride and hydrophilic anions) (Burmistrova et al., 2020) with the aid of multivariate analysis. The evaluation of heparin powder samples by FTIR alone can distinguish between heparin calcium and heparin sodium and between samples of different species of origin, as well as between pure heparin, heparinoids, and contaminated heparin (Burmistrova et al., 2021).
The focus on analysis of heparin arising from the contamination episode, and also comparisons of biosimilar/generic LMWH products, has raised the level of detail to which heparin samples are now inspected. Process-related structural impurities arising from harsh manufacturing conditions such as high pH and high temperature can, for example, give rise to 2-O-desulfation of heparin samples that can be monitored by disaccharide analysis (Anger et al., 2018).
C. Introduction of Heparin from Other Species
Structural and functional differences between heparin from different sources have implications for regulatory matters. For example, the Brazilian Pharmacopeia has separate monographs for bovine and porcine heparin from intestinal mucosa, with different acceptance criteria for the two heparin types (Vilanova et al., 2019b). Methods for distinguishing between BLH, BMH, OMH, and PMH are discussed in Section 2.9. Surveys of recently manufactured BMH have shown that overall levels of impurities (whether protein, nucleic acid, or galactosamine containing GAGs) in BMH are comparable to those observed in PMH (Workman and Carrick, 2020); molecular weight distributions for the same set of BMH samples vary more than do current PMH samples (Bertini et al., 2017c).
The detection of blended heparin samples, from more than one species/tissue, is now a necessity. PCR methods are sensitive and rapid (Houiste et al., 2009; Concannon et al., 2011; Auguste et al., 2012), and have already shown signs of ruminant DNA in a number of crude industrial porcine heparin samples (Huang et al., 2012). While disaccharide profiling is less sensitive than qPCR (Houiste et al., 2009), quantitative analysis of mixtures of one type of heparin in another can be achieved by multivariate analysis of NMR spectra and disaccharide or tetrasaccharide analysis (after digestion with heparin lyase II) (Ouyang et al., 2019). The application of time-of-flight secondary ion mass spectrometry with multivariate analysis gives particularly sensitive results, both for the detection of contaminants such as OSCS and for the detection of traces of BLH or BMH in PMH (Hook et al., 2021). Both this technique, and principal component analysis of NMR spectra, yield surprisingly good but as yet incompletely analyzed correlations between spectral features and anticoagulant activity of heparin samples (Monakhova et al., 2019; Hook et al., 2021).
IV. Molecular Interactions of Heparin
A. Heparin Interactions with Proteins
The number of heparin-binding proteins identified so far is now large enough to form a dataset suitable for analysis using bioinformatic techniques (Ori et al., 2011; Gómez Toledo et al., 2021; Vallet et al., 2021). This dataset is referred to as the heparin interactome. Rather than thinking of each individual heparin-protein interaction as a simple one-to-one phenomenon (or very occasionally as a ternary complex), it is now possible to describe intricate functional networks that include protein-protein interactions as well as protein-heparin interactions. Though this field is still young, it might in time be a useful tool for both basic research and drug discovery.
Ori et al. (2011) were able to put together a list of 435 human heparin-binding proteins, with later researchers assembling 530 human proteins (Gómez Toledo et al., 2021) or 580 mammalian proteins (Vallet et al., 2021). This information can then be combined with databases of protein-protein interactions to generate a combined network of heparin-protein and protein-protein interactions, which can then be sorted into subnetworks (clusters) of functionally related interactions. The bioinformatics protocols to achieve this vary between groups, but the overall conclusions drawn agree that the major functional clusters associated with the heparin interactome involve the immune and inflammatory responses, signaling, and developmental biology. Though proteases of the coagulation cascade form an identifiable cluster, it is relatively minor in size, emphasizing the very wide range of heparin/HS functions as compared with the limited range of current therapeutic uses of heparin.
Heparin interactomes have also been described for subsets of human proteins, such as a comparison between the heparin interactomes of healthy and diseased pancreas. Heparin-binding proteins unique to the acute pancreatitis or pancreatic ductal adenocarcinoma interactomes could be of value as a source of potential biomarkers or drug targets (Nunes et al., 2013). MCF-7 cancer cells cultured in serum-free medium and treated with heparin showed alterations of expression of 105 of 1357 genes potentially related to breast cancer pathogenesis, resulting in a less tumourigenic phenotype. This was attributed to the ability of heparin to interfere with the interactome of cell surface HS (Chen et al., 2013).
Studies using systematic proteomics-based protocols to enlarge the known heparin interactome will inevitably identify novel heparin-binding proteins. The heparin interactome of human and mouse endothelial cells, including membrane proteins, as well as soluble proteins, has been studied using partial proteolysis of live cells, heparin affinity chromatography, and liquid chromatography with tandem mass spectrometry. Among several other examples, the C-type lectin 14a, a modulator of angiogenesis, was identified and its heparin binding site characterized (Sandoval et al., 2020).
The GAG interactome of E. coli has also been investigated using a proteome chip incorporating about 4300 purified E. coli proteins. Among the 185 heparin-binding proteins found, one outer membrane protein YcbS has micromolar affinity for heparin and is crucial for invasion of host cells (Hsiao et al., 2016). A later study concentrating on the iduronic acid-containing GAGs found an additional outer membrane protein MbhA, also involved in the interaction between E. coli and the host cell surface (Hsiao et al., 2019).
The broad sweep of interactomics does not obviate the need for detailed characterization of individual heparin-protein interactions. Structural biology in this area is however running behind the sheer number of interactions now identified, and the experimental techniques employed for solving GAG-protein complex structures are not by and large suited to high throughput protocols. It is therefore necessary to deploy computational chemistry methods, in particular molecular docking and molecular dynamics protocols, to fill in the gaps in the database of experimentally defined binary and ternary complex structures involving heparin/HS. Paiardi and colleagues have recently published a very readable overview of this field that describes both the scope and the limitations of structural in silico studies of GAG-protein interactions (Paiardi et al., 2021); another survey describes the role of molecular dynamics in defining levels of selectivity for oligosaccharide sequences in protein-GAG binding, ranging from highly selective (heparin-AT for example) through moderate and plastic selectivity, to entirely charge-based nonselective interactions (Nagarajan et al., 2022).
Where the three-dimensional structure of a protein is already known, the approximate location of a heparin binding site on its surface is sometimes not difficult to find, as positively charged areas on a protein surface will inevitably be attracted to the negatively charged polysaccharide. Calculations that generate a model of a heparin oligosaccharide ligand docked into a binding site on the protein surface can give more detailed predictions (an easy-to-use example is provided by the ClusPro server) (Kozakov et al., 2017). However, a short oligosaccharide is not always an adequate model for a full-length GAG polysaccharide, and modeling a whole, heterogenous heparin/HS molecule is currently impractical. One way around this may be to use a grid-based calculation protocol in which a surface map of binding probability density is generated using a small fragment ligand to trace on the protein surface a likely extended polysaccharide binding site (Grad et al., 2018). Such a method is less computationally expensive than molecular dynamics and gives results in line with experimental data for the interaction between the morphogen sonic hedgehog and heparin (Grad et al., 2018).
Where experimental data give incomplete three-dimensional structures, as is often the case in NMR and/or site-directed mutagenesis studies of heparin-protein interactions, molecular docking calculations with experimentally derived restraints can generate plausible three-dimensional models. This has been achieved for example in a study of the matrix metalloproteinase 7 (matrilysin; MMP7) for which heparin/HS promotes maturation of proMMP7 to the active form. Besides chemical shift perturbations on heparin titration, paramagnetic techniques yielded relaxation enhancements that were used in addition to mutagenesis data as the basis for docking restraints. This led to the identification of two basic heparin binding tracks on the protein surface, one involving the pro-domain of proMMP7 and the other the catalytic domain and C-terminus (Fulcher et al., 2017). Molecular docking has also been used to illustrate the interactions between several GAGs and the MMP2 complex with tissue inhibitor of metalloproteinase 3 (Ruiz-Gómez et al., 2019).
B. Neutralization of Heparin by Protamine and Other Compounds
The approved neutralizing agent for heparin is protamine, a mixture of highly cationic peptides extracted from fish (Pai and Crowther, 2012). It is used clinically in cases of heparin overdose or to reduce excess anticoagulation after cardiac surgery (see also Section 6.1). The constituent peptides of protamine are rich in arginine residues and can be separated by high performance liquid chromatography to control identity and purity (Awotwe-Otoo et al., 2012). The interaction between heparin and protamine is charge based, between cationic peptide and anionic polysaccharide, and the neutral macromolecular salt formed has no anticoagulant activity. Binding is not dependent on any element of fine structure in heparin, and it is likely that protamine also neutralizes its other biologic activities. Protamine varies in its quantitative capacity to neutralize anticoagulant activity depending on the specific activity and molecular weight profile of the heparin sample (Hogwood et al., 2017). Protamine does not completely neutralize the anti-Xa activity of LMWHs containing short heparin oligosaccharides (Schroeder et al., 2011), but anti-IIa activity of LMWH, as it is exhibited by longer heparin polysaccharide chains, can be neutralized by protamine (Kouta et al., 2021).
Besides its limitations for neutralization of LMWH, protamine has the disadvantage of several potential adverse side-effects (Sokolowska et al., 2016) and can form large immunogenic complexes with heparin reminiscent of the PF4/heparin complexes that cause HIT (Bakchoul et al., 2016; Sommers et al., 2017). Alternatives to protamine such as PF4 have yet to receive regulatory approval, and another approach using recombinant human FVIIa that increases procoagulant activity has also been considered (Pai and Crowther, 2012). New heparin-neutralizing agents are in development at various preclinical and clinical stages (Sokolowska et al., 2016) (see Section VI.A.1), in part attempting to address the recent difficulties encountered due to protamine shortages (Maneno and Ness, 2021).
Low molecular weight protamine, a product of enzymatic digestion of salmon protamine sulfate, has been found to neutralize both UFH and LMWH and to exhibit less antigenic potential than unfractionated protamine (He et al., 2014). Low molecular weight protamine may also be used in other applications of protamine, for example as an excipient in insulin formulations, but its use cannot address the problem of protamine shortage. Another cationic peptide, poly-L-lysine, can also bind to and neutralize heparin, and a poly-L-lysine fraction with MW 15,000 g/mole has been identified as a promising substitute for protamine (Muralidharan-Chari et al., 2017). As poly-L-lysine can be prepared by bacterial fermentation or synthetically, it may not be as vulnerable as protamine to supply problems. A different readily available cationic macromolecule is the polysaccharide chitin, which can be converted to quaternized chitosan with similar neutralizing effects to protamine (Drozd et al., 2019). Completely synthetic block copolymers, consisting of one neutral and one cationic block, have been made and optimized for heparin binding in vitro (Välimäki et al., 2016); the complexes so formed have a neutral outer surface and do not aggregate. Another di-block copolymer termed HBC (heparin binding co-polymer) neutralizes LMWH effectively and was well tolerated in animal studies (Kalaska et al., 2020).
Poly-L-lysine may also be incorporated into dendrimer format, and a G2 dendrimer can be designed that is able to neutralize UFH as well as can protamine. In addition, this molecule can provide better neutralization of the anti-Xa activity of LMWH and even, to some extent, fondaparinux (Ourri et al., 2019). Other dendrimers under development as protamine substitutes include self-assembling cationic dendrimers (Marson et al., 2019) and the “universal heparin reversal agents” in which a cationic dendrimer is substituted with an outer brush of methylated polyethylene glycol, partially shielding the charged dendrimer and so preventing multivalent aggregation (Kalathottukaren et al., 2017).
Other strategies for generating heparin-neutralizing cationic structures involve the design of recombinant virus-like particles using a two-plasmid expression system to incorporate heparin-binding peptides (Choi et al., 2018) or using a simple single T to R mutation to enhance the heparin binding of a bacteriophage virus-like particles (Cheong et al., 2017).
Not all of the recently proposed heparin neutralizing agents depend on charge-based non-specific interactions of heparin with a cationic polymer. For example, a recombinant inactive AT was as efficient as protamine at neutralizing heparin after cardiopulmonary bypass in rats (Bianchini et al., 2018). A similar strategy has been adopted in the design of andaxanet, a recombinant inactivated factor Xa (FXa) already approved as an antagonist to the Xa inhibitors apixaban and rivaroxaban and shown to also neutralize the activity of heparin (Maneno and Ness, 2021). These inactivated proteins of the coagulation system act as decoy molecules, binding either to the high affinity motif in heparin (for inactivated AT) or to heparin-activated AT (for andaxanet).
Ciparantag is a small (MW 512 g/mol), polybasic molecule that is currently in clinical trials as a heparin antidote. It was designed specifically to interact with heparin on the basis of charge, and, it seems by chance, has also been found to bind to direct oral anticoagulants (DOACs) and neutralize their activity (Ansell et al., 2021). At a much earlier stage of development for medicinal application, the use of a dynamic covalent selection approach has led to the identification and synthesis of a dialkylated spermine with low micromolar affinity for heparin, capable of neutralizing anti-Xa activity in a chromogenic assay (Corredor et al., 2018).
C. Heparin Sensors
Measurement of the concentration of heparin (and other highly sulfated polysaccharides) in aqueous solution, in terms of weight rather than units of activity, has for many years been possible by dye binding assays (Templeton, 1988). More recently, the development of improved UV/visible absorbing or fluorescent heparin-binding molecules and complexes has given rise to a substantial literature, with a view to the design of heparin-sensing systems for use in monitoring heparin concentration in plasma (Fan et al., 2021). Clinical heparin monitoring generally uses clotting times, commonly the APTT (see Section V.I), but there are circumstances in which a direct measurement of heparin substance might be useful. For example, the concentrations of heparin mimetics and derivatives with reduced anticoagulant activity cannot be estimated by their effects on coagulation (Warttinger et al., 2016). However, there is one fluorescent dye assay currently available in a kit formulation (Heparin Red) (Warttinger et al., 2016; Rappold et al., 2017), and a considerable number of heparin-sensing fluorescent systems are still at the development stage and have been reviewed elsewhere (Fan et al., 2021). A few recent examples follow.
Several heparin-sensing systems with good sensitivity make use of gold nanoparticles (AuNPs) (Qi et al., 2019, 2021). A particularly sensitive heparin sensor uses the fluorescence of a lead halide perovskite on aggregation in aqueous solution; the fluorescence is quenched by AuNPs at low concentration, restored by addition of protamine to sequester the AuNPs, then requenched in the additional presence of heparin to neutralize the protamine. Though it may seem elaborate, this strategy gave a low limit of detection in the subnanogram range (Qu et al., 2021). Much simpler, though less sensitive, is the use of thiazole orange, for which heparin-induced aggregation causes a 100 nm red shift in its absorption maximum combined with enhancement of fluorescence (Pandey et al., 2021). In another study, a fluorophore bearing a diethylaminocoumarin donor and a pyridinium acceptor was synthesized that detects heparin by reduction of fluorescence (Jana et al., 2018).
The use of protamine in heparin-sensing systems is common. As described, sensors can be designed that give enhanced fluorescence in the presence of protamine, which can then be quantitatively reversed by heparin (Aparna et al., 2019; Chan et al., 2019; Ghosh et al., 2019; Jiang et al., 2020), or vice versa, with sensors that “turn on” with heparin and “turn off” with subsequent addition of protamine (Maity and Schmuck, 2016; Gong et al., 2017; Qi et al., 2019; Cui et al., 2020). Fluorescence is not the only read-out; electrochemical methods have also been reported (Rengaraj et al., 2019). A particularly innovative approach uses protamine inhibition of the rolling circle amplification of DNA, turned off quantitatively by heparin (Lin et al., 2021).
D. Interaction of Heparin with Chemokines, Cytokines, and Growth Factors
The cytokines are a structurally diverse group of small proteins that provide communication between inflammatory and hematopoietic cells. They are released from immune cells and have their effect on other cells by interacting with cell surface receptors, after diffusing through the extracellular matrix. Interactions with cell- and matrix-bound HS can influence the diffusion, stability, and cell surface reception of cytokines. Certain subgroups of cytokines are referred to as growth factors, chemokines, interleukins, or interferons, and in this update, we focus on recent structural studies of some of the chemokines, in particular PF4. These are covered in Sections 4.4.1 and 4.4.2. The mechanisms underlying the anti-inflammatory activities of heparin are as yet not fully understood (see Section VII.A) but are likely to involve cytokine interaction, though the extent of the heparin interactome is so great (see Section IV.A) that analysis of the contribution of individual cytokines to clinical observations is far from simple. On the other hand, it is clear that some of the adverse effects of heparin are the consequence of binding to specific cytokines such as PF4 (see Section IV.D.2). In addition, cytokine binding to heparinized matrices forms the basis for a number of drug delivery strategies and biomaterials for use in regenerative medicine, as recently reviewed (Ishihara et al., 2019; Anderegg et al., 2021).
The structural biology of the interactions between heparin/HS and growth factors, particularly the FGFs, has been the subject of much study, as summarized in the earlier version of this article and elsewhere (Mulloy et al., 2016; Pomin, 2016; Zulueta et al., 2018; Ghiselli, 2019). It is interesting to note that the structural characteristics of FGF interactions, in terms of preferred sulfation patterns of the heparin/HS partner and location and architecture of the heparin binding site on the protein, are correlated with phylogenetic relationships between the FGFs (Li et al., 2016).
The structural biology of the FGF-7 family (FGFs 3, 7, 10, and 22) has been reviewed; differing affinities for HS between members of this family could contribute to biologic action by controlling local diffusion (Zinkle and Mohammadi, 2019). A useful study has provided a comparison of the surface plasmon resonance binding affinities of several FGFs along with HGF and transforming growth factor-beta1 for heparin; heparin fragments, selectively desulfated heparins, and other GAGs were then compared by competition experiments (Zhang et al., 2019a).
Structures of the complexes between FGF-1 and FGF-2 with heparin/HS fragments are sufficiently well documented that they are often used as model systems for the development of new theoretical and experimental techniques. For example, the evaluation of computational approaches such as docking and molecular dynamics for the simulation of heparin-protein complexes have used the experimental FGF-1/heparin complex as a benchmark (Babik et al., 2017). Molecular dynamics simulations of the FGF-1 complex with a heparin hexasaccharide have been carried out, extending to the microsecond scale, with a detailed analysis of the results that can be expected to have impact on theoretical approaches to heparin-protein interactions in general (Bojarski et al., 2019).
A mass spectrometric method has made use of the FGF-1-heparin complex as a model system for identification of high affinity sequences for the protein within heparin, by subjecting the complex to collisionally induced dissociation. Those parts of the heparin molecule not directly involved in interaction with the protein suffer sulfate loss and breakage of glycosidic bonds, leaving behind only the minimal protein-binding motif within the heparin chain (Zhao and Kaltashov, 2020). Electrospray ionization mass spectrometry has been applied by the same group to investigate interactions between FGF-1 and heparin oligomers of defined length, identifying the overall extent of sulfation as the major determinant of binding efficiency. Sulfation level controls the affinity of heparin oligomers toward single FGF-1 molecules and also promotes their multimerization (Minsky et al., 2017). This emphasis on the importance of local dynamics and electrostatic interactions was echoed in a study of polyanion binding to FGF-1 by hydrogen-deuterium exchange mass spectrometry (Angalakurthi et al., 2018).
It is interesting to note that the thermal stability of FGF-1 is strongly affected by structural changes in and near the heparin binding site. Nullification of charges in the heparin binding pocket by mutagenesis was found to significantly increase the stability of wtFGF-1 (Agrawal et al., 2021), whereas the introduction of a basic residue to extend the heparin binding site (D82R) increased backbone flexibility and reduced biologic activity, in spite of increased affinity for heparin (Davis et al., 2018).
The selectivity of FGF-1 for patterns of sulfation within the HS sequence has been explored by NMR methods such as transferred NOEs and saturation transfer difference spectroscopy using a library of variously sulfated GlcN-IdoA-GlcN trisaccharides (García-Jiménez et al., 2017). The authors were able to confirm both that the oligosaccharides interact with FGF-1 in an extended fashion, involving the reducing and nonreducing monosaccharides and that a 6-sulfate on the reducing GlcN is particularly important for binding.
The interactions between heparin/HS and the cytokines of the transforming growth factor-beta family have been reviewed elsewhere (Rider and Mulloy, 2017). The largest group within this family are the bone morphogenetic proteins (BMPs) and some of their antagonists, several of which are known to bind to heparin/HS. Osteoporosis is a recognized adverse side-effect of heparin therapy (Alban, 2012; Signorelli et al., 2019), and though the mechanisms are as yet ill-understood, it is likely that BMPs and their antagonists play a part in the bone remodelling process (Zou et al., 2021). A recent study has concluded that long-term enoxaparin treatment may impair bone healing through suppressing the differentiation of bone marrow-derived stem cells toward osteoblasts, with concomitant reduction in expression of BMP-2 (Li et al., 2022).
BMP-2 is a prospective therapeutic agent in the treatment of bone defects and fractures, and the minimal size and sulfation pattern of heparin oligosaccharide that can potentiate BMP-2 bone formation has been defined as an N-sulfated decamer with additional 6-O-sulfation but reduced 2-O-sulfation (Smith et al., 2018). BMP-2 promoting heparin mimetics such as sulfated chitosan has also been described (Zheng et al., 2021). Difficulties in expressing BMP-2 in prokaryotic systems have been addressed by the design of a modified BMP-2 protein with increased solubility (due to hydrophilic mutations) and enhanced heparin binding (due to extension of the N-terminal heparin-binding sequence) (Heinks et al., 2021). The BMP-2 heparin interaction has also been made use of in a mineralized ECM/heparin scaffold loaded with a BMP-2 peptide, designed for guided regeneration of osteoporotic lesions (Sun et al., 2018). BMP-4, like BMP-2, has a heparin-binding site in the N-terminal sequence; truncation of this sequence results in reduced heparin binding and altered type II receptor binding profile (Aykul et al., 2022).
In contrast, the heparin-binding domains of BMP-5, BMP-6, and BMP-7 appear to be located in the C-terminal tail. Peptides corresponding to the C-terminal sequence of BMP-5 or the N-terminal sequence of BMP-2 or BMP-4 were able to stimulate chondrogenesis, perhaps by dislodging HS-immobilized BMPs at the cell surface or in the matrix (Billings et al., 2018). For BMP-6, cooperative binding with contributions from basic amino acid residues in both the N-terminal and C-terminal unstructured tails has been proposed based on site direct mutation studies supported by molecular dynamics calculations; BMP-6 is a hepcidin inducer, so modulation of its activity by heparin/HS may influence iron availability (Asperti et al., 2019; Denardo et al., 2021).
For the CAN family of BMP antagonists, heparin/HS binding sites are located within the cysteine knot region (characteristic of transforming growth factor-beta family structures), rather than the N- or C-terminal unstructured tails (Rider and Mulloy, 2017). Heparin binding sites have been characterized for both gremlin-1 (Tatsinkam et al., 2015) and gremlin-2 (Kattamuri et al., 2017), both of them made up of basic amino acids in a linear arrangement along finger 2 of the cysteine knot structure. This is distinct from the BMP binding site, and in the bound complex of grem-2 and BMP-2 the heparin-binding sites form a single continuous site with enhanced affinity for heparin (Kattamuri et al., 2017). Another CAN family member, sclerostin, has been the subject of a systematic surface plasmon resonance study of binding to GAGs, showing that an oligosaccharide at least 18 monosaccharides in length is required to compete effectively with whole heparin (Zhang et al., 2020a).
As a co-crystal of interleukin-10 with heparin could not be obtained, the structure of the interleukin-10/heparin complex has been determined by innovative protein NMR techniques using not only chemical shift perturbations but also introducing the use of pseudocontact shifts in the presence of lanthanides to protein-GAG complex studies. Heparin-binding sites on the domain-swapped dimer are located so that a single long heparin molecule could bridge the two monomers (Künze et al., 2016).
Interleukin-12 (IL-12) is a heparin-binding cytokine of the immune system, made up of two disulfide-bridge subunits, resembling a 4-a helix bundle cytokine (subunit p35) covalently prebound to a soluble class I cytokine receptor chain (subunit p40) (Garnier et al., 2018). The presence of heparin also positively modulates the bioactivity of human IL-12 (Jayanthi et al., 2017). The location of the heparin-binding site has been found near the C-terminus of the p40 unit in both human and murine IL-12 (Garnier et al., 2018). The mutation of a sequence of basic residues near the C-terminus of murine IL-12 removes heparin-binding ability and reduces biologic activity (Luria-Pérez et al., 2019). A heparin-based complex coacervate formulation significantly improved the bioactivity of IL-12 and provided protection from proteolytic cleavage; a single injection of IL-12 coacervate inhibited tumor growth in a syngeneic B16F10 mouse melanoma model (Hwang et al., 2020). In human NK cells, heparin was found to increase interferon-gamma production in synergy with IL-12, although the mechanism remained elusive (Rossi et al., 2020).
Another cytokine of the IL-12 family, IL-27, is also affected by heparin/HS; though cell surface HS is a positive modulator for IL-27 activity, soluble heparin or HS inhibit the activity of this cytokine (Cavé et al., 2020).
1. Chemokines
The inflammatory chemokines are small heparin-binding proteins, similar to each other in tertiary structure, that are involved in the recruitment and chemotaxis of leukocytes from the circulation, toward locations of infection or injury (Stone et al., 2017). The structural biology of interactions between chemokines and heparin (or other GAGs) has recently given rise to several strategies for combining experimental data such as NMR spectroscopy with computational predictions of binding geometry (Künze et al., 2021; Préchoux et al., 2021). On the whole, computational chemistry has so far been more successful in locating the heparin-binding sites of proteins than in identifying specific saccharide sequences in heparin with enhanced affinity for a particular protein (Winkler et al., 2019). Alternative approaches have used a combination of surface plasmon resonance to pull down high-affinity heparin oligosaccharides onto a cytokine derivatized chip, followed by MALDI-mass spectrometry direct from the chip surface (Przybylski et al., 2020).
Members of a subgroup of these proteins, the ELR chemokines (so-called because of the ELR sequence in the receptor binding site of CXCLs 1, 2, 3, 5, 6, 7, and 8) have a partly conserved heparin-binding site (Rajarathnam and Desai, 2020). Heparin, here acting as a mimetic of HS, promotes dimerization of these chemokines, stabilizes the protein structure, and protects it from proteolysis. The GAG-bound dimeric form may have reduced affinity for the chemokine receptor (in this case CXCR1 or CXCR2) as the heparin and receptor binding sites tend to overlap (Sepuru et al., 2016; Brown et al., 2017b; Joseph et al., 2017). The contribution of HS or other GAGs may well not be preferential recognition of the GAG-bound chemokine by the receptor but may lie rather in local control of chemokine concentration. The formation of a chemotactic gradient of chemokine concentration must necessarily involve an equilibrium between HS-bound and free chemokine, leading to the idea that HS may encourage the formation of a localized “chemokine cloud” in which a high proportion of the chemokine in the vascular glycocalyx and in extracellular matrix is in the free monomeric form and able to bind to the leukocyte cell surface receptor (Majumdar et al., 2014; Graham et al., 2019).
Heterodimerization can also occur between these structurally closely related chemokines; the ELR chemokine CXCL7 (NAP-2) can form heterodimers with CXCL1 and CXCL4 but not so well with CXCL8; an engineered disulfide-linked CXCL7-CXCL1 heterodimer has biologic activity (Brown et al., 2017a). The formation of these somewhat asymmetric heterodimers has an effect on GAG binding in terms of geometry and stoichiometry as also found recently for a trapped CXCL1/CXCL2 dimer (Sepuru and Rajarathnam, 2021), as expected considering that CXCL1 and CXCL2 have distinct heparin binding sites (Sepuru et al., 2018).
The ELR chemokines also offer an opportunity to examine details of specific amino acid residues involved in GAG and receptor binding, uncovering the significant observation that lysine and arginine residues, both of which are long side-chain basic amino acids, are not interchangeable (Joseph et al., 2018).
CXCL12 (stromal cell-derived factor 1a) and CXCL13 (B-lymphocyte chemoattractant) are important in tissue regeneration and play roles in the migration of T- and B-lymphocytes to their positions in secondary lymphoid organs, where they are involved in the formation of the germinal centers during the adaptive immune response. A study of CXCL-12 GAG binding by NMR chemical shift perturbation titration identified a high affinity heparin binding site and a second lower affinity site overlapping the receptor binding area (Panitz et al., 2016). More recently this information has been used to engineer mutant CXCL12 with reduced or enhanced GAG-binding ability, as shown by rate of release from a heparin-substituted hydrogel (Spiller et al., 2019). A relatively recently described chemokine, CXCL14, has also been investigated using a similar strategy to show more than one heparin-binding location on the protein surface. An unexpected loss of NMR signal during the titration was attributed to the formation of higher oligomers than a simple dimer (Penk et al., 2019).
The structure of CXCL13 with a heparin tetrasaccharide has been solved, showing that part of the heparin binding site is made up of basic residues in a disordered C-terminal extension, not present in the ELR chemokines (Monneau et al., 2017). In that study the question of whether CXCL12 and CXCL13 recognize different sequences in their HS ligands remains tantalizingly out of reach, but very recently reported technological advances in the form of 13C labeled semi-synthetic heparin/HS oligosaccharides have been designed with a view to their use in NMR studies of interactions with chemokines such as CXCL12 (Préchoux et al., 2021). Though the number of oligosaccharides studied so far is low, this method, especially when used together with 15N-labeled proteins, allows detailed atom-by-atom investigations of protein-GAG interactions. Using this approach, the two isoforms, CXCL12α and CXCL12γ, were shown to prefer tetrasaccharide ligands with specific patterns of sulfation, rather than simply a higher overall degree of sulfation (Préchoux et al., 2021).
The chemokines CCL3 (MIP-1a) and CCL5 (RANTES) can self-assemble into very large oligomeric structures, a process that is promoted by interaction with GAGs. Structures of oligomers in complex with a synthetic heparin octasaccharide have been studied by crystallography, small angle X-ray scattering, and molecular modeling, to suggest that the basic hexameric asymmetric unit can be extended to produce long double-helical oligomeric structures with an overall rod-like shape (Liang et al., 2016). The same study offers the possible formation of a hetero-oligomer as an explanation for the ability of the chemokine CXCL4 (also known as PF4) to arrest CCL5-stimulated monocytes.
2. Platelet Factor 4
An occasional negative consequence of heparin treatment is HIT, in which multivalent interactions between heparin and PF4 give rise to ultra-large complexes (ULCs) that induce an immune response (see Section VI.C.1). The resulting HIT IgG antibodies bind to the complexes and also to FcgRIIA on the surface of platelets and monocytes, resulting in platelet activation and aggregation, thus causing thrombocytopenia, as well as monocyte-mediated activation of coagulation through the release of tissue factor (Arepally and Cines, 2020).
Several recent studies have helped to elucidate details of the molecular interactions underlying HIT ULC formation (Khandelwal and Arepally, 2016), such as the crystal structures of PF4 complexed with the synthetic heparin pentasaccharide, fondaparinux, and complexed with the monoclonal antibody KKO, a model for (polyclonal) HIT antibodies. In the PF4-fondaparinux crystal structure, one fondaparinux molecule binds to a groove in the PF4 tetramer formed by three monomers that is shared by another PF4 tetramer by binding to its C-terminal helix. The ability of such a short heparin fragment to bridge two protein tetramers provides insight into the ability of longer heparin molecules to induce ultra-large, multivalent complexes. A model was proposed in which the KKO antibody interacts with the PF4 tetramer tightly clustered around a central heparin molecule, in which heparin stabilizes the tetrameric structure and increases the avidity of the antibody interaction by clustering (Cai et al., 2015). This model also shows how heparin can be a crucial element of the ULCs while not contributing directly to the epitope of the HIT antibodies. Indeed, ULCs can assemble in the absence of heparin, due to the presence of other polyanions such as nucleic acids and polyphosphates (Greinacher et al., 2017) or extended strings of von Willebrand factor released from endothelium following injury (Johnston et al., 2020).
Formation of HIT ULCs is dependent on heparin chain length, with several physicochemical techniques indicating that small heparin oligosaccharides bound to PF4 less strongly than longer fragments and induced less conformational change in PF4 (Delcea and Greinacher, 2016; Nguyen et al., 2020). However, not all antibodies raised by PF4-heparin complexes are capable of activating platelets; small-molecule force spectroscopy can distinguish several types of antibody and has been able to demonstrate that there exists a particular class of HIT antibody that can cluster PF4 in the absence of heparin, providing a plausible mechanism for autoimmune thrombocytopenia in patients with no history of heparin treatment (Bui and Nguyen, 2018).
E. Heparin and Neurodegeneration
1. Repair of Nervous Tissue after Injury
The development and repair of nervous tissue is known to be modulated by the GAG sidechains of extracellular proteoglycans, in particular chondroitin sulfate PGs (Djerbal et al., 2017; Hussein et al., 2020; Mencio et al., 2021). Current thinking regards CS as being inhibitory toward neuronal regeneration in adult CNS (Rauvala et al., 2017), although it has also been shown that a CSPG binding factor pleiotrophin can work with CS or HS to enhance neurite outgrowth both in vitro and in a mouse model (Rauvala et al., 2017). Neural regeneration requires both neurite growth and myelination; sulfated heparin/HS-like polysaccharides have been screened for both of these processes in a mixed cell co-culture system, with some heparin mimetics able to promote growth and others myelination (McCanney et al., 2019a,b). HS and its mimetics have been suggested as part of a therapeutic approach to CNS injury based on cell transplantation (Lindsay et al., 2020).
2. Alzheimer’s Disease and Other Protein Misfolding Related Conditions
In Alzheimer’s disease, abnormally folded microtubule associated protein tau (known simply as tau) forms insoluble neurofibrillary tangles within neurons of the CNS. In addition, a misfolded peptide known as amyloid beta (Aβ) forms extracellular plaques. The spread of misfolded tau and Aβ through the brain as the disease progresses is thought to happen through a prion-like mechanism; tau is transferred from cell to cell through a synaptic route and acts as a template for misfolding. This pathway involves HS, which is known to bind to tau monomers, oligomers, and has long been known to exist in tangles in vivo (Mah et al., 2021). The involvement of HS in both aggregation and transport of misfolded tau and Aβ has raised much recent interest in the potential of HS mimetics such as heparin to interfere in the biology that lies behind these dementia-causing conditions (Alavi Naini and Soussi-Yanicostas, 2018).
The presence of heparin (or other sulfated polysaccharides such as dextran sulfate) (Masuda-Suzukake et al., 2020) encourages aggregation and fibril formation of tau, and the structure of heparin-induced tau fibrils has been studied by numerous means such as solid-state NMR (Dregni et al., 2020, 2021), nanopore sensors for particle-size distribution (Giamblanco et al., 2020), near infra-red spectroscopy for the interaction of tau with water and its influence on folding (Sun et al., 2020), and hydrogen/deuterium exchange mass spectrometry for conformational dynamics of folding (Huang et al., 2018). Protocols have been published for the study of heparin-induced fibrils by FTIR spectroscopy, UV resonance Raman spectroscopy, and atomic force microscopy (Ramachandran, 2017). Some of the structural studies, however, find differences between heparin-induced aggregates and naturally formed fibrillar structures that are induced by other mechanisms (Fichou et al., 2018). Differences between heparin-induced and phosphorylation induced aggregates have also been noted in FRET and NMR studies (Despres et al., 2019); cryo- and immune-electron microscopy have also shown that heparin-induced filaments of tau are not identical in structure with those formed in Alzheimer’s or Pick’s disease (Zhang et al., 2019b).
Details of the influence of polysaccharide fine structure on heparin-tau interactions have shown a dependence on 6-O-sulfation (Zhao et al., 2017). Transmission of misfolded tau between two cells involves HS as a cell surface receptor; cells lacking HS biosynthetic enzymes (notably the 6-O-sulfotransferase) have impaired ability to take up tau aggregates (Stopschinski et al., 2018). This process is inhibited by HS mimetics such as the synthetic heparinoid SN7-13 (a polydisperse mixture of linked fondaparinux-like pentasaccharides) (Stopschinski et al., 2020), as well as quite short synthetic heparin oligosaccharides (Wang et al., 2018).
The enzyme β-secretase-1 (BACE-1) cleaves amyloid precursor protein to give the Aβ peptide and is therefore a target for Alzheimer’s disease therapy. Several sulfated polysaccharides can inhibit BACE-1 including GAGs from marine sources (Mycroft-West et al., 2020a, 2021) and long oligosaccharide (up to 26-mer) products of chemoenzymatic synthesis (Li et al., 2019b). It is also possible to extract low anticoagulant HS and LMWH from crude porcine mucosal heparin that have BACE-1 inhibitory activity, positively correlated with increasing size and increasing degree of sulfation (Zhang et al., 2016).
Heparin can accelerate or inhibit formation of Aβ fibrils in a concentration-dependent fashion, acting essentially as a polyelectrolyte (So et al., 2017), though it has been observed that 6-O-sulfation and N-sulfation of heparin are both necessary for interaction with Ab40 fibrils, whereas 2-O-sulfation is not (Stewart et al., 2017). Heparin can also slow the zinc-induced aggregation of Aβ peptides, possibly by means of an interaction with the metal-binding domain of Aβ (Radko et al., 2018). The precursor protein of Aβ, amyloid precursor protein, and relations amyloid precursor-like proteins 1 and 2 bind to heparin/HS through a conserved domain known as E2. The structure of the amyloid precursor-like protein-1/heparin dodecasaccharide co-crystallized complex has identified two distinct heparin binding modes, one of which involves tight and specific binding of the protein to a nonreducing end 2-sulfated iduronic acid and the second of which is simply charge-based binding to a linear hexasaccharide sequence (Dahms et al., 2015).
The contribution of protein misfolding to human disease is wide, and a number of other proteins can be induced to form extracellular aggregates in the presence of heparin, such as peptides from amyloidogenic mutants of apolipoprotein-1 (Mikawa et al., 2016; Townsend et al., 2020) and the naturally amyloidogenic neuropeptide β-endorphin (Nespovitaya et al., 2017). In both cases heparin appears to not only be an accelerator of aggregation but is also incorporated into the aggregated structures. In type II diabetes mellitus, amyloid plaques formed from islet amyloid polypeptide (sometimes known as amylin) are found in the pancreas containing matrix components including heparin/HS; these have recently been the focus of computational chemistry simulations predicting a strong dependence on oligosaccharide length on the interaction between heparin and peptide (Asthana et al., 2018).
F. Mast Cells and Heparin
Mast cells, derived from bone marrow progenitors, play a role in defense against pathogens and are found particularly in tissues exposed to foreign antigens such as the respiratory and gastrointestinal tracts (Krystel-Whittemore et al., 2016). These cells may be activated by many types of stimuli, including but not limited to the cross-linking of IgE receptors by allergens that plays a crucial role in the allergic response (Olivera et al., 2018). The cytoplasm of mast cells contains granules from which prestored factors such as proteolytic enzymes, peptides, amines, and GAGs from the proteoglycan serglycin are released on activation, and in addition mast cells may be stimulated to release a range of chemokines, cytokines, and growth factors without degranulation (Theoharides et al., 2019; Elieh Ali Komi et al., 2020). The presence of heparin and other highly sulfated GAGs attached to the serglycin core in cells generating such a wealth of heparin-binding proteins may not be a coincidence. Though the interactions between heparin and the inflammatory and immune systems are usually thought of as mimetic of cell surface HS, it is also possible that they may reflect something of the biologic role of mast cell heparin (Mulloy et al., 2017). A recent study of the intestinal mucosa of baby pigs with a depleted intestinal microbiome (raised in a clean animal facility) found fewer mast cells than expected and recorded the absence of heparin and chondroitin sulfate E, two GAGs particularly associated with mast cells. This may be simply an age effect or might result from the lack of challenge from potential pathogens in the gut (Yu et al., 2017), supporting the idea that mast cell GAGs, directly or indirectly, form part of the organism’s host defense response to infection. Another less predictable biologic role for mast cell heparin has been proposed as a promoter of adipogenesis in superficial fascia (Chen et al., 2021). Mast cell derived heparin, by binding to many proinflammatory mediators, often leads to neutralization of their biologic activity and has been previously proposed as a “natural braking mechanism” homeostatically regulating inflammatory responses (Page, 1991). The wide range of anti-inflammatory effects of mast cell derived heparin has recently been discussed (Lever et al., 2016), and these effects can be mimicked with exogenous heparin and some heparin mimetics to alleviate a correspondingly wide range of inflammatory conditions (see Section VII).
The monosaccharide composition and sequences of heparin derived from mast cells varies according to the tissue and species of origin, as is clear from recent investigations into the introduction of nonporcine heparin into the US market (Mulloy et al., 2017) (as discussed in Section 3.3). Both heparin and dermatan sulfate have been identified in granule contents from rat peritoneal mast cells (Lever et al., 2016) that, when purified, inhibited leukocyte recruitment in response to an inflammatory insult. Heparin is cleaved from serglycin by the endo-beta-glucuronidase, heparanase, and partially depolymerized to give heparin chains of roughly the same size as clinically used UFH (Lindahl and Li, 2020). However, this enzyme cannot depolymerize any GAGs of the chondroitin family, and indeed chondroitin sulfate E is capable of heparanase inhibition (Higashi et al., 2019). It has recently been shown that in human mast cells hyaluronidase 1 and the more unusual hyaluronidase 4 are present and can cleave proteoglycan-linked chondroitins to smaller oligosaccharides (Farrugia et al., 2019). The expression of a serglycin core and of heparin biosynthetic enzymes can be controlled by Mitogen-activated protein kinase (MAPK) kinase signaling; thus, inhibition of MEK1/2 (a MAPK kinase) leads to increased serglycin and GAG concentration in mast cells (Hu Frisk et al., 2018).
G. Contact and Complement Systems
Factor XIIa is a major part of the “contact system” and activates the proteases factor XI and prekallikrein to initiate both the intrinsic coagulation cascade and the kallikrein-kinin system, respectively (Bender et al., 2017). Sulfated polysaccharides can modulate the activity of factor XIIa in vitro depending on their structure and concentration (Schoenfeld et al., 2016), the best-known example being potentiation of kallikrein formation by OSCS (see Section V.H). Heparin does not share this property, but surprisingly it also fails to promote FXIIa inhibition by the serpin C1 inhibitor (Schoenfeld et al., 2016). Heparin does, however, potentiate the activity of the C1 inhibitor on complement factors C1s, as shown in a study in which a library of sulfated polysaccharides was screened for their C1-inhibitor modulating properties (Schoenfeld et al., 2016). Heparin and LMWH have also been found to inhibit all three arms of the complement system (classic pathway, lectin pathway, and alternative pathway) both directly and through enhancement of C1 inhibitor activity (Poppelaars et al., 2016).
Complement factor H (FH) is a large, extended protein made up of 20 globular domains linked in a relatively flexible way. FH regulates the progress of the complement system by binding to and inactivating complement factor 3b when bound to cell surface HS, thus protecting host cells from attack. A recent study has confirmed the existence of two heparin/HS binding sites on FH, located in domains 6 to 7 and 19 to 20 and in addition supports the suggestion that a third site in domains 11 to 13 binds heparin with lower affinity (Haque et al., 2020). X-ray scattering and ultracentrifugation analysis have indicated bivalent interaction between heparin/HS and fH involving cooperative binding to two distinct sites in FH, so that malfunctioning of either site could lead to loss of affinity between FH and C3b (Perkins et al., 2014). Mutations in each of the heparin/HS binding sites lead to tissue-specific effects; damage to the domains 6 to 8 site increases susceptibility to age-related macular degeneration and the domain 19 to 20 site is similarly linked to atypical hemolytic uremic syndrome in the kidney (Clark et al., 2013; Parente et al., 2017).
H. Neutrophil Proteins
The recruitment of neutrophils to a site of infection is an early event in the innate immune response, leading to the release of cationic proteases aimed at killing the pathogen. In excessive or prolonged inflammation [e.g., chronic obstructive pulmonary disease (COPD) resulting from smoking], host tissues can also be damaged by a range of neutrophil derived mediators such as elastase and various metalloproteinases. Neutrophils can also give rise to neutrophil extracellular traps (NETs) in which cell compartments not normally exposed to the extracellular space such as DNA and histones form a network to trap pathogens and hold them within range of the antibacterial proteases (Li and Tablin, 2018; Niu et al., 2021).
Sevuparin, a LMWH with low anticoagulant activity, has been found to prevent neutrophil-induced lung plasma leakage in a mouse model of systemic streptococcal-induced inflammation; a proteomics approach has identified a number of sevuparin-binding proteins in neutrophil secretions, including histone H4 and serprocidin proteases such as cathepsin G, neutrophil elastase, and the inactive elastase known simply as heparin binding protein (Rasmuson et al., 2019). Sevuparin does not reduce degranulation or adhesion of neutrophils but neutralizes the cationic proteins they release that cause vascular hyperpermeability (Rasmuson et al., 2019). Heparin binding protein concentration in plasma rises quickly in sepsis, before the onset of hypotension or organ dysfunction, and for that reason is a useful marker for the diagnosis of sepsis (Fisher and Linder, 2017; Yang et al., 2019).
The interaction of heparin and its mimetics with the highly proinflammatory enzyme neutrophil elastase has been the subject of much study, as heparin is a potent elastase-neutralizing agent with the disadvantage (in this context) of high anticoagulant activity. The potential of heparin and its mimetics in the treatment of cystic fibrosis has been reviewed (Voynow et al., 2020), and previous studies have suggested that heparin may be of benefit in treating COPD and emphysema by virtue of the ability to inhibit the tissue-damaging effects of elastase (Lafuma et al., 1991). Modified heparins, among them a 2-O-, 3-O-desulfated heparin (ODSH) preparation, neutralize elastase in a purified system but not in the presence of cystic fibrosis sputum; computational chemistry indicates that ODSH and DNA compete for binding to elastase and indeed in the presence of the DNA degrading enzyme dornase-a, the anti-elastase activity of ODSH is restored (Kummarapurugu et al., 2018).
Characterization of the interaction by native mass spectrometry and molecular dynamics calculations reveal that heparin and neutrophil elastase can form complexes in which the stoichiometry is not simply 1:1. This is likely because there is a sufficiently large cationic surface area on the protein to accommodate more than one heparin decamer, and heparin-stabilized dimers have also been identified. These observations have been made in spite of heparin’s sequence heterogeneity and the many possible glycoforms of elastase (Niu et al., 2021).
Fucoidan and xyloglucan partial depolymerization and fractionation has yielded preparations of these plant-derived heparin mimetics that neutralize elastase activity with similar effectiveness to that of heparin itself but with much lower anticoagulant activity (Lahrsen et al., 2018, 2019). A sulfonated heparin mimetic elastase inhibitor with a noncarbohydrate backbone has also been described. As for heparin, more than one binding mode is predicted by molecular modeling of the heparin-elastase complex (Al-Horani et al., 2021). Systematic screening methods for the identification of optimal nonsaccharide heparin mimetics specifically targeted at neutrophil elastase have also been described (Morla et al., 2019).
The roles of histones and heparin in sepsis have been reviewed (Zhang and Li, 2022), pointing out that heparin can reduce histone-mediated cytotoxicity, inflammation, and platelet binding, while calling for further basic structural work in this field. In vitro studies (using whole blood) have established that both heparin itself, and partially desulfated heparin derivatives, can reduce histone-induced markers for inflammation such as interleukin-6 (IL-6), interleukin-8 (IL-8), tissue factor, and complement factor 3a (Hogwood et al., 2020). Extracellular histones and AT compete for binding to vascular GAGs, thus modulating both coagulation and inflammation processes (Biswas et al., 2021).
I. Heparin and Bacteria
1. Bacterial Adhesins
The use of cell surface GAGs as attachment factors for bacterial adhesins is commonplace among many species of bacteria, whether Gram +ve or -ve, pathogenic, or harmless (García et al., 2016; Zimmermann et al., 2016; Lin et al., 2017; Rajas et al., 2017; Martín et al., 2019; Shi et al., 2021). Panels of bacterial species have been screened for GAG-mediated adherence to both lung-derived (Rajas et al., 2017) and corneal-derived cells (García et al., 2016), demonstrating the involvement of both CS and HS, particularly HS carried by the proteoglycan syndecan (Zimmermann et al., 2016). Bacterial adherence to host proteins and glycans may be part of the process of host cell invasion by the bacterium and may also hinder its mechanical clearance from host tissue (Paulsson and Riesbeck, 2018). The ability of GAGs and GAG mimetics to interfere with bacterial adhesion, and the consequent therapeutic potential for treatment of infectious disease has been discussed for the tick-borne Lyme disease causing spirochaete Borrelia burgdorferi (Lin et al., 2017). HS competitors such as N-acetyl heparin and glycol-split heparin have also been shown to have promise in the treatment of pseudomonal infections of the lung (Lorè et al., 2018). Some species of bacteria (and viruses) in the circulation can be reduced by extracorporeal blood filters based on immobilized heparin, and this approach has recently been reviewed (Seffer et al., 2021).
2. Mycobacterial Heparin-Binding Hemagglutinin
Besides several adhesins with host cell protein targets, mycobacteria including M. tuberculosis display a heparin-binding hemagglutinin (HBHA) on the outer side of the cell wall; its interaction with cell surface HS is the basis for its adherence to epithelial cells and its role in extrapulmonary dissemination of M. tuberculosis (Squeglia et al., 2018). The complex of HBHA with HS has been studied by NMR, unusually making use of a synthetic 13C, 15N labeled HS octasaccharide and demonstrating that HS binds the C-terminal domain of HBHA by both charge-based and hydrophobic interactions (Huang et al., 2017). The interaction has also been explored by atomic force microscopy and single-molecule force microscopy, as summarized in a recent review (Viljoen et al., 2021); single-molecule force microscopy using heparinized or HBHA functionalized probe tips can map the localization of the opposite partner on the cell or mycobacterial surface.
The potential of HBHA in the diagnosis, prevention, and treatment of mycobacterial infections has been pointed out. However, difficulties in production of recombinant HBHA with correct post-translational modifications (methylated lysines in the C-terminal region) may be hampering its further exploitation (Pu et al., 2020).
2. Bacterial Degradation of Heparin/HS
The major energy source for gut bacteria consists of a mixture of dietary carbohydrate and host glycans. Some species are particularly well equipped to use specific polysaccharides, with enzymes of polysaccharide degradation coded for by clustered genes in a polysaccharide utilization locus (PUL) (Brown and Koropatkin, 2021). The GAGs of the intestinal mucosa are no exception, and PULs for both heparin/HS and CS/HA have been identified for example in Lactobacillus (Kawai et al., 2018) and Bacteroidetes (Brown and Koropatkin, 2021). Transporter systems have been identified that internalize the GAG polysaccharide whole prior to degradation (Oiki et al., 2017). The enzymes subsequently involved in GAG degradation include sulfatases, glycosyl hydrolases, and the lyases that have proved useful for the manufacture of low molecular weight fractions of heparin and for use in the exhaustive digestion and disaccharide analysis of GAGs (see Section III). The mechanism and counter-ion dependence of one of these lyases, heparinase 1 from F. heparinum, has been explored (Córdula et al., 2014), and a new class of heparin lyases with a reducing-end exolytic mode of action has recently been described and characterized (Zhang et al., 2021).
A study of the enzymes of the heparin-degrading PUL of Bacteroides thetaiotaomicron concluded that the backbone of heparin/HS is degraded before the action of the sulfatases (Cartmell et al., 2017) and that the principal source of the polysaccharide is host HS (rather than from a dietary source). Presumably any released intestinal heparin is included in this, though most mucosal heparin is stored in mast cell granules until these cells are triggered to degranulate.
The consumption of intestinal mucosal glycans by bacteria can have consequences for host health, and if excessive can trigger colitis (Brown and Koropatkin, 2021).
J. Heparin and Viruses
Cell-surface HS acts as an attachment receptor for a wide range of viruses, including some very significant human pathogens for which there may be limited therapeutic options (Kim et al., 2017b; Tamhankar et al., 2018; McAllister et al., 2020). Host HS/heparin binds to either viral envelope proteins or directly to the capsid proteins of nonenveloped viruses (Agelidis and Shukla, 2020; Huang et al., 2014; Kim et al., 2017b). Viruses that bind to HS can be purified efficiently using heparin affinity chromatography (Du et al., 2017; Liu and Moon, 2016; Auricchio et al., 2020b; Pereira Aguilar et al., 2020).
The following survey of recent studies of heparin interactions with viral proteins shows a variety of different modes of interaction and in addition brings to the foreground a few common themes relevant to the potential therapeutic use of heparin to interfere in virus-host cell attachment. Implementation of heparin-based antiviral treatments is appealing, but there are obstacles such as for some species, the rapidity with which mutations can modulate HS binding in adaptation to the environment (Tee et al., 2019), and for others the reported enhancement of infection in response to exogenous heparin (Kim et al., 2019).
The use by a virus of HS as a viral attachment factor can also be a consequence of adaptation to cell culture conditions, so reading too much significance into results from such culture-adapted strains should be avoided (Cagno et al., 2019). However, it is clearly the case that some human viruses can use cell surface HS, as well illustrated by an otherwise extinct human endogenous retrovirus K, that has been locked for some considerable time into the human genome (Robinson-McCarthy et al., 2018). A vesicular stomatitis virus encoding human endogenous retrovirus K envelope protein as its sole attachment and fusion protein requires HS for viral attachment (Fig. 3). There are, however, a wide range of other virus families that interact with HS and/or heparin:

HS as an attachment factor in viral infections and potential use of heparin as a competitive decoy therapy. (A) Proposed mechanism of HS as sole attachment factor and receptor for human endogenous retrovirus-K (HERV-K). HERV-K binds HS on the cell surface to attach to the cell, and the virus is taken up by endocytosis (Robinson-McCarthy et al., 2018). (B) Proposed mechanism of attachment and receptor-mediated uptake of SARS-CoV-2 virus: spike protein of SARS-CoV-2 binds to cell surface HS, which promotes its interaction with high-affinity receptor ACE2. This receptor is activated by transmembrane serine protease 2 (TMPRSS2) leading to viral uptake by the cell (Clausen et al., 2020). (C) Exogenous heparin (or heparin mimetic) binds to exposed viral protein (SARS-CoV-2 spike protein or HERV-K envelope in this example) in competition with cell surface HS, reducing the ability of the virus to attach to and enter the cell.
1. Papovaviridae
Following earlier reports of several distinct heparin binding sites on the capsid protein L1 of human papillomavirus (Dasgupta et al., 2011; Richards et al., 2013), a recent cryo-electron microscopy study has identified only a single heparin binding site, organized so that the polysaccharide chain encircles the fivefold symmetry axis of the capsid (Guan et al., 2017).
2. Parvoviridae
Adeno-associated viruses (AAVs) are dependent on helper adenovirus to complete their lifecycle but are not known to cause any human diseases. However, recombinant AAVs are used as vectors in gene therapy, the viral genes in their capsids being replaced by therapeutic genes (Wang et al., 2019a). The affinity of strains such as AAV-2 and rAAV-DJ for heparin allows simple chromatographic purification using a heparin affinity column (Liu and Moon, 2016; Auricchio et al., 2020). Vector design can include modulation of affinity for HS (Boye et al., 2016; Gorbatyuk et al., 2019).
High-resolution cryo-electron microscopy of the complex between rAAV-DJ and fondaparinux has identified a heparin binding site near the threefold symmetry axis of the capsid (Xie et al., 2017), in agreement with earlier, low-resolution studies of AAV-2 with full-length heparin (O'Donnell et al., 2009) and rAAV-DJ with sucrose octasulfate, a highly sulfated disaccharide (Xie et al., 2013).
In their review of parvovirus glycan interactions, Huang and coauthors compare HS binding sites of AAV2, 3AAV3B, AAV6, and AAV13, indicating that although they are all in the same area, the specific amino acid residues involved are only partly conserved (Huang et al., 2014). Although fondaparinux binds weakly to each single binding site, the complete capsid contains 60 such sites so that cooperative binding can increase the effective affinity by several orders of magnitude.
3. Picornaviridae
Heparin affinity chromatography has also been applied to the recovery and purification of foot-and-mouth disease virus (FMDV) from cell culture (Du et al., 2017). Though the high-affinity host cell surface receptors for FMDV are integrins (Kotecha et al., 2017), tissue culture adapted strains can acquire the ability to bind HS and gain entry to the cells by caveola-mediated endocytosis without requiring a high-affinity protein receptor (O'Donnell et al., 2008).
The enterovirus EV-D68 can use either sialic acid or HS as a cell surface receptor, though as for FMDV it may be the case that HS binding is associated with culture-adapted strains. For this particular strain, the use of HS as an attachment factor changes the dependence of the virus on protein receptors to gain entry to the cell (Baggen et al., 2019).
The enterovirus A71 causes hand, foot, and mouth disease and uses HS as an attachment receptor. The HS/heparin binding sites involve basic residues of the VP1 protein, near the fivefold axis. Mutations that abolish heparin binding (such as K242A, K244A) can be compensated for by mutations elsewhere (T100K, E98A) that restore heparin binding capacity (Tan et al., 2017). A later study of the same strain used systematic mutation of heparin binding determinant residues to demonstrate that mutants with little affinity for heparin showed increased neurovirulence in mice (Tee et al., 2019). It may be that HS-binding viruses are more readily cleared from a living host organism, encountering high concentrations of heparin binding proteins in sites away from the viruses target cell type.
The acquisition of HS binding capacity in cell-culture adapted viruses, and the relative ease with which mutations in the viral capsid can modulate HS affinity, indicate that the interaction is unlikely to be highly selective in terms of either conserved amino acid residues or specific structures within HS. In addition, the choice of HS binding as a target for antiviral therapeutics may prove to be less effective than might be hoped in some cases.
4. Circoviridae
A porcine circovirus (PCV-2) Cryo-EM study of the PCV-2, a very small virus of great veterinary importance, has revealed that there are five sites per PCV-2 capsid subunit capable of heparin binding, such that a PCV-2 virus-like particle can possess a maximum of 60 sites occupied by heparin (Dhindwal et al., 2019).
5. Poxviridae
The poxvirus vaccinia virus binds HS and has both enveloped virus and nonenveloped mature virus forms. The infectivity of both forms is inhibited by heparin and its mimetics (Khanna et al., 2017). The H3 protein, located on the surface of the mature virus but not exposed in the enveloped virus, has been found to bind heparin (Singh et al., 2016), as has the envelope protein A27L (Hsiao et al., 1998). The exact contribution made by HS to the binding and internalization of vaccinia virus remains unclear.
6. Togaviridae
The chikungunya virus (CHIKV) is a mosquito-transmitted pathogen that causes debilitating disease. CHIKV is known to use cell-surface GAGs as attachment factors, and glycan microarray analyses suggest that CHIKV most efficiently binds longer, sulfated GAGs, with a preference for HS and heparin (McAllister et al., 2020). A heparin binding sequence motif (XBXXBX) on the envelope protein E2 of CHIKV has been defined using theoretical and experimental methods; this motif is common to a number of related alphaviruses (Sahoo and Chowdary, 2019).
7. Herpesviridae
Recent surveys of herpesvirus surface glycoprotein ligands and their cell surface receptors (Madavaraju et al., 2021; Huang et al., 2022) distinguish between binding to cell-surface HS as a simple attachment factor, and subsequent specific interaction between glycoprotein D of herpesvirus-1 and 3-O-sulfated HS as part of the fusion process. However, an array of immobilized synthetic HS hexasaccharides including several 3-O-sulfated sequences was not able to demonstrate high affinity between glycoprotein D and any 3-O-S oligosaccharide (Chopra et al., 2021).
It is interesting to note that chronic post-herpetic neuralgia after infection with human herpesvirus 3 is associated with a single-nucleotide polymorphism of the heparan sulfate 3-O-sulfotransferase 4 gene (Nishizawa et al., 2021) and enhances virus-mediated fusogenic activity (Ohka et al., 2021).
8. Flaviviridae
Like CHIKV, the flaviviruses, dengue virus (DENV) and Zika virus (ZIKV), pose threats to human health as their geographical ranges expand into new areas of the world. All the pathogenic flaviviruses [including those of veterinary importance such as classic swine fever (Cheng et al., 2019) and duck tembusu virus (Wu et al., 2019)] bind to cell surface GAGs and several heparin mimetics have been examined for their potential as antiviral agents (Kim et al., 2017b).
Host cell dependencies of DENV and ZIKV have been explored by orthologous functional genomic screening, identifying among others the HS biosynthetic enzymes NDST and exostosin-1 (Savidis et al., 2016). It is interesting to note that though added heparin reduces DENV replication in Vero cells, ZIKV replication was promoted (Kim et al., 2019). However, though heparin does not significantly reduce replication of ZIKV in human neural progenitor cells, it is capable of preventing ZIKV-induced necrosis in this cell type (Ghezzi et al., 2017).
9. Rhabdoviridae
Heparin can inhibit rabies virus infection of cells, both by competing with the virus for its protein receptor neural cell adhesion molecule and also by direct interaction with the virus envelope, in competition with cell surface HS attachment factor (Sasaki et al., 2018).
10. Filoviridae
A study of ebolavirus infection of Caco-2 cells, a polarized cell type, indicated that the virus binds preferentially to the basolateral side of the cell layer. This basolateral infection bias may be dependent on polarized distribution of cell surface HS (Tamhankar et al., 2018).
11. Arteriviridae
Equine arteritis virus infection of equine endothelial cells was reduced by 90% in the presence of heparin (Lu et al., 2016). The viral binding site was localized within an amino acid sequence near the C-terminus of the E minor envelope protein by site-directed mutagenesis. A double arginine to glycine mutant eliminated the interaction but did not completely abolish infection.
12. Retroviridae
Heparin/HS interactions with HIV are not restricted to the well-documented binding to the envelope glycoprotein gp120 (Mulloy et al., 2016). The HIV matrix protein p17 is released by the virus and acts in the manner of a cytokine; it binds to heparin/HS through a sequence of basic amino acids near the N-terminus (Caccuri et al., 2016). HS-induced modulation of p17 oligomerization may be instrumental in p17-induced lymphoid dysregulation during AIDS (Bugatti et al., 2019).
13. Hepadnaviridae
The effect of heparin on viral infection is not necessarily negative. Heparin at relatively low concentration (1-5 µg/mL) can enhance HepB infection of hepatocytes, whereas heparin at higher concentrations (40 µg/mL and higher) inhibited infection (Choijilsuren et al., 2017).
14. Coronaviridae
Coronaviruses, such as the human pathogens SARS-CoV and SARS-CoV-2, are enveloped viruses with a surface-exposed spike protein that mediates cell attachment through its S1 subunit and cell entry through its S2 subunit. Both SARS-CoV and SARS-CoV-2 require protease cleavage between the two subunits for successful internalization via the protein receptor angiotensin converting enzyme-2 (ACE-2) (Chu et al., 2021). SARS-CoV and SARS-CoV-2 spike proteins also bind to heparin/HS (Clausen et al., 2020; Kim et al., 2020), and SARS-CoV-2 requires HS as an attachment factor (Clausen et al., 2020; Zhang et al., 2020b; Chu et al., 2021), enhancing the interaction between spike protein and ACE-2 (Clausen et al., 2020). The heparin binding sites of SARS-CoV-2 spike protein have been located near the ACE-2 binding site in the receptor binding domain (Clausen et al., 2020; Mycroft-West et al., 2020b) at the S1-S2 cleavage sequence PRRARS (Kim et al., 2020) and/or in the N-terminal domain (Schuurs et al., 2021). The preference of the protein for long-chain heparin over shorter oligomers (Kim et al., 2020) indicates that more than one of these binding sites may be involved in the interaction. Two recent theoretical studies both identify potential paths along the spike protein surface that could accommodate long-chain heparin or HS linking two heparin binding sites (Schuurs et al., 2021; Paiardi et al., 2022). Both studies model the spike protein with intact N-glycosylation. Though such studies imply that interference with proteolytic activation of the spike protein or indirect, allosteric hindering of ACE2 binding are both possible mechanisms by which heparin might inhibit infectivity (Bugatti et al., 2019), experimental evidence indicates that a probable mechanism is simple competition with cell surface HS for the virus (Liu et al., 2021) (Fig. 3). The synthetic heparin mimetic pixatimod has been shown to inhibit SARS-CoV-2 spike protein to ACE-2 directly; this synthetic compound is made up of a sulfated oligosaccharide and a lipid tail (Guimond et al., 2022).
Heparin also inhibits the infection of cells in culture by SARS-CoV-2 (Mycroft-West et al., 2020b; Zhang et al., 2020b); the potential exploitation of this property for therapeutic application has been pointed out by numerous groups, including those cited here (Cheng et al., 2019; Clausen et al., 2020; Conzelmann et al., 2020; Kim et al., 2020; Liu et al., 2020; Mycroft-West et al., 2020b; Yang et al., 2020; Tree et al., 2021). Heparin mimetics such as synthetic sulfated fucan oligosaccharides (Koike et al., 2021), pentosan polysulfate (Ennemoser et al., 2021; Zhang et al., 2022), and a sulfated rhamnan (Song et al., 2021) may also have potential as anti-COVID agents as they bind to the spike protein and neutralize viral infectivity by SARs-CoV-2.
V. Mechanism of Anticoagulant Action
A. Overview: Via Potentiation of Endogenous Coagulation Inhibitors
Heparin, it should be noted, is not anticoagulant itself but rather potentiates the mechanism of action of a variety of endogenous-clotting cascade inhibitors, thereby maintaining the fluidity of blood. Heparin also possesses an antithrombotic effect, which can be considered an interaction with the cellular components of the coagulation system. The process of coagulation can be split into two steps: (1) primary, which involves cellular components, and (2) secondary, which involves the soluble clotting factors (Versteeg et al., 2013) depicted in Fig. 4. The in vivo process of thrombosis can be described broadly as follows: surface damage exposes the endothelium and/or subendothelium to blood; thrombogenic cell surfaces activate platelets leading to their adherence; platelets localize activation of the coagulation cascade (Hoffman and Monroe, 2001); and activation of the coagulation cascade leads to formation of insoluble fibrin around platelets. Given that heparin potentiates an array of coagulation inhibitors, a brief description of the coagulation system is provided next.

Heparin potentiation of coagulation cascade inhibitors. Triggering of the coagulation cascade occurs through tissue factor and damage surface exposure. The inhibitory action of heparin is through potentiation of a range of inhibitors with interactions linking to the kinin and complement pathways. Coagulation factors: FXII, factor XII; red, activated factor; blue, inhibitor.
The initiation of coagulation is typically through surface damage, due to trauma or injury, to the endothelial cell layer of the vasculature. The underlining subendothelium and extracellular matrix is highly thrombogenic, with fibroblasts expressing tissue factor (Mandal et al., 2006), which affects the coagulation cascade (see Fig. 4) and the matrix itself containing collagen, a potent activator of platelets (Roberts et al., 2004). Exposure of the subendothelium will lead to platelet adherence, first via transient interactions of platelet expressed glycoprotein Ia/IIa to collagen, which releases von Willebrand factor from platelets (Peyvandi et al., 2011). The released von Willebrand factor enhances platelet binding, allowing other glycoprotein interactions to occur, which leads to activation of platelets (Bryckaert et al., 2015). This activation alters localized calcium levels (a critical cofactor in the coagulation cascade), which enables the modification of platelet glycoprotein IIb/IIIa to bind with increased affinity to fibrinogen (Swieringa et al., 2018). Activation of platelets also leads to the exposure of negatively charged phospholipids, which provides a surface for activation of the coagulation cascade (Swieringa et al., 2018).
Vascular damage also leads to the exposure of tissue factor presenting cells, such as fibroblasts. Tissue factor will interact with the small amounts of FVIIa naturally present in blood and will initiate the coagulation cascade (see Fig. 3), primarily through activation of FX and some FIX (Hoffman, 2003). The small amounts of FXa will bind to Fva, released from platelets, forming the prothrombinase complex on tissue factor presenting cells and platelets. The small amounts of thrombin converted from prothrombin will feedback into the coagulation cascade, activating FV, VIII, and XI. These enzymes will localize on platelet surfaces with FXIa activating FIX; the formed FIXa will then bind to FVIIIa as the tenase complex converting FX to FXa at a higher rate than the TF/FVIIa complex (Versteeg et al., 2013). This increase in the level of FXa will then form larger amounts of the prothrombinase complex on the surface of platelets, greatly increasing levels of thrombin. The thrombin produced will result in fibrin formation and subsequently blood clots.
The different endogenous coagulation inhibitors—AT, HCII, TFPI, C1-esterase inhibitor, and protein C inhibitor (PCI)—act as a balance ensuring localization of the clotting response. Of these inhibitors the major inhibitor is the serine proteases inhibitor (serpin) AT, which targets many of the activated coagulation factors (Fig. 4). As described earlier, the interaction of heparin and AT requires a specific pentasaccharide sequence, whereas the other serpins require no highly defined sequence (Huntington, 2011). The nonserpin inhibitor, TFPI, is also potentiated by heparin (Ellery and Adams, 2014), and the interactions of heparin with the different coagulation inhibitors is briefly summarized next.
B. Potentiation of Antithrombin
In 1982, the interaction mechanism between heparin and AT was partially elucidated by Björk and Lindahl (Björk and Lindahl, 1982), with more refined descriptions following advances in analytical techniques (Olson et al., 2010; Huntington, 2011). A pentasaccharide sequence was determined to be the minimal antithrombin-binding structure within heparin and HS (Choay et al., 1983). In the in vivo setting, AT interacts with HS in the cell-surface glycocalyx (Chappell et al., 2009), although it is thought that this may exert an anti-inflammatory rather than an anticoagulant effect (Shworak et al., 2010). It should be noted that a longer octasaccharide sequence, incorporating the pentasaccharide, binds with higher affinity to AT (Lindahl et al., 1984) and that heparin without the pentasaccharide can still potentiate AT (Streusand et al., 1995).
Native AT is a slow inhibitor as the reactive center loop (RCL) is partially folded within a beta-sheet structure (Huntington, 2003), thus limiting access by the serine protease target. The interaction of the pentasaccharide with AT releases the RCL by a two-step process; the initial binding involves the first three monosaccharides and induces conformational changes in AT, then the interaction is stabilized with the last two monosaccharides (Desai et al., 1998). Structural changes are transmitted through AT, with the expelled RCL-enabling increased interaction with the target proteases (Huntington, 2003; Izaguirre et al., 2021). The protease partially cleaves the RCL but is then caught in a stable covalent intermediate state, which results in “entrapment” within AT, thereby inactivating the enzyme (Huntington, 2011). Heparin is then released to catalyze further interactions (Carlström et al., 1977; Huntington, 2006). For each of the target proteases there are some different requirements of heparin and AT.
1. Factor Xa Inhibition
In addition to expulsion of the RCL in AT following the binding of heparin, an exosite that binds to FXa is also exposed (Izaguirre et al., 2014). This protein-protein interaction provides the specificity of AT to FXa (Gettins and Olson, 2009). Therefore, the pentasaccharide is sufficient for potentiation of AT inhibition of FXa, and this was translated to the development of the synthetic oligosaccharide fondaparinux (Choay et al., 1983; and see Section II.B). It should be noted (Gray et al., 2012) that, in the presence of calcium, longer heparin chains bind both AT and FXa in a template effect, which further enhances inhibition (Rezaie, 1998). Therefore, a calcium-based molecular weight dependent inhibition of FXa exists (Lin et al., 2001) and may have some relevance in vivo (Barrowcliffe and Le Shirley, 1989).
2. Thrombin (Factor IIa) Inhibition
The inhibition of thrombin (FIIa) is through a template mechanism in which thrombin interacts with the same heparin molecule that is bound to AT. There is a minimum length requirement of 13 additional saccharides at the nonreducing end of the pentasaccharide for this interaction (Hoylaerts et al., 1984). The thrombin-heparin interaction is rather nonspecific; the monosaccharides involved requiring merely a negative charge to interact with the thrombin exosite II (Johnson et al., 2010; Mosier et al., 2012). The 18 saccharides required for thrombin-AT potentiation equates to 5400 daltons in weight and explains some of the differences in anti-Xa and anti-IIa activity of low molecular weight heparins (Gray et al., 2012).
3. Factor IXa Inhibition
The interaction of FIXa with AT is similar to FXa as FIXa interacts with the same exosite on AT, exposed by binding to heparin (Huntington, 2006). Refined structural analysis has indicated that FIXa/AT association on a single heparin molecule is also required (Johnson et al., 2010). As with the interaction of heparin-antithrombin-factor Xa, the presence of calcium enhances affinity for longer heparin chains thereby increasing inhibition (Wiebe et al., 2003) indicating a template mechanism similar to heparin-antithrombin-thrombin.
4. Factor XIa, Factor XIIa, and Kallikrein Inhibition
Structural analysis of the interaction between AT and the “contact” factors (FXIa, FXIIa, and kallikrein) is limited compared with the main targets for AT, FXa, and thrombin. Heparin plays a dual role with kallikrein, where it has been shown to directly enhance kallikrein action (about eightfold) on converting FXII to FXIIa but also marginally potentiates AT inhibition (about threefold) of kallikrein (Gozzo et al., 2006). However, these activities are likely marginal contributors in coagulation. The inhibition of both FXI and FXIIa by heparin-antithrombin is via a bridging mechanism whereby longer chains show an increase in potentiation (Olson et al., 2004).
5. Factor VIIa Inhibition
Antithrombin, in the presence of calcium, can inhibit FVIIa and the tissue factor/FVIIa complex, although this is very weak (Olson et al., 2004; Martínez-Martínez et al., 2011). Interaction with heparin enhances this inhibitory action, with a binding site for heparin determined on FVIIa (Martínez-Martínez et al., 2011) suggesting a similar template mechanism as for thrombin and FIXa.
C. Potentiation of Heparin Cofactor II
The thrombin-specific serpin, HCII, is present in plasma at similar levels to AT, although its contribution in prevention of clotting is considered to be minimal relative to AT (Tollefsen and Blank, 1981). Deficiency of HCII has no effect on coagulation, but there is an increase in arterial thrombus risk following endothelium damage (He et al., 2002). In vivo HCII is potentiated by dermatan sulfate (Tovar et al., 2005) with some specificity in this interaction; a hexasaccharide containing 2-O-sulfated iduronic acid and 4-O-sulfated N-acetyl-galactosamine (Maimone and Tollefsen, 1990). Interaction with heparin requires no specific sequence (Huntington, 2006) with other polyanions also able to bind and potentiate HCII (Colwell et al., 1999; Bano et al., 2022). Interaction with heparin induces conformational changes in HCII, similar to that of AT leading to exposure of the RCL (O’Keeffe et al., 2004). Additionally, heparin (and dermatan sulfate) binding releases a high affinity thrombin-binding domain in the N-terminal tail of HCII accelerating inhibition of thrombin (Baglin et al., 2002). In a similar manner to AT, heparin is released following HCII-thrombin binding (Huntington, 2006).
D. Potentiation of Protein C Inhibitor
Protein C inhibitor regulates the activity of activated protein C (APC), which is an anticoagulant through inactivation of FVa and FVIIa (Comp et al., 1982). Therefore, PCI by inhibiting an inhibitor of coagulation actually acts in a manner that promotes coagulation. When compared with other serpins, PCI has a flexible RCL close to where the heparin binding region, helix H, is located (Li and Huntington, 2008). However, while binding to heparin potentiates PCI inhibition of APC, high concentrations of heparin (>2IU/ml) are needed (Pratt and Church, 1992); therefore the physiologic role of PCI-heparin is unclear given its wide distribution in tissues (Wahlmüller et al., 2017).
PCI has been found to also inhibit thrombin, FXa and FXIa, with calcium-dependent heparin potentiation of this activity (Sun et al., 2009; Van Walderveen et al., 2010). The inhibition of coagulation enzymes by PCI is dependent on the size and concentration of heparin, indicating that both PCI and the protease need to bind to the same heparin molecule. The minimal length of heparin needed to enhance the APC inhibitory activity of PCI is 7 saccharides (Aznar et al., 1996) with the rate of inhibition of APC (also FXa) increasing with saccharide length (Pratt and Church, 1992).
E. Interaction with C-1-Esterase Inhibitor
C1 esterase inhibitor, a serpin that inhibits intrinsic pathway proteases (kallikrein, FXIIa, FXIa), is also involved in the regulation of complement activation (Davis et al., 2010). A deficiency in C1inh results in hereditary angioedema through continuous overactivity of the contact system (Konings et al., 2013). Heparin marginally potentiates the ability of C1inh to inhibit kallikrein (Gozzo et al., 2003) but paradoxically neutralizes C1inh inhibition of FXIIa (Pixley et al., 1987). The interaction of heparin with C1Inh is more effective on the complement system, via potentiation of its inhibition of C1s (Poppelaars et al., 2016) (see Section IV.G). Resolution of the crystal structure of C1inh indicates a novel “sandwich” mechanism as the mode of action (Beinrohr et al., 2007), which is different from the action of AT. Several key sites for polyanion interaction have been identified (Hor et al., 2020), which highlight the potential for sulfated heparin-like material such as OSCS to enhance C1inh activity (Poppelaars et al., 2016).
F. Interaction with Tissue Factor Pathway Inhibitor
Unlike the inhibitors discussed previously, TFPI is not a serpin (Mast, 2016; Sandset et al., 1988) but rather a polypeptide with several domains involved in heparin binding. The inhibitor activity of TPFI is twofold—first, injection of heparin releases HS bound TFPI from the endothelium into the bloodstream to act as an inhibitor of the tissue factor pathway, and, second, binding of heparin potentiates its inhibitory activity on FXa; TFPI can inhibit free FXa and FXa in the FVIIa/TF/FXa complex (Broze et al., 1988; Peraramelli et al., 2016; Xu et al., 2002). While in vivo the concentration of TFPI is low (2.5 nM), it acts as a major FXa inhibitor (Adams, 2012) preventing the progression of the coagulation cascade. Heparin interacts with the C-terminal domain in TFPI (Ye et al., 1998), but structural features in heparin are not clear as LMWH has a reduced ability to release TFPI and potentiate FXa inhibition. This indicates that there is either a molecular weight dependency to bind/release TFPI (Ma et al., 2007) or that heparin total sulfate content and charge localization is important (Valentin et al., 1994).
G. Antithrombotic Nature of Heparin
The anticoagulant activity of heparin in vitro is through potentiation of coagulation inhibitors, but in vivo reduction of clot formation is not exclusively through actions on the clotting factors but also due to an action on platelets, which form thrombi (Periayah et al., 2017). The anticoagulant and antithrombotic activities of heparin are not mutually inclusive as hemostasis is far more complex than the clotting cascade and involves cellular blood elements (Versteeg et al., 2013) as shown in Fig. 4. Heparin is known to interact with a number of cell surface binding proteins involved in hemostasis as described previously (Mulloy et al., 2016). Furthermore, heparin fractionated to possess no anticoagulant activity can still reduce thrombus formation in a thrombogenic challenge model (Gray et al., 1994). Similarly, in an experimental venous and arterial model of thrombosis, a HS structurally very similar to heparin was found to be efficacious (Nader et al., 2004), and heparin has been known to possess antithrombotic activity in vivo for many years (Barrett et al., 1984).
The challenge when using heparin clinically as an anticoagulant is to ensure that the correct dosing regimen is applied with the aid of appropriate in vitro assays (Dougherty et al., 1992) while recognizing that they may not correspond well to antithrombotic activity. For example, in a deep vein thrombosis model that used in vitro assays for dose adjustment, UFH was more effective than LWMH at limiting the formation of thrombi (Morris et al., 2000). The heterogenous nature of heparin and the ability to bind and interact with a wide range of proteins (Capila and Linhardt, 2002) may be the reason for this superior efficacy. One interaction of relevance is the release of cell surface TFPI by heparin (Sandset et al., 1988), where TFPI has multiple anticoagulant activities (Mast, 2016) (Section 5.6). Another plausible reason may be that the nonanticoagulant portion of heparin interacts with other heparin binding proteins “freeing” the anticoagulant portion to interact with AT (Merton et al., 1984), effectively meaning that one part of the heparin molecule potentiates the action of another part of heparin (Barrowcliffe et al., 1984).
The discovery of NETosis (Brinkmann et al., 2004) and then subsequent observations of the close link between inflammatory and thrombotic responses (Stark and Massberg, 2021) has led to another possible mechanism by which heparin can be antithrombotic. The clear interplay of neutrophils, platelets, and endothelial cells in inflammation and thrombosis (Iba and Levy, 2018; Rayes and Jenne, 2021), along with the observed inhibition/interaction of heparin with a number of neutrophil proteins (see Section IV.H) demonstrates a further role for how heparin is antithrombotic by disruption of this thromboinflammatory interaction. Furthermore, heparin is able to neutralize extracellular histones (Wang et al., 2015), which have both procoagulant and proinflammatory effects (Ammollo et al., 2016; Gould et al., 2016) thereby further limiting the effect of NETosis. Heparin has also been found to disrupt histone-mediated fibrin formation (Longstaff et al., 2016; Komorowicz et al., 2021), which will further reduce localized thrombosis. Importantly these inhibitory effects on thrombosis are also retained by modified nonanticoagulant heparins (Wildhagen et al., 2014; Hogwood et al., 2020; Sharma et al., 2022), drugs that have the potential to offer novel therapeutic uses as discussed below (see Sections VII and X).
H. Heparin-Like Materials and Their Anticoagulant Activity
As discussed previously (Section 3.2), the adverse effects observed when using heparin contaminated with OSCS resulted in rapid revisions of pharmacopeia monographs. An issue at the time of the contamination was the lack of selectivity in the plasma pharmacopeial potency assays (Kishimoto et al., 2008) and, as described earlier, will therefore incorporate all the interactions of heparin or heparin-like materials with plasma coagulation inhibitors. The contaminant, OSCS, which lacks the antithrombin-binding pentasaccharide, was considered to act through HCII (Fareed et al., 2008) with detailed activity-based analysis demonstrating how oversulfation influenced CS potentiation of HCII (Hogwood et al., 2018). This highlighted the role that HCII has as an anticoagulant and that sulfated heparin-like materials can possess antithrombin-independent anticoagulant activity and/or antithrombotic activity.
Anticoagulant activity that is not through AT is the primary mode of action for the clinical product danaparoid (Ibbotson and Perry, 2002), which is a mixture of HS, DS, and CS. Danaparoid with anticoagulant and antithrombotic activity is a treatment option in the event of heparin-induced thrombocytopenia (Nilius et al., 2021) (see Section VI.C). With interest in modified heparin and heparin-like materials it is critical to consider anticoagulant activity outside of the heparin potency assessment assays as described in the pharmacopeia monographs. As shown with LMWH, fractionation of heparin alters its anticoagulant profile, so that each LMWH has a unique ratio of anti-Xa to anti-IIa activity (Gray et al., 2008) and should not necessarily be considered clinically interchangeable. Furthermore, selective desulfation of heparin alters the anticoagulant profile but can retain the nonanticoagulant activity of interest (Hogwood et al., 2020). It is therefore prudent when considering modified heparins for new indications (see Section X) not to limit anticoagulant testing to antithrombin-based assays as described in the various monographs but to include a broader profile of anticoagulant tests.
I. Measurement of the Anticoagulant Activity of Heparin Preparations
Accurate measurement of the anticoagulant activity of heparin is important for labeling of therapeutic products and clinical monitoring of their use. UFHs and LMWHs are extracted from animal sources and are complex polydisperse molecules, and as such gravimetric mass units obtained using physicochemical methods (see Section III) do not provide adequate information on the anticoagulant action of these drugs. Similar to other biologicals, the measurement of anticoagulant activity requires comparison with a reference standard, in a bioassay; results are expressed as relative potency or relative activity to the standard. Both World Health Organization international and pharmacopoeial reference standards are available to assign potency in International Units to heparin products. The history and development of heparin and LMWH units, standardization landmarks and statistical considerations for bioassays have been discussed in detail elsewhere (Gray, 2012). Bioassays, using citrated plasma or purified reagents, are designed based on the ability of heparin to potentiate the inhibitory action of plasma coagulation factor inhibitors such as AT and HCII.
1. Plasma-based Assays
The plasma-based assays are global assays and measure the potentiation of the inhibitory effect of coagulation factor inhibitors by heparin on activated coagulation factors such as FXIa, FIXa, FXa, and FIIa (thrombin). The endpoint of these assays is clot formation, and, with increasing amounts of heparin, there is an increase in the prolongation of clotting times. A number of assays including APTT and protamine sulfate titration have been used for measurement of heparin, especially in clinical settings, and the final readout from these assays is influenced by the quality of the plasma in use; for example, concentrations of PF4 and the presence of other anticoagulants may vary between samples. Although commonly used for the clinical measurement of UFH, these plasma-based assays are seldom used for LMWHs.
APTT is used as a screening test for detection of clotting factor deficiency. It is highly sensitive to heparin and is currently the method of choice for clinical monitoring of UFH treatment. This method involves activation of plasma via the intrinsic pathway with a negatively charged activator (e.g., ellagic acid), in the presence of phospholipid, and the clotting time is recorded following the addition of calcium. Although the APTT is easily adapted to run on automated instruments, results are variable and highly dependent on the APTT reagent used. It is recommended that therapeutic APTT ranges should be determined locally against therapeutic heparin levels obtained using anti-Xa assay or protamine titration (Hirsh and Raschke, 2004; Baglin et al., 2006). Until recently, variations on this test, using sheep plasma instead of human plasma, were used by the EP and USP as the pharmacopoeial monograph methods for potency labeling of therapeutic UFH. These methods were revised following the contamination of heparin with OSCS and the current EP and USP monograph methods are based on the potentiation of the inhibitory action of AT on FXa and thrombin. The establishment of these new monograph assays may help detect any attempts to adulterate heparin preparations with contaminants in the future to prevent another clinical crisis like that seen with the use of heparin contaminated with OSCS.
The protamine sulfate titration assay has also been used for measurement of heparin in patient plasma samples and is based on the ability of protamine sulfate, a highly positively charged protein, to neutralize the anticoagulant activity of heparin (Refn and Vestergaard, 1954; Newall, 2013). The principle of the assay is based on the normalization of the heparin prolonged thrombin clotting times by protamine sulfate. However, this assay is not easily automated, and, since protamine can also act as an anticoagulant (Kresowik et al., 1988), addition of excess protamine can lead to an incorrect estimation of heparin potency. This assay is therefore not recommended for potency labeling of heparin products.
2. Purified System Assays
The purified reagent methods are the methods of choice for potency labeling of therapeutic heparin products. The current EP and USP potency assays for both UFH and LMWHs are based on the ability of heparin to potentiate the inhibition of thrombin (FIIa) or FXa by AT (US Pharmacopeial Convention, 2014; European Pharmacopeia, 2015) and are known as the anti-Xa or anti-IIa assay. These AT-dependent assays are highly specific for heparins as only heparin, LMWHs, and HS (and the synthetic pentasaccharide) are known to possess the essential pentasaccharide sequence that binds to AT (see Section II). These assays employ purified proteins (AT, FXa, and FIIa) and are carried out by incubation of the heparin/AT mixture with either FXa or FIIa for a specified length of time. The residual FIIa or FXa cleaves a chromogen from chromogenic substrates that are specific for FIIa or FXa. Color development is inversely proportional to the concentration of heparin.
Anti-Xa assays are also commercially available for monitoring LMWH treatment, and the source of AT may come from the patient’s own plasma or exogenous AT may be included in the kit to avoid low level or depletion of AT in the patient’s plasma, which may lead to an underestimation of heparin concentration.
VI. Clinical Use of Heparin as an Anticoagulant/Antithrombotic
A. Treatment and Prophylaxis of Venous Thromboembolism
As previously reviewed in Mulloy et al. (2016) and elsewhere (Bates et al., 2018; Anderson et al., 2019; Ortel et al., 2020; Lyman et al., 2021), heparins in the form of both UFH and LMWH remain central to the prophylaxis and the treatment of venous thromboembolism (VTE) across a range of clinical settings. LMWH treatment is a standard approach in the initial management of VTE, although UFH may be more suitable in selected patients, including those considered to be at a high risk of bleeding (Garcia et al., 2012; Cohen et al., 2014), or where renal function is significantly impaired (Cohen et al., 2014), due to the comparatively rapid cessation of anticoagulant effects upon withdrawal and the relative sensitivity to protamine reversal where rapid reversal is likely to be required (Garcia et al., 2012; Pai and Crowther, 2012). Generally, however, the more predictable pharmacokinetic profile of LMWHs, and the associated convenience of fixed-dosage regimens, makes these agents more attractive in terms of routine clinical use and LMWH can be used in patients with significant renal impairment or disease with appropriate dose adjustment in place (Leung and MacRae, 2019). Furthermore, regular monitoring of the effects of UFH, usually by APTT measurement (Marlar et al., 2017), is required, whereas routine monitoring of the effects of LMWH therapy, usually achieved by assays of anti-FXa activity, is less strictly necessary (Gray et al., 2008; Weitz and Weitz, 2010; Garcia et al., 2012; Babin et al., 2017). In situations where relatively protracted thromboprophylaxis is also required in an outpatient setting, these advantages become particularly prominent, along with the generally reduced propensity of LMWHs, compared with UFH, to cause side-effects including osteoporosis and thrombocytopaenia (Bates et al., 2012; Lussana et al., 2012). Current guidelines for the initial treatment of VTE in patients with cancer recommend the use of LMWH (Lyman et al., 2021), which are also the mainstay of VTE management in pregnancy, with weight-adjusted dosing and anti-FXa activity monitoring recommended to ensure adequate dosing where the risk of VTE remains high, and anticoagulation continued for at least 6 weeks post-partum (Brenner et al., 2021). In terms of monitoring, when recommended, the therapeutic effect of LMWH (including fondaparinux) is monitored through use of anti-FXa assays, with approaches such as thromboelastography used in special circumstances (Babin et al., 2017). However, it has been suggested that body weight and renal function should take precedence in guiding dosage adjustment beyond the need to monitor (Witt et al., 2018). Anti-FXa activity has additionally been suggested to be a plausible method for establishment of the therapeutic range of UFH, with potentially greater accuracy than the standard approach of APTT measurement (Baluwala et al., 2017), although the latter is the preferred assay in most clinical settings for the monitoring of UFH therapy and as a surrogate marker for estimation of heparin concentration (Marlar et al., 2017).
The key indications for heparins in the prophylaxis of VTE are in hospitalized medical and surgical patients, in cancer patients, in management of acute coronary syndromes, and in pregnancy where an enhanced risk of thrombosis has been established. In the latter setting, the inability of heparins to cross the placenta (Flessa et al., 1965; Forestier et al., 1984, 1987) and their established safety profile (Lepercq et al., 2001; Rodie et al., 2002; Greer and Nelson-Piercy, 2005; Kher et al., 2007) make these agents uniquely suitable. LMWH is generally accepted to present a safe means of prophylaxis in pregnancy, with bleeding risk similar to background levels (Lu et al., 2017) and is preferred to UFH for this purpose (Bates et al., 2018). In hospitalized patients considered to be at risk of VTE due to the presence of one or more risk factors, heparins are likely to be given unless the risk of hemorrhage outweighs the thrombotic risk. In this respect, both UFH and LMWH regimens are safe and effective in preventing VTE in acutely ill medical patients, and in surgical patients both pre- and post-discharge, following procedure, whereby an enhanced risk of thrombosis may persist for several weeks (Leclerc et al., 1998; White et al., 1998, 2003).
Critically ill patients present an additional challenge, in terms of both thromboprophylaxis and the management of active thrombosis, for heterogenous reasons. Significant renal impairment is common in intensive care unit (ICU)-admitted patients and is associated with increased risk of VTE but also with an increased risk of developing bleeding complications (Cook et al., 2008). In addition, the risk of VTE in critically ill patients shows significant individual variability dependent on underlying pathology and treatments and the consequences of even relatively minor pulmonary embolism (PE) in these patients can be severe on account of reduced cardiopulmonary function (McLeod and Geerts, 2011).
B. Heparin in Relation to Alternative Anticoagulants
The increasing availability of alternative, non-heparin anticoagulants, for the majority of the clinical indications for heparin, has in recent years seen a reduced reliance on heparin-based regimens across a range of key clinical settings, with DOACs that target either FIIa or FXa currently recommended for both the prophylaxis and the primary treatment of DVT and PE (Ortel et al., 2020). However, while clear advantages exist over heparin-based regimens with respect to the convenience of oral administration, advantages associated with safety and the relative lack of therapeutic-monitoring requirements apply more clearly in relation to vitamin K antagonist drugs than to heparin. For example, while DOACs may be preferable to heparins for thromboprophylaxis in orthopedic surgery patients (Anderson et al., 2019; Khatri et al., 2021), LMWH or UFH is recommended in hospitalized patients for major general surgical indications (Anderson et al., 2019). Similarly, thromboprophylaxis with LMWH in hospitalized medical patients was found to be associated with a reduced bleeding risk, in comparison with DOAC therapy, without inferior efficacy (Neumann et al., 2020), and the results of a 2019 meta-analysis support the use of thromboprophylaxis with LMWH (7–10 days) in medical patients, following discharge from hospital, in favor of an extended (>30 day) DOAC regimen on the basis of bleeding risk (Alshouimi et al., 2019). In the initial treatment of cancer-associated VTE, DOACs were reported in a 2019 meta-analysis to have greater efficacy but to be associated with an increased risk of major and clinically significant bleeding (Li et al., 2019a). However, DOACs may represent a more convenient alternative to the standard therapy with LMWH in prevention of VTE in patients with cancer, with only a modest increase in bleeding risk compared with LMWH, suggesting that an individualized approach may be taken with respect to bleeding risk and convenience, with respect to longer-term therapy in this setting (Brea et al., 2021).
Prior to the availability of licensed reversal agents for the DOACs, namely idarucizumab in respect of the FIIa-inhibitor dabigatran and andexanet alfa in the case of the FXa inhibitors rivaroxaban and apixaban, one perceived disadvantage of these agents over heparins and vitamin K antagonist was the absence of an “antidote,” such as protamine or phytomenadione, respectively. In practice, however, protamine is far from an ideal agent for the reversal of heparin therapy, both due to the intrinsic adverse effects of protamine itself (Park, 2004) and the insensitivity of the non-FIIa-mediated effects of heparin to protamine reversal, which significantly limit its efficacy with respect to LMWHs (see Section IV.B). However, with potentially safer and more effective agents to reverse the effects of heparin on the horizon, which have arisen in tandem with the development of such entities for DOAC reversal, the prominence of heparins in the antithrombotic drug arsenal should be reinforced.
The pharmacology of agents developed for the reversal of DOACs is reviewed in detail elsewhere (Dobesh et al., 2019), although two current examples are of particular relevance also to the clinical use of heparin (see also Section 4.2). Andexanet alfa, a modified, recombinant, inactive factor Xa (“decoy” factor Xa) is approved for reversal of the activity of certain direct FXa inhibitors but additionally can reverse the actions of indirect FXa inhibitors and indeed was initially developed also for this purpose (Lu et al., 2013; Apostel et al., 2021). Hence, following reversal of direct FXa inhibitor therapy with andexanet, subsequent heparin resistance, mediated by binding of andexanet to heparin-activated AT may manifest (Erdoes et al., 2021). This effect has been reported to be managed by administration of exogenous AT (Apostel et al., 2021), although the consideration of alternative reversal approaches to andexanet has been suggested for management of DOAC-induced bleeding in situations where subsequent anticoagulation with heparin may be required (Levy and Connors, 2021)—while andexanet is currently approved for reversal of rivaroxaban or apixaban activity in the event of life-threatening bleeding, there are reports of off-license preoperative use with unclear benefit (Levy and Connors, 2021). Nonetheless, andexanet presents as a plausible future alternative to protamine for reversal of the anti-FXa-mediated effects of heparin therapy (Maneno and Ness, 2021).
Ciraparantag (formerly PER977) also neutralizes the activity of heparin (see Section IV.B), and that of the DOACs, through charge-charge interactions, without affecting physiologic coagulation factors or the efficacy of other commonly used (nonanticoagulant) drugs (Ansell et al., 2022). Ciraparantag was found to reverse the effects of apixaban and rivaroxaban in a dose-related manner, in dose-ranging trials in healthy elderly subjects, and was well tolerated (Ansell et al., 2022; Chan and Weitz, 2022). Ciraparantag was also demonstrated to reverse the bleeding effects induced by UFH and a LMWH (enoxaparin) in a preclinical (rat) model, whereas protamine did not (Ansell et al., 2021). However, protamine did restore the APTT to control levels whereas ciraparantag had no effect on this measurement, with similarly contradictory effects on an anti-FXa assay (Ansell et al., 2021; Siegal, 2021). Hence, standard plasma-based assays are not suitable tools to assess the effect of ciraparantag on anticoagulant reversal (Ansell et al., 2022), with whole-blood clotting time being used successfully for this purpose in animal and human studies (Ansell et al., 2014, 2016, 2021, 2022).
C. COVID-19-Associated Thrombosis
The challenges associated with management of coagulation in critically ill patients are highlighted by the prominent association between COVID-19 disease and thrombotic complications. Management of COVID-19-associated coagulopathy, underpinned by a severe, infection-induced inflammatory response and including disseminated intravascular coagulation (Connors and Levy, 2020), has been a major recent consideration with respect to the anticoagulant activity of heparins, in addition to likely further benefit derived from nonanticoagulant activities discussed elsewhere in this review (see Sections IV.I.4 and VII).
Two relatively early meta-analyses of clinical trials, investigating the incidence of VTE in patients with COVID-19 and the impact of anticoagulant therapy, reported a composite VTE rate of 21% in hospitalized COVID-19 patients (Lu et al., 2020) and a rate of major VTE events of 12.5% in hospitalized patients, rising to 17.2% in those admitted to the ICU (Sridharan et al., 2020), respectively. However, a retrospective study reported that radiographically confirmed PE was prevalent among ambulatory patients, suggesting the risk of thrombosis to be present prior to hospitalization (Daughety et al., 2020). Moreover, a further systematic review with meta-analysis revealed that fewer than half of COVID-19 patients with PE had evidence of DVT, with a rate of PE events in patients admitted to ICU that exceeds that seen in ICU patients with non-COVID viral pneumonia or with acute respiratory distress syndrome (ARDS) (Suh et al., 2021). Pulmonary artery occlusion risk is high in patients with COVID-19 and reflects the development of intrapulmonary thrombosis rather than VTE (Birocchi et al., 2021).
COVID-19-related coagulopathy can be broadly summarized as involving a combination of enhanced coagulation with decreased endogenous anticoagulant and fibrinolytic mechanisms (Corrêa et al., 2020). A specific coagulopathy in this setting is supported by the abnormal APTT response that can be observed in COVID-19 patients approximately two weeks post-infection, in a manner that appears to be unrelated to disease severity and that is not seen in non-COVID patients with disseminated intravascular coagulation but which is partially mimicked in patients with lupus anticoagulant or coagulation factor IX deficiency (Shimura et al., 2021). Reviews of the mechanisms underpinning thrombosis associated with COVID-19 have been extensively reviewed elsewhere (e.g., Colling and Kanthi, 2020; Hanff et al., 2020; Iba et al., 2020a,b; Ali and Spinler, 2021; Bonaventura et al., 2021; Castro and Frishman, 2021). However, in the context of this review, COVID-19 presents key challenges, not only for the prevention and management of thrombosis but also in the monitoring of hemostasis and evaluation of thrombosis risk, against the backdrop of significant systemic inflammation and derangement of coagulation parameters. This extends further to complicate the effective monitoring of heparin therapy, particularly with respect to measuring the response to UFH by APTT in patients with COVID-19 (Hardy et al., 2020). This highlights that APTT is inappropriate given the change in levels of coagulation acute phase proteins (such as factor VIII and fibrinogen) due to COVID infection (Devreese, 2021).
Interestingly, the benefits of heparin treatment in severely ill patients with COVID-19, while clearly reflective of effective anticoagulation, do not relate as clearly to an anticoagulant effect (Magnani, 2021) in terms of dose: following intense focus on the most appropriate dosage level in this setting, thromboprophylactic dosing schedules are currently recommended over therapeutic (active-treatment) regimens (REMAP-CAP, ACTIV-4a, and ATTACC Investigators, 2021). It seems highly likely that the benefit of heparins in the management of COVID-19 extend beyond anticoagulant activity to encompass effects relevant to the underlying inflammatory response and indeed mechanisms of viral infection (see Sections VII and IV, respectively).
Smaller studies carried out relatively early in the pandemic tended to suggest a more aggressive antithrombotic approach to be warranted in the management of COVID-19-related coagulopathy. In the small, randomized, open-label HESACOVID trial, which compared therapeutic and prophylactic-dose anticoagulation in severely ill COVID-19 patients, improvements in gas exchange and the need for mechanical ventilation were associated with the higher-dose regimen (Lemos et al., 2020). Similarly, therapeutic anticoagulant dosing for primary prevention of VTE in hospitalized COVID-19 patients was found to have greater efficacy than prophylactic dosing regimens, in a meta-analysis of 11 studies, although bleeding risk was not assessed in this analysis (Sridharan et al., 2020). However, the RAPID randomized clinical trial (RCT), comparing the effect of therapeutic and prophylactic heparin dosing on mortality, need for mechanical ventilation, or ICU admission, in moderately ill COVID-19 patients, did not find a statistically significant difference in clinical outcome, though did report a low incidence of major bleeding in both groups (Sholzberg et al., 2021).
A further systematic review with meta-analysis addressed the prevalence of VTE in COVID-19 patients admitted to ICU and receiving anticoagulation, with a subgroup analysis indicating a higher rate of thrombosis in those receiving prophylactic anticoagulant regimens than in those receiving mixed (prophylactic and treatment) dosage regimens, concluding that individualized schedules based on clinical monitoring parameters may be preferable to protocol-based regimens in these patients (Hasan et al., 2020). However, a multicenter, open-label RCT of prophylactic compared with intermediate-dose enoxaparin, in COVID-19 patients admitted to the ICU with evidence of coagulopathy, indicated a lack of difference in 30-day outcomes between groups (Perepu et al., 2021) and a recent meta-analysis (Kuno et al., 2022) reported similar mortality outcomes in corticosteroid-treated ICU patients with COVID-19, irrespective of the anticoagulant regimen employed. With respect to safety, a systematic review with meta-analysis investigating all-cause mortality in hospitalized COVID-19 patients reported both therapeutic and prophylactic anticoagulant approaches to reduce all-cause mortality, with a greater effect attributed to therapeutic dosing but with an associated increase in bleeding risk (Parisi et al., 2021). Similarly, in a retrospective cohort study, standard-dose fondaparinux in noncritically ill COVID-19 patients was found to confer a greater bleeding risk without clinical benefit over a standard regimen of enoxaparin (Prandoni et al., 2020). Additionally, the use of LMWH was found to have no effect on hypercoagulability of patients but was associated with reduced mortality and curtailment of virus persistence in an observational study (Pereyra et al., 2021).
Importantly, the multicentre INSPIRATION RCT, which compared standard and intermediate-dose anticoagulant prophylaxis in ICU patients with COVID-19, reported no significant differences in thrombotic outcomes, mortality, or the need for extracorporeal membrane oxygenation (Sadeghipour et al., 2021). Furthermore, the multicentre ACTION RCT, in hospitalized COVID-19 patients with elevated D-dimer concentrations, found an increase in bleeding without improvement in clinical outcome following treatment with therapeutic, as opposed to prophylactic, doses of rivaroxaban, compared with a standard prophylactic heparin regimen (Lopes et al., 2021). Ultimately, the combined REMAP-CAP, ACTIV-4a, and ATTACC RCT outcomes established that there is no therapeutic benefit to applying an initial strategy of therapeutic dosing with heparin in critically ill COVID-19 patients, over and above standard prophylactic regimens (Sadeghipour et al., 2021). Current guidelines recommend in general the use of prophylactic, rather than intermediate or treatment-level, anticoagulant use, in critically ill patients with COVID-19 but without suspected or confirmed VTE (Cuker et al., 2021a,b).
D. Adverse Reactions/Risk
The vast array of interactions that heparin has with various proteins, as described in Section 4, can give rise to the risk of adverse reactions or a degree of risk when using this drug. The main risk associated with heparin treatment is bleeding, but the level of risk can be difficult to determine due to the factors involved in the use of heparin in patients—their indication, procedure, level of heparin required, and any comedication. The adverse reactions to heparin are also associated with its interactions with proteins outside of the coagulation system. The most well-known and common of these adverse reactions is HIT with an incidence of about 2.5% with UFH and 0.2% with LMWH (Martel et al., 2005). Other reported adverse incidents are skin lesions, osteoporosis, alopecia, and increase in liver enzymes. The risk of bleeding and adverse events was covered in our previous review (Mulloy et al., 2016), and herein only a brief summary/update of HIT is described; some similarities have been observed between HIT and the vaccine-induced immune thrombotic thrombocytopenia associated with SARs-coronavirus vaccines (Makris et al., 2021).
1. Heparin-Induced Thrombocytopenia
There are two types of HIT, type 1 and type 2, which both result in a reduction of circulating platelet numbers in response to heparin therapy, but they arise through slightly different mechanisms. Type 1 is described as a mild thrombocytopenia that occurs at the onset of treatment but stabilizes with continued treatment (Warkentin et al., 2008). The reduction in platelet numbers is caused by heparin directly affecting platelet activation and can be referred to as heparin-associated thrombocytopenia (Chong and Castaldi, 1986). This is the most common type of HIT and occurs in 10% to 30% of all patients administered heparin but does not require the cessation of treatment (Shantsila et al., 2009). Type 2 HIT is an immune-related reaction occurring after repeated exposure to heparin and is more serious (Martel et al., 2005). A feature of this condition is thrombosis due to the activation of platelets, which has recently been reviewed (Arepally and Padmanabhan, 2021).
The immunologic nature of the more serious type 2 HIT is due to the generation of antibodies that recognize complexes of heparin and PF4. As described earlier (Section 4.4.2) heparin can bind with high affinity to PF4, which is present in large quantities in platelets and is released upon activation. Natively, PF4 released by platelets binds to GAGs, such as CS and HS, on endothelial cells, which alters the cell surface to be more prothrombotic by release of surface bound AT. However, PF4 binds with higher affinity to heparin than surface GAGs, and this interaction can give rise to large complexes of heparin-PF4 in the circulation (Bertini et al., 2017b). The size of these complexes is dependent on overall charge, and therefore heparin size is crucial, with larger complexes (>670 kDa) associated with the pathogenesis of the disease (Rauova et al., 2005). These large complexes can then induce an immune response (see Section IV.F).
Broadly, the antibodies formed against PF4/heparin complexes are IgG isotypes (Greinacher et al., 2007), although not all antibodies give rise to HIT (Nazi et al., 2015). The antibodies are bound to PF4/heparin complexes, and it is the Fc receptor that can bind to the FcγIIa receptor on platelets that “crosslinks” platelets together leading to their activation and aggregation (Kelton et al., 1988). This response gives rise to thrombocytopenia and a thrombotic state due to the release of procoagulant elements from platelets (Tardy-Poncet et al., 2009). The HIT antibodies have also been shown to activate endothelial cells (Cines et al., 1987), monocytes (Pouplard et al., 2001), neutrophils (Xiao et al., 2008), and the complement system (Khandelwal et al., 2018). Due to the prothrombotic state, alternative anticoagulant therapy is required following cessation of heparin during which platelet levels should recover (Warkentin and Kelton, 1996).
2. Vaccine-Induced Thrombotic Thrombocytopenia
In response to the COVID-19 pandemic, vaccines against coronavirus were developed at pace showing high effectiveness at preventing hospitalization from the disease. One vaccine, which used an adenoviral vector, has been reported to cause an incidence of thrombocytopenia in a small number of patients several days after vaccination (Schultz et al., 2021; Wolf et al., 2021). So-called HIT antibodies have been detected, which cause platelet activation indicating similarities to HIT given the reduction in platelets count and an observable thrombotic state. The term vaccine-induced immune thrombotic thrombocytopenia was coined to describe the condition (Greinacher et al., 2021). However, it should be noted that similarities to HIT are limited to the presence of activating antibodies as the underlining mechanisms (Dotan and Shoenfeld, 2021) are likely to be different. At the time of writing, investigations are underway (Goldman and Hermans, 2021) to determine the mechanisms involved in generating this rare immune response.
VII. Nonanticoagulant Effects of Heparin
It is now well accepted, as discussed earlier, that more than 400 key proinflammatory mediators and adhesion molecules involved in inflammatory cell recruitment into tissues have heparin binding regions in their structure (Mulloy et al., 2016; Paluck et al., 2016; Mulloy, 2019) (see Section IV). In many cases when heparin binds to these inflammatory proteins, the function of the protein is inhibited. This effect may well contribute to the ever-increasing number of observations that heparin is anti-inflammatory in many experimental and clinical settings, which has been reviewed extensively elsewhere (Cassinelli and Naggi, 2016; Beurskens et al., 2020). In many cases this anti-inflammatory effect of heparin is mimicked by heparin-like molecules lacking anticoagulant activity (Cassinelli and Naggi, 2016; Oduah et al., 2016; Mohamed and Coombe, 2017). Many of these activities of heparin are now considered to be independent of anticoagulant actions and as such are ripe for exploitation as novel approaches to treating a wide range of diseases (see Section X). This has led to interest in developing heparin-like molecules lacking anticoagulant activity for controlling the progression of cancer, particularly metastasis (Bendas and Borsig, 2020; Liebsch and Schillers, 2021), which shares many similarities with leukocyte diapedesis into tissues during inflammatory responses. Furthermore, there is increased interest in heparin and related drugs in controlling infectious diseases caused by prions (Vieira et al., 2014), viruses (de Boer et al., 2012; Tree et al., 2021), or bacteria (McCrea et al., 2014)(see Section IV.I). Heparin is thought to exert many of its nonanticoagulant actions through binding of proteins such as chemokines and growth factors that are functionally dependent upon binding to HS (see Section IV.D). However, the exact structural characteristics that mediate the anti-inflammatory effects of heparin are, in the majority of cases, not fully known. Interactions between heparin and proteins can vary from highly sequence specific, such as the binding of AT, to relatively nonspecific (see Section III). A significant number of proteins that can be classed as heparin binding are fundamentally associated with the inflammatory response, including, but by no means limited to, cytokines, growth factors, adhesion molecules, cytotoxic, and tissue-degrading enzymes such as elastase and metalloproteinases (Mulloy et al., 2016).
The heparin-binding sites of many proinflammatory proteins are either known or can now be predicted (see Section IV.A). This then provides a clear rationale for the development of polysaccharides that recognize these heparin binding domains such as oligosaccharides isolated from a marine organism, Holothuria forskali, that can recognize the adhesion molecule P-selectin (Panagos et al., 2014). Understanding how heparin binds to certain proteins has also led to the rational development of synthetic oligosaccharides and novel sugars that specifically bind to certain cytokines involved in inflammatory responses (Roy et al., 2014; Paluck et al., 2016; Mohamed and Coombe, 2017; Mulloy, 2019) and key proteins found on viruses used to infect cells (Tree et al., 2021) (see Section IV.J).
A. Effects of Heparin on Inflammatory Responses
Heparin is now known to be of use in the treatment of a number of inflammatory diseases (see the following discussion) (Mousavi et al., 2015; Mulloy et al., 2016), where the anticoagulant effects are not always necessary and indeed are often perceived as providing a potential safety concern, thereby limiting the wider use of this drug. Thus, a greater understanding of the interactions between heparin and specific mediators involved in the inflammatory response is facilitating the discovery and development of a number of novel anti-inflammatory drugs lacking anticoagulant activity, as, for example, has been described for novel heparin analogs isolated from the ascidian Styela plicata that were able to reduce colitis in rats with a lower risk of hemorrhage (Belmiro et al., 2009) (see Section X) and novel anti-inflammatory polysaccharides isolated from the sea squirt, Ascidiela aspersa (Thomson et al., 2016).
Heparin has been reported to inhibit the activation of a number of inflammatory cell types that we have previously reviewed (Slungaard et al., 1990; Ahmed et al., 1992; Rohrer et al., 1992; Bazzoni et al., 1993; Inase et al., 1993; Teixeira et al., 1996; Piccardoni et al., 1996; Brown et al., 2003; Lever et al., 2007). This can be the result of binding and neutralization of various mediators and enzymes released during the inflammatory response that would otherwise lead to activation of inflammatory cells and via inhibition of the release of inflammatory mediators from different inflammatory cell type (as previously reviewed) (Mulloy et al., 2016; Mulloy, 2019).
Heparin is now recognized to be highly effective in limiting the recruitment of many inflammatory cell types into a variety of tissues, through modulation of interactions between leukocytes and vascular endothelial cells at a number of levels, including binding to important adhesion molecules preventing them from recognizing their counterligands as we previously reviewed (Mulloy et al., 2016). We and others have previously shown that the infiltration of leukocytes into various tissues is known to be dependent on platelet activation following allergen challenge of allergic animals (Pitchford et al., 2004), following exposure to LPS (Kornerup et al., 2010), acute lung injury (Zarbock et al., 2006), and following pulmonary infection with pseudomonas aeruginosa (Amison et al., 2018). We have recently demonstrated that the platelet dependent recruitment of leukocytes is inhibited by pretreatment with heparin or a nonanticoagulant heparin fraction further supporting the idea that this important anti-inflammatory effect of heparin is unrelated to anticoagulant activity (Riffo-Vasquez et al., 2016).
The glycocalyx is now seen as an important part of the endothelial surface that is heavily involved in regulating the trafficking of various inflammatory cell types from blood into tissues. The importance of the glycocalyx in health and disease has been discussed in several recent manuscripts, particularly in the context of sepsis (Schmidt et al., 2012) and in COVID-19 (Wadowski et al., 2021). The most important components of the glycocalyx include heparan sulfate proteoglycans, chondroitin sulfate, hyaluronan, and sialic acid. Alterations in the glycocalyx exposes receptors (adhesion molecules) to allow leukocyte and platelet activation and adhesion and also allows for altered vascular permeability (LaRivière and Schmidt, 2018). A recent study has shown that the infusion of LMWH was able to reduce the shedding of glycans from the glycocalyx following stimulation of endothelial cells by N-formyl-met-leu-phe (Lipowsky and Lescanic, 2017). This was attributed in part to the ability of heparin to inhibit heparanase and supports earlier work from our laboratory showing that recombinant heparanase can induce inflammatory cell recruitment by promoting adhesion to the vascular endothelium (Lever et al., 2014). Lipowsky also suggested that LMWH may have been anti-inflammatory by binding certain components of the glycocalyx and the endothelium such as heparan sulfate proteoglycans and P-selectin leading to a dose-dependent inhibition of leukocyte adhesion to the endothelial surface supporting earlier work in vitro demonstrating that heparin can reduce the adhesion of leukocytes to vascular endothelial cells (Lever et al., 2000) and supports our observations that exogenous heparin can replace HS enzymatically removed from the surface of endothelial cells by heparanase (Lever et al., 2016). Furthermore, a very recent study has shown that plasma from patients with COVID-19 can disrupt the glycocalyx, which can be prevented by both UFH and LMWH (Potje et al., 2021). These observations would support the suggestion that the release of heparin from mast cells located anatomically in close proximity to blood vessels is to provide a mechanism for the endogenous homeostatic regulation of inflammation (neutralizing excess proinflammatory mediators and topping up the damaged endothelial glycocalyx), rather than being released primarily for its anticoagulant activity (Page, 1991; Lever et al., 2016).
We have previously reviewed the various studies supporting the ability of heparin and nonanticoagulant heparins to reduce allergic inflammation (Mulloy et al., 2016) and other studies have confirmed these observations using house dust mite sensitized mice following chronic intranasal treatment with heparin (Fu et al., 2013). In addition, a nonanticoagulant heparin (S-NACH) has been shown to inhibit TH2-driven allergic inflammation in sensitized mice through an effect on IL4 mediated signal transduction involving the Janus kinase 1 pathway (Ghonim et al., 2018).
B. Trauma and Lung Injury
A hallmark histologic feature of acute lung injury (ALI) that can lead to ARDS is a fibrin mesh in the air sacs of the lung known as a hyaline membrane, which leukocytes attach to and that contributes to the development of diffuse alveolar damage. Another early manifestation of the inflammatory response is fibrin accumulation in pulmonary capillaries and venules, which lead to microvascular thrombosis as another feature of ALI. Several clinical trials have investigated the effect of nebulized heparin to target alveolar coagulopathy and fibrin deposition in patients with ALI and related conditions. These studies have suggested that nebulized heparin significantly reduces pulmonary dead space, activation of the coagulation system, and microvascular thrombosis in the lung, as well as preventing a deterioration of the Murray Acute Lung Injury score and providing increased time free from ventilatory support (Dixon et al., 2010, 2011, 2016). Heparin has been demonstrated to have a number of actions that may be beneficial in producing these effects against ALI (Dixon et al., 2021). Thus, heparin can bind to a number of bacterial and viral pathogens (see Section IV.I) to reduce the ability of the pathogens to initiate an inflammatory response in the lung that has been confirmed by the ability of heparin to demonstrate efficacy in a range of animal models of pneumonia and ALI (Dixon et al., 2021) (and see later discussion). This type of observation has recently been extended with the observation that various UFH preparations can bind the spike protein of the recently identified SARS-CoV-2 coronavirus responsible for causing the COVID-19 pandemic, thereby inhibiting the ability of the virus to infect a mammalian cell line (Tree et al., 2021). This effect was most obvious with UFH. In addition to the antiviral effects of heparin, clearly the anticoagulant effects of this drug against alveolar coagulation and microthrombi are likely to contribute to the benefit of heparin in patients with ALI and ARDS (reviewed by van Haren et al., 2020), and in reducing mortality in patients with COVID-19 who have met the sepsis induced coagulopathy criteria (Gozzo et al., 2020; Thachil, 2020; Shen et al., 2022). Furthermore, the ability of heparin to inhibit the recruitment of various inflammatory cells into tissues such as the lung, as well as inhibiting the activation of inflammatory cells and bind to key adhesion molecules and cytokines involved in inflammatory cell recruitment (Mulloy et al., 2016; van Haren et al., 2020 for reviews), undoubtedly contributes to the ability of heparin to reduce the inflammatory sequelae following pneumonia induced by various pathogens.
A recent landmark multicentre clinical study (CHARLI) has reported that nebulized heparin is well tolerated in patients with ALI or who are at risk of ARDS (Dixon et al., 2021). In this study, while nebulized heparin did not improve self-reported performance and daily physical activities at day 60 following treatment, it did nonetheless have a significant impact on a range of exploratory end points in this population. Thus, nebulized heparin administered on top of standard of care, which included the use of systemically administered heparin, reduced the number of patients developing ARDS, with less deterioration in the Murray Acute Lung Injury scores and a faster recovery, allowing more survivors being able to reside at home at day 60 compared with placebo-treated patients.
Importantly these beneficial effects were found with only modest increases in APTT in patients who concomitantly received systemic UFH and had no effect on APTT in patients who received treatment with concomitant LMWH. These findings suggest that the additional benefit of nebulized heparin is likely to be due to actions local to the lung and supports the safety of using nebulized heparin as has been reported in other clinical trials in patients with other diseases of the lung (Ledson et al., 2001; Yildiz-Pekoz and Ozsoy, 2017; Shute et al., 2018a; Ashoor et al., 2020). The findings from the CHARLI study suggest that further research is justified to establish whether nebulized heparin can accelerate recovery in patients who have or are at risk of developing ARDS. Additionally, a recent case series has reported the use of nebulized UFH in patients with COVID-19 and suggested that the use of this drug can improve a number of important physiologic and clinical parameters (van Haren et al., 2022). While this was an uncontrolled series, the impressive benefit observed is being investigated in a meta-trial to better understand whether nebulized heparin has a use in the treatment of the lung injury resulting from infection with SARs-CoV-2 (Dixon et al., 2021).
The results presented in the CHARLI study are also consistent with an earlier double-blind trial of intubated patients with acute exacerbations of COPD that reported significantly more ventilator-free days following treatment with nebulized heparin (Ashoor et al., 2020). Furthermore, a case control study of patients with ALI following burns also reported that nebulized heparin increased the number of ventilator-free days (McIntire et al., 2017). Another study has also reported that the use of a LMWH (nadroparin) for one week on top of standard of care significantly reduced the mean duration of mechanical ventilation, and length of stay in ICU and hospital, in patients having an acute exacerbation of COPD requiring ventilatory support (Qian et al., 2014). However, another study investigated the effect of prophylactic nebulized heparin in the management of pneumonia in ventilated patients in ICU and found no significant difference when using this treatment compared with sodium chloride on top of standard of care (Bandeshe et al., 2016). However (as pointed out by Dixon et al., 2021), this study used a much lower nebulized dose of heparin than other studies and the nebulization methodology was not standardized (Dixon et al., 2021).
LMWH has also been shown to reduce the systemic inflammation and acute lung injury induced by endotoxin in rats (Luan et al., 2014). At least with UFH the inhibitory effect on endothelial barrier dysfunction is via the induction of high mobility group box 1 and regulation of the P38 pathway (Luan et al., 2018). Moreover, 2-O,3-O-desulfated heparin has been shown to inhibit neutrophil elastase-induced secretion of high mobility group box 1 and resulting airways inflammation further supporting the suggestion that many of the anti-inflammatory effects of heparin are independent of its anticoagulant activity (Griffin et al., 2014). The ability of heparin to inhibit LPS-induced inflammation has recently been reported to be secondary to inducing caveolin-1 and subsequent activation of the p38/mitogen-activated protein kinase pathway in macrophages (Liu et al., 2015). Furthermore, heparin has been shown to inhibit the activation of human alveolar macrophages, alveolar type II cells, and fibroblasts activated by LPS, by reducing the expression of IRAK1 and MyD88 in these important cell types implicated in the pathogenesis of ALI (Camprubí-Rimblas et al., 2017). Moreover, self-assembling lipid modified glycol-split heparin nanoparticles have recently been reported to suppress LPS-induced inflammation via an effect on TLR4-NF-KB signaling (Babazada et al., 2014). This in vitro work has been extended to show that nebulized heparin reduces both inflammation and coagulation in an ALI model in rats induced by intratracheal administration of HCl and LPS, although there was no further inhibition when AT was used with heparin (Camprubí-Rimblas et al., 2020). Another recent study has suggested that UFH can alleviate sepsis-induced ALI by reducing the levels of IL-6 in bronchoalveolar lavage fluid and improving the tight junctions in human lung microvascular endothelium by inhibiting the ERK1/2 MAPK pathway and downregulating the expression of claudin 5, occluding and ZO-1 (Liu et al., 2019). Furthermore, the use of UFH as a lock solution in catheters in patients undergoing hemodialysis also reduces the levels of IL-6 (Ezzat et al., 2021). Extracellular histones are known to be major contributors to organ dysfunction and death in patients with sepsis as they cause problems in the microcirculation. It is therefore of considerable interest that UFH has been demonstrated to inhibit histone-induce cytoxicity in vitro and to prevent the microcirculatory disturbances in the gastrointestinal tract of rodents infused with histones (Zhu et al., 2019).
These latter observations support our own studies where we have demonstrated that histone-treated whole blood showed elevation in the inflammatory markers IL-6, IL-8, and tissue factor and an increase in the level of a complement component, C3a. Heparin and selectively desulfated heparins were found to have antihistone properties, reducing the level of all the biomarkers measured. The selectively desulfated heparins, which have reduced anticoagulant activities, retained a high degree of effectiveness relative to unmodified heparin as an antihistone agent, whereas a fully desulfated heparin was no longer effective. This suggests that modified heparin, with reduced anticoagulant activity, may be a useful compound to treat inflammatory conditions where there is an increase in the level of histones (Hogwood et al., 2020). This suggestion has been supported by the observation that a nonanticoagulant heparin that binds histones can also provide protection against sterile inflammation and sepsis (Wildhagen et al., 2014).
C. Other Inflammatory Conditions
Heparin has been demonstrated to inhibit the proliferation of fibroblast-like synoviocytes found in rheumatoid arthritis that are thought to contribute to cartilage destruction in this disease. This antiproliferative activity was via inhibition of the NF-kB pathway (Qi et al., 2016). In addition, a recent study has reported the ability of a sustained release LMWH preparation to reduce the lung inflammation and subsequent fibrosis following exposure of mice to the profibrotic agent bleomycin (Saito et al., 2020). This raises the possibility of heparin being of value in fibrotic conditions such as idiopathic pulmonary fibrosis and long COVID where there is a large unmet clinical need.
Heparin has also been demonstrated to reduce cerebrovascular inflammation and brain edema and to accelerate cognitive recovery following severe traumatic brain injury (Nagata et al., 2016), supporting earlier work with LMWH under similar circumstances (reviewed by Stutzmann et al., 2002). This protective effect was due in part to inhibition of leukocyte adhesion and vascular permeability in the pericontusional cerebral vasculature (Nagata et al., 2016).
A very interesting recent clinical study has evaluated the effect of prophylactic administration of low dose LMWH in women with risk factors associated with placental inflammation. In a study of 300 pregnant women, prophylactic low-dose LMWH was significantly able to prevent metabolic and immunologic disorders causing placental inflammation contributing to various obstetric complications (Beksac et al., 2022). Inflammation is known to be a hallmark of cervix remodeling, and HS has been shown to be of possible value in inducing an inflammatory-driven ripening of the cervix as HS has been shown to be elevated in late pregnancy (Åkerud et al., 2021). Another experimental study in mice has reported that heparin and a glycol-split LMWH with low anticoagulant activity enhances myometrial contraction and the production of IL-8, leading to a marked infiltration of neutrophils and macrophages into the cervix. These effects were reduced in TLR4- and IRF3-deficient mice, and the authors have suggested that glycol-split LMWH acts as a novel TLR4 agonist that may find therapeutic use in ripening of the cervix for initiation of labor (Åkerud et al., 2021).
The role for using heparin in the treatment of patients with sepsis has recently been reviewed elsewhere, although the clinical data are conflicting (Li and Ma, 2017). This group suggested that if heparin is to be of use in the treatment of sepsis, it should probably be used in more severe patients. This conclusion is supported by the findings of a systematic review and meta-analysis that showed that heparin is able to reduce 28-day mortality in patients with severe sepsis (Wang et al., 2014).
A recent open-label clinical study has shown that use of subcutaneous LMWH on top of standard care in 100 patients with acute pancreatitis showed that this treatment was safe and produced clinical benefit suggesting this approach should be investigated further in controlled trials (Tozlu et al., 2019).
D. Eyes
Interestingly heparin coatings have been used to reduce signs of postoperative inflammation after extracapsular cataract extraction (Borgioli et al., 1992). Thus in 524 patients, a heparin surface modified posterior chamber intraocular lens was compared with a conventional polymethylmethacrylate intraocular lens and shown to be able to reduce inflammation one-year post-surgery (Borgioli et al., 1992). A further study in an Asian population confirmed the earlier clinical work and showed that heparin surface modification of intraocular lenses significantly reduced the inflammatory response to conventional polymethylmethacrylate lenses (Lai and Fan, 1996). However, a more recent study reported that a heparin-coated intraocular lens provided no benefit compared with a conventional lens up to three months postoperatively (Maedel et al., 2013). Furthermore, LMWH has been shown to be safe and effective as a treatment of the postoperative inflammation associated with phacomorphic glaucoma (Zarei et al., 2006), although enoxaparin was not found to be of benefit when added to the infusion fluid of children undergoing cataract surgery in an attempt to reduce postoperative inflammation in one study (Sukhija and Ram, 2012), although another study showed some benefit of heparin sodium (Bayramlar et al., 2004). A further study assessed a heparin surface-modified hydrophobic acrylic intraocular lens in comparison with the same lens that was not heparin coated and found that the heparin-coated lens showed less inflammation in the perioperative stage (Krall et al., 2014). Heparin added to intraocular irrigation solution has also been shown to reduce postoperative inflammation associated with cataract surgery in children (Ozkurt et al., 2009).
Experimentally topically administered heparin has been shown to reduce allergic conjunctivitis in mice, associated with an inhibitory effect on mast cell infiltration into the eye (Kocatürk et al., 2013). The effect of heparin in this model was as good as topical dexamethasone and so it is plausible that topical heparin could be of use in treating allergic inflammatory conditions of the eye, consistent with its known antiallergic effects in other tissues (reviewed by Mulloy et al., 2016; Mulloy, 2019).
E. Cancer
Heparin and LMWH are recommended options for use in the prevention and treatment of venous thromboembolisms in cancer (Key et al., 2020; Lyman et al., 2021) but have also been indicated as a potential treatment of metastasis (Mohamed and Coombe, 2017; Mulloy, 2019), which has led to the development of new agents mimicking heparin but that lack anticoagulant activity. The ability of heparin to inhibit P-selectin, as a key adhesion molecule involved in metastasis, and to inhibit heparanase, a critical enzyme in allowing tumor cells to leave blood and enter tissues, has been the driving force behind this area of pharmacology (Vlodavsky et al., 2007; Bendas and Borsig, 2020). In addition, heparin has been well described as an inhibitor of angiogenesis, which is critical for the survival of solid tumors (Folkman and Shing, 1992). Indeed, it is now recognized that heparin will mimic HS, which can bind to almost all known angiogenic growth factors (Lanzi and Cassinelli, 2018). These observations have led to the identification of a large number of heparin-like molecules that bind different proangiogenic growth factors selectively, including chemically modified heparins, sulfated K5 derivatives, heparan sulfate mimetics, and a wide variety of naturally occurring polysaccharides (Chiodelli et al., 2015).
A number of other drugs have been identified as heparin-like for the treatment of metastasis, such as the series of partially desulfated heparin derivatives that inhibit galectin-3-mediated metastasis (Duckworth et al., 2015) and heparin-containing cryogel microcarriers as a delivery device for doxorubicin (Newland et al., 2020). Other drugs are antimetastatic secondary to inhibition of heparanase such as roneparstat (Alekseeva et al., 2017) and PI-88 (Liao et al., 2016), the latter drug, which has also been demonstrated to have effects on angiogenesis (reviewed by Kudchadkar et al., 2008). PI-88 is now in clinical trials for the treatment of different types of cancer after promising phase 1 and 2 clinical trial data (see Mohamed and Coombe, 2017). However, several recent clinical trials investigating the effects of heparin or LMWH have not been encouraging in this clinical setting. Thus, the very large FRAGMATIC trial of daily subcutaneous dalteparin failed to show any clinical benefit in the treatment of patients with lung cancer, although a significant effect was seen in the heparin arm against venous thromboembolic events (Macbeth et al., 2016). Furthermore, two more recent clinical trials investigating the effects of LMWHs in treating lung cancer were both negative. In the RASTEN study, supratherapeutic doses of enoxaparin were administered subcutaneously on top of standard of care and found to have no significant effect on survival times (Ek et al., 2018). In addition, a phase 3 trial investigated the effect of treatment with tinzaparin for 12 weeks in patients with non-small cell lung cancer and showed to have no overall benefit on survival (Meyer et al., 2018). The nonanticoagulant heparin derivative, necuparanib, also failed to show benefit in a phase 2 trial in patients with pancreatic cancer (O'Reilly et al., 2020). A recent systematic review and meta-analysis of the efficacy and safety of LMWH also concluded that existing data do not support the use of this drug in patients with cancer to improve survival (Montroy et al., 2020). Other developments in potential exploitation of the anticancer properties of heparin have recently been extensively reviewed (Atallah et al., 2020; Ma et al., 2020), as has the use of heparin, in comparison with other anticoagulants, for treating cancer-associated thrombosis (Moik et al., 2020). Another interesting approach is the recent description of a tumor microenvironment-responsive PEGylated heparin-pyropheophorbide, a nanoconjugate that is photosensitive (Wu et al., 2021).
VIII. Heparin in Biomaterials and Regenerative Medicine
Since our previous review in 2016 (Mulloy et al., 2016) there have been considerable advances in the development of new approaches to delivering heparin immobilized on medical devices for use in regenerative medicine. Many biocompatible medical devices intended for contact with the circulation are treated with heparin so as to diminish their prothrombotic and increasingly their anti-inflammatory properties. The technologies that have been developed for coating blood-contacting devices such as stents, vascular grafts, and extracorporeal circulation components with heparin have been reviewed elsewhere (Biran and Pond, 2017).
Ischemia-reperfusion injury is a major complication of many thrombotic conditions and arising from whole-organ transplantation. Activation of the vascular endothelium and shedding of the glycocalyx is known to increase during ischemia-reperfusion injury, and so it is of interest that recent work in vitro has shown that a heparin conjugate immobilized to the endothelium and the collagen in the basement membrane of the vessel wall protects the endothelium from the impact of ischemia-reperfusion injury (Nordling et al., 2015). Inflammation-associated thrombosis has also been successfully inhibited by the use of nanoparticles containing copolyoxalate vanillyl alcohol and heparin deoxycholic acid, without leading to excessive bleeding (Xiang et al., 2019). Another approach to promote endothelialisation, antithrombotic and anti-inflammatory activity has been to covalently immobilize heparin on the surface of small-diameter grafts manufactured from polytetrafluoroethylene (Gao et al., 2017). Heparin has also been widely investigated as a component of bioactive wound dressings for accelerated wound healing (reviewed by Biran and Pond, 2017). Recently an N-acetylated heparin-poly(N-isopropylacrylimide) has been investigated as a thermoresponsive hydrogel for delivering ibuprofen locally as an anti-inflammatory agent for treating wounds (Andrgie et al., 2020).
Another recent development for the potential treatment of wound healing is the development of heparin-based hydrogels incorporated with Cu5.40 ultrasmall nanozymes. This product outperformed the standard of care in terms of reducing inflammation and increasing the regeneration and vascularization of cutaneous wounds (Peng et al., 2021). Recently, the description of widely used polycaprolactone/gelatin nanofiber scaffolds that release heparin has provided a novel approach to induce anti-inflammatory and antithrombotic activity (Wang et al., 2019b). The nanofibers have been designed to allow controlled release of heparin by use of reactive oxygen species-responsive poly(ethylene glycol)-based B-thioester copolymers and mesoporous silica nanoparticles in the nanofibers (Wang et al., 2020a). Heparin has also been used with PDGF-containing porous microspheres to provide an anti-inflammatory and tendon healing effect in a model of rotator cuff tendinitis in rabbits (Kang et al., 2019). Furthermore, the use of poly(lactic-co-glycolic acid) microparticles to produce a sustained release formulation of LMWH has been reported to have anti-inflammatory and antifibrotic activity in mice (Saito et al., 2020).
Heparin-loaded liposomes formulated with phospholipid, cholesterol, and stearylamine have been used as an enema to exert an anti-inflammatory effect in an experimental model of colitis (Ahmad et al., 2021), as have heparin-coated albumin nanoparticles for targeting inflammation in the gastrointestinal tract (Zhang et al., 2020c). Another recently published study has investigated the anti-inflammatory effect of covalently immobilized hyaluronan with heparin on different surfaces using EDC/NHS cross-linking chemistry that could reduce the adhesion of macrophages and reduce their activation (AlKhoury et al., 2020).
A hydrogel dressing encapsulating heparin and basic fibroblast growth factor has been described that has been prepared by the Michael addition of four-arm acrylated polyethylene glycol and dithiothreiotol has been described. This dressing accelerated wound healing in a cutaneous model, as well as reducing inflammation (Peng et al., 2021). Heparin-incorporated star-PEG nanofilms have recently been described as bioengineered surfaces to protect pancreatic islet cells to improve cell survival after implantation (Lou et al., 2017). Furthermore, heparin has been shown to improve the effectiveness of bone marrow-derived mesenchymal stem cells as cytotherapy (Liao et al., 2017).
IX. Novel Formulations and Drug Delivery Technology for Heparin
Heparin has traditionally been administered by injection, either subcutaneous or intravenous, and there have been multiple attempts to develop oral formulations to make the drug easier to take and because it has been appreciated for more than 50 years that heparin has poor bioavailability (see Jaques, 1979). Some of the various attempts to improve the bioavailability of heparin have been discussed elsewhere (Schluter and Lamprecht, 2014; Mulloy et al., 2016) and include the use of sodium N-[8(-2-hydroxybenzoyl)amino]caprylate (Baughman et al., 1998), chitosan nanoconstructs (Paliwal et al., 2012), and polyaminomethacrylate coacervates (Viehof and Lamprecht, 2013). Attempts have also been made to create solid formulations of heparin for oral delivery by use of heparin conjugated with deoxycholic acid, formulated with the polymer Poloxamer 407 (Park et al., 2010).
The increasing awareness of the ability of heparin and related drugs to be of value in treating a range of diseases where the anticoagulant effects of this drug would not be required has increased the interest to find other routes of administration, particularly for the management of chronic inflammatory diseases. For example, there is now growing evidence for the effectiveness and safety of administering heparin by inhalation which has recently been reviewed (Yildiz-Pekoz and Ozsoy, 2017). Thus, heparin has been administered safely to humans by inhalation for up to 28 days (Markart et al., 2010) and in most studies in patients, inhaled delivery of heparin is not associated with adverse effects, and indeed does not cause systemic changes in coagulation (Shastri et al., 2014). Novel formulations of inhaled heparin have also been developed such as large inhalable microspheres (Rawat et al., 2008), lactose formulations (Bai et al., 2010), and co-sprayed with L-leucine as a dry powder for the treatment of COPD and cystic fibrosis (Shur et al., 2008), as heparin has been shown to have effects as a mucolytic agent and to cause the breakdown of DNA tangles (Broughton-Head et al., 2007) and to be of clinical benefit in the treatment of patients with COPD (Shute et al., 2018a; Ashoor et al., 2020) and cystic fibrosis (Ledson et al., 2001; Shur et al., 2008). A recent review has summarized the various clinical studies investigating the effectiveness of inhaled heparin in the treatment of a range of respiratory diseases (Yildiz-Pekoz and Ozsoy, 2017 ;Shute et al., 2018b).
Others have developed transdermal approaches to deliver heparin (Lanke et al., 2009) as an alternative method to parenteral administration for anticoagulant use.
X. Novel Drugs Based on the Nonanticoagulant Actions of Heparin
There is now increasing interest in developing drugs that mimic some of the wide range of pharmacological effects of heparin but that have reduced or no anticoagulant activity. Some of these approaches have been extensively reviewed elsewhere (Smith and Bertozzi, 2021), which is a wide-ranging review discussing new therapies inspired by glycan and carbohydrate research, including work with heparin. Novel approaches to mimicking aspects of the pharmacology of heparin include synthetic mimics based on small molecules, peptides, polysaccharides, and polymers (reviewed by Paluck et al., 2016), as well as other drugs that are chemically modified heparin or LMWH fractions (Mohamed and Coombe, 2017). Two small molecule mimics have already been approved, suramin as an antiparasitic drug and carafate as an antiulcer medicine (reviewed in Paluck et al., 2016). Other mimetics such as sulfated tetrapeptide, which binds to EGF (Maynard and Hubbell, 2005) and polymers/polysaccharides, which act as anticoagulants or interact with HS binding proteins such as FGF (Paluck et al., 2016), are “designed” to target specific interactions that heparin has with the aim to have a therapeutic benefit focused on these specific interactions. Furthermore, there are now a range of drugs in development that are agents mimicking HS, particularly for use in regenerative medicine such as OTR3 (Barritault et al., 2017). Another recently described nonanticoagulant heparin like GAG from the China white jade snail has been shown to have wound healing properties in diabetic mice (Wu et al., 2020).
Furthermore, a heparosan heptasaccharide obtained by partial desulfation of LMWH has been described that retains good anti-inflammatory activity (Pan et al., 2020). The antitumor properties of heparin have also been exploited with the identification of a new nonanticoagulant heparin analog isolated from the mollusc Nodipecten nodusus that is an inhibitor of P-selectin and heparanase, that experimentally is able to reduce metastasis and inflammatory cell recruitment (Gomes et al., 2015).
Different approaches to prepare derivatives of heparin lacking anticoagulant activity by the periodate cleavage of 2,3 vicinal diols in nonsulfated uronate residues (so called glycol-split technology) and replacement of N-sulfamido with N-acetomido- groups in glucosamine residues has proved successful at identifying compounds that can inhibit elastase, IL-8, and tumor necrosis factor-alpha with minimal anticoagulant activity. This type of approach has been reviewed elsewhere and looks promising as a way of identifying novel compounds that mimic the anti-inflammatory actions of heparin (Veraldi et al., 2015). 2-O,3-O desulfated heparin is a selectively desulfated molecule that retains the anti-inflammatory effects of heparin but without the anticoagulant effects of heparin (Rao et al., 2010) that has been evaluated in a number of clinical conditions (reviewed elsewhere by Cassinelli and Naggi, 2016). This drug has been shown to also be of benefit in reducing lung infections due to pseudomonas aeruginosa by enhancing bacterial clearance and reducing the lung injury associated with pneumonia (Sharma et al., 2014), an observation more recently confirmed with a range of synthetic heparan sulfate competitors such as N-acetyl heparin (Lorè et al., 2018). Furthermore, a recent study has reported that a novel oxidized sulfated ultra-low molecular weight heparin, S-NACH, which is devoid of anti-factor Xa and IIa activities and with limited systemic anticoagulant effects, showed enhanced binding to endothelial cells compared with UFH and LMWH (Darwish et al., 2021).
XI. Summary
Heparin has been in continuous clinical use for more than 100 years, but there is still much to learn from this remarkable molecule. It is now clear that heparin exhibits a wide range of pharmacological properties beyond the well-recognized anticoagulant and antithrombotic activity. The anticoagulant activity of heparin has been shown to be due to a particular pentasaccharide sequence contained within the heparin polymer, and it is now becoming clear that other regions of the heparin molecule are responsible for other nonanticoagulant functions. Furthermore, a wide range of novel agents are in development to mimic particular pharmacological actions of heparin for the treatment of a wide range of conditions where the anticoagulant effect of heparin is not required. It is anticipated that in the coming decade some of these experimental approaches will be translated into new approaches for the treatment of inflammatory disorders, cancer and infectious diseases.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Hogwood, Mulloy, Lever, Gray, Page.
Footnotes
- Received July 7, 2022.
- Revision received November 4, 2022.
- Accepted November 8, 2022.
This work received no external funding.
No author has actual or perceived conflict of interest with the contents of this article.
An earlier version of this paper appears in Pharmacology of Heparin and Related Drugs under the DOI dx.doi.org/10.1124/pr.115.011247.
Abbreviations
- AAV
- adeno-associated viruses
- ACE-2
- angiotensin converting enzyme-2
- ALI
- acute lung injury
- APC
- activated protein C
- API
- active pharmaceutic ingredient
- APTT
- activated partial thromboplastin time
- ARDS
- acute respiratory distress syndrome
- AT
- antithrombin
- BACE-1
- enzyme β-secretase-1
- BLH
- bovine lung heparin
- BMH
- bovine mucosa heparin
- BMP
- bone morphogenic proteins
- CHIKV
- chikungunya virus
- COPD
- chronic obstructive pulmonary disease
- CS
- chondroitin sulfate
- DENV
- dengue virus
- DexS
- dextran sulfate
- DS
- dermatan sulfate
- EP
- European Pharmacopeia
- FGF2
- fibroblast growth factor
- FH
- factor H
- FMDV
- foot-and-mouth disease virus
- FTI
- Fourier transform infrared
- FXa
- factor Xa GAG, glycosaminoglycan
- GlcA
- β-D-glucuronic
- GlcNAc
- N-acetyl α-D-glucosamine
- GlcNS
- N-sulfamido α-D-glucosamine
- HARE
- hyaluronic acid receptor for endocytosis
- HBHA
- heparin-binding hemagglutinin
- HCII
- heparin cofactor II
- HIT
- heparin-induced thrombocytopenia
- HS
- heparan sulfate
- IdoA
- α-L-iduronic
- ICU
- intensive care unit
- IL-6
- interleukin-6
- IL-8
- interleukin-8
- IL-12
- interleukin-12
- LMWH
- low molecular weight heparin
- MAPK
- mitogen-activated protein kinase
- MW
- molecular weight
- NET
- neutrophil extracellular traps
- NMR
- nuclear magnetic resonance
- OMH
- ovine mucosa heparin
- ODSH
- 2-O-, 3-O-desulfated heparin
- OSCS
- over-sulfate chondroitin sulfate
- PE
- pulmonary embolism
- PF4
- platelet factor 4
- PMH
- porcine mucosa heparin
- PPS
- pentosan polysulfate
- PCI
- protein C inhibitor
- PCV-2
- porcine circovirus
- PT
- prothrombin time
- PUL
- polysaccharide utilization locus
- RCL
- reactive center loop
- RCT
- randomized controlled trial
- TFPI
- tissue factor pathway inhibitor
- VTE
- venous thromboembolism
- UFH
- unfractionated heparin
- ULC
- ultra-large complexes
- USP
- US Pharmacopeia
- VTE
- venous thromboembolism
- ZIKV
- Zika virus
- Copyright © 2023 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
{kind=link}
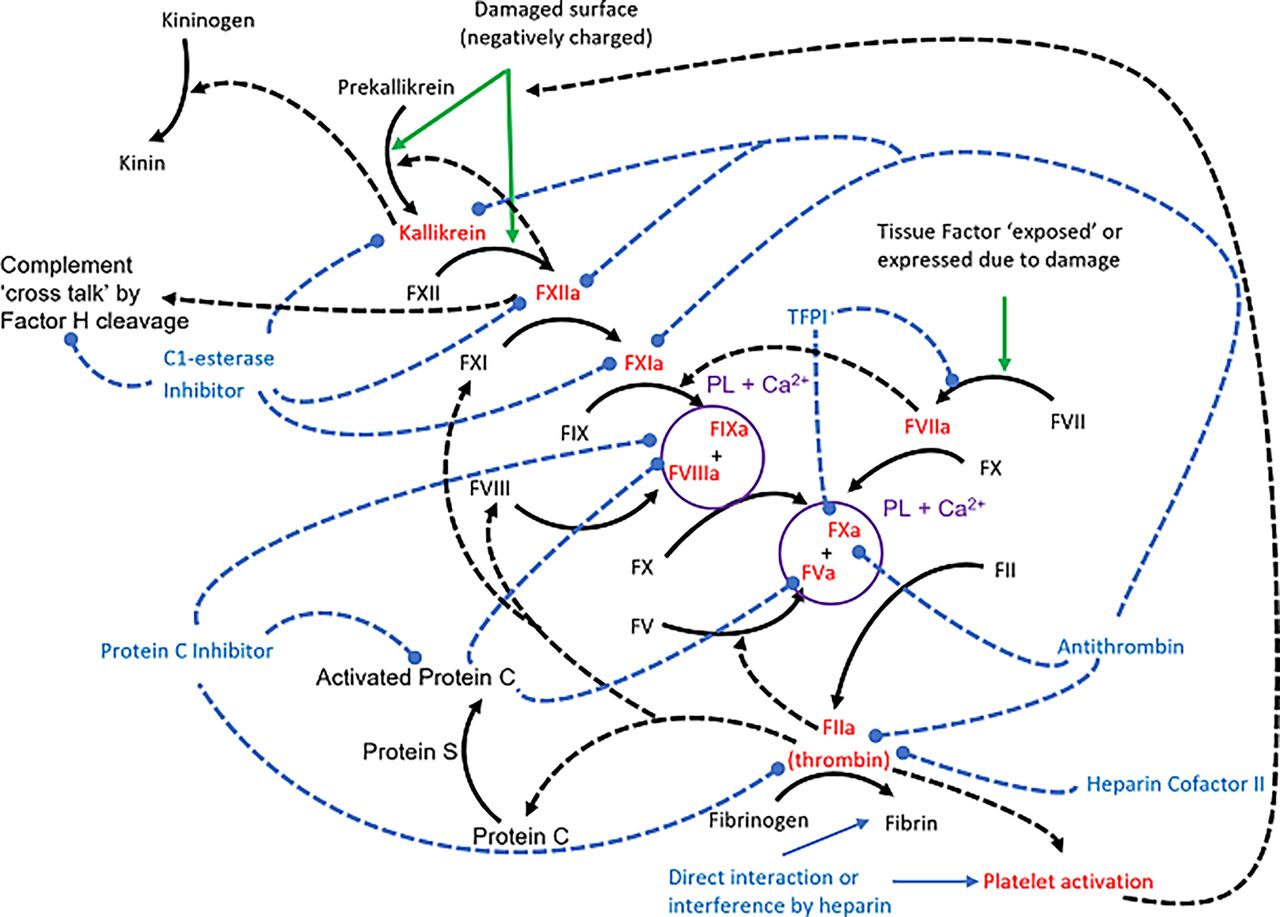{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Structural Aspects of Heparin and Related Drugs
- III. Analysis of Heparin
- IV. Molecular Interactions of Heparin
- V. Mechanism of Anticoagulant Action
- VI. Clinical Use of Heparin as an Anticoagulant/Antithrombotic
- VII. Nonanticoagulant Effects of Heparin
- VIII. Heparin in Biomaterials and Regenerative Medicine
- IX. Novel Formulations and Drug Delivery Technology for Heparin
- X. Novel Drugs Based on the Nonanticoagulant Actions of Heparin
- XI. Summary
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters