


Abstract
Chemokine receptors comprise a large family of seven transmembrane domain G protein-coupled receptors differentially expressed in diverse cell types. Biological activities have been most clearly defined in leukocytes, where chemokines coordinate development, differentiation, anatomic distribution, trafficking, and effector functions and thereby regulate innate and adaptive immune responses. Pharmacological analysis of chemokine receptors is at an early stage of development. Disease indications have been established in human immunodeficiency virus/acquired immune deficiency syndrome and in Plasmodium vivax malaria, due to exploitation of CCR5 and Duffy, respectively, by the pathogen for cell entry. Additional indications are emerging among inflammatory and immunologically mediated diseases, but selection of targets in this area still remains somewhat speculative. Small molecule antagonists with nanomolar affinity have been reported for 7 of the 18 known chemokine receptors but have not yet been studied in clinical trials. Virally encoded chemokine receptors, as well as chemokine agonists and antagonists, and chemokine scavengers have been identified in medically important poxviruses and herpesviruses, again underscoring the importance of the chemokine system in microbial pathogenesis and possibly identifying specific strategies for modulating chemokine action therapeutically. The purpose of this review is to update current concepts of the biology and pharmacology of the chemokine system, to summarize key information about each chemokine receptor, and to describe a widely accepted receptor nomenclature system, ratified by the International Union of Pharmacology, that is facilitating clear communication in this area.
I. Overview
The aim of this article is to describe the nomenclature system for chemokine receptors, as approved by the Nomenclature Committee of the International Union of Pharmacology (NC-IUPHAR),2and to update their main molecular and biological properties. A general overview is given first, followed by a synopsis of key information about each receptor, with an emphasis on recent discoveries and new concepts.
Chemokine receptors are defined by their ability to signal on binding one or more members of the chemokine superfamily of chemotactic cytokines (Premack and Schall, 1996; Baggiolini et al., 1997; Yoshie et al., 1997; Luster, 1998; Zlotnik et al., 1999). To date, 18 human proteins have met this definition, and they have been designated CXCR1 through 5, CCR1 through 11, XCR1, and CX3CR1 based on their specific chemokine preferences, as described in subsequent sections. Together, chemokine receptors comprise a large branch of the rhodopsin family of cell surface, seven-transmembrane domain (7TMD), G protein-coupled receptors (GPCRs). In addition, D6 and Duffy (sometimes called the Duffy antigen receptor for chemokines, or DARC) are 7TMD mammalian chemokine-binding proteins that apparently do not signal and therefore are excluded from the systematic nomenclature (Horuk, 1994; Nibbs et al., 1997a).
To date, chemokine receptor-like sequences have been identified in mammals, birds (Gupta et al., 1998a), and fish (Daniels et al., 1999) but not in invertebrates, plants, yeast, or bacteria, suggesting a relatively recent origin. Common features include conserved structure [25–80% amino acid (aa) identity], coupling to the Gi class of G proteins, expression in leukocytes, and chemotactic signaling. The major shared biological function is leukocyte trafficking and dependent processes such as immune surveillance, innate and adaptive immune responses, and various forms of pathological inflammation (Springer, 1994; Foxman et al., 1997). Within this general area, however, each chemokine receptor appears to have a specific role, determined by its expression pattern on specific subsets of leukocytes, and by the temporal and spatial specificity of cognate ligand expression. Specific roles have also been delineated in hematopoiesis (Broxmeyer et al., 1996, 1999; Reid et al., 1999), angiogenesis (Salcedo et al., 1999), development (Forster et al., 1996;Nagasawa et al., 1996; Ma et al., 1998; Tachibana et al., 1998; Zou et al., 1998), and, counterintuitively, facilitation of certain infectious diseases.
With regard to the latter, two major themes have been defined. In the first, cellular chemokine receptors are exploited as cell entry and disease transmission factors by intracellular pathogens. Rigorously proved examples of this are the human immunodeficiency virus (HIV) coreceptor CCR5 in acquired immune deficiency syndrome (AIDS) and Duffy in the form of malaria caused by Plasmodium vivax; CXCR4 and other chemokine receptors also function as HIV coreceptors, but their importance in disease is not established (Horuk et al., 1994; Rucker et al., 1997; Berger et al., 1999). The second theme, which is not as well understood, involves herpesvirus- and poxvirus-encoded chemokines and chemokine receptors, evidently acquired as copied host genes, which may subvert the immune response or dysregulate cell growth (reviewed inPease and Murphy, 1998).
Apart from Duffy in malaria and CCR5 in HIV/AIDS, disease indications have not yet been unequivocally established for chemokine receptors. Rapid progress in this area can be anticipated in the near future as receptor knockout mice and receptor-blocking agents are tested in animal models of disease. To date, only CXCR4 has been shown to be essential for life. Phenotypes of knockout mice for other chemokine receptors are more subtle in the absence of specific stresses (Gerard, 1999). Many types of chemokine and chemokine receptor-blocking agents of high and low selectivity have been discovered, including viral chemokine scavengers, viral chemokine antagonists, antagonistic chemokine variants, small molecules, ribozymes, intrakines, and monoclonal antibodies (mAbs) (Schwarz and Wells, 1999). Moreover, a novel HIV vaccine has been discovered in which CCR5 is a critical component (Lacasse et al., 1999). However, as of this writing, none of these has been tested in a clinical trial.
II. Introduction
A. Historical Background
To understand chemokine receptors, first their ligands must be explained. Chemokines are perhaps the most complex of GPCR ligands because of their large number, overlapping receptor specificity, and extensive phylogenetic divergence. To date, more than 40 different human chemokines have been identified, with the first identified in 1977, when Walz et al. (1977) sequenced native platelet factor 4, a procoagulant and angiostatic factor stored in platelet α-granules. Subsequently, from 1984 through 1989, cDNAs for structurally related proteins, including IP-10 (see Table 1 for chemokine acronyms; synonyms and chemokine classes are given in Table 2), JE, Mig, RANTES (regulated on activation, normal T cell expressed and secreted), I-309, KC, and macrophage inflammatory protein-1α (MIP-1α), were cloned by investigators looking mainly for cell differentiation- and activation-associated genes, establishing the existence of a gene family before identifying any functions (Wolpe and Cerami, 1989;Schall, 1991; Oppenheim et al., 1991).
Chemokine acronyms
The chemokine family
The discovery of the neutrophil-targeted chemokine interleukin (IL)-8 represents a landmark in immunology because it was the first leukocyte subtype-selective chemoattractant to be found (Yoshimura et al., 1987;Walz et al., 1987). The discovery of IL-8 also focused the search for functions for other chemokines on leukocyte chemotaxis and stimulated a search for new family members. Interest in the field grew with subsequent reports of MCP-1, RANTES, and eotaxin, the first important monocyte-, T cell-, and eosinophil-directed chemokines, respectively (Matsushima et al., 1989; Yoshimura et al., 1989; Schall et al., 1990;Jose et al., 1994). Methods of chemokine discovery expanded to include purification of chemoattractant activity as well as cDNA cloning by signal sequence trapping, homology hybridization, and, most recently, bioinformatics and expressed sequence tag (EST) analysis (Tashiro et al., 1993, 1999; Wells and Peitsch, 1997). Chemokines are particularly easy to find in EST databases because their coding sequences are sufficiently small, typically 70 to 90 codons, to be captured by a single EST and because their conserved sequence motifs are easy to recognize (see later). As the number of family members expanded, various short-lived collective terms for them were used, including “the platelet factor-4 family” (Wolpe and Cerami, 1989), “the small inducible cytokine family” (Schall, 1991), and “the intercrines” (Oppenheim et al., 1991). Finally, in 1992 at the Third International Symposium on Chemotactic Cytokines in Baden, the term “chemokine,” a neologism short for “chemotactic cytokines,” was coined and accepted as the standard (Lindley et al., 1993).
With respect to leukocyte specificity, both broad- and narrow-spectrum chemokines have been identified. Together they cover the full spectrum of leukocytes, acting through a signaling pathway that includes a pertussis toxin-sensitive G protein (Gi/Go), calcium flux, and chemotaxis. This fact pointed to use of GPCRs and suggested homology hybridization as a strategy to identify them (reviewed in Murphy, 1996), which has been highly successful.
In 1995, an NC-IUPHAR subcommittee on chemokine receptor nomenclature was organized. Recommendations developed at the second Gordon Conference on Chemotactic Cytokines held in Plymouth, NH, in 1996, were accepted unanimously by meeting participants, ratified by NC-IUPHAR in January 1997, and widely used since. The nomenclature is based on the subclassification of the chemokine superfamily, delineated in the next section. In 1998, a second nomenclature committee, led by O. Yoshie and A. Zlotnik, was formed to address the proliferation of chemokine aliases that has accompanied the codiscovery of chemokines by multiple groups using bioinformatics (Table 1). A nomenclature system that parallels the receptor nomenclature was proposed at the Keystone Symposium on Chemokine and Chemokine Receptors, January 18 to 23, 1999, in Keystone, CO (Table2).
B. Chemokine Classification
Chemokines can be subclassified by structure according to the number and spacing of conserved cysteines into four major groups, given the preferred names CXC, CC, C, and CX3C, which are used in the systematic nomenclatures (Tables 2 and3). Less commonly these groups are referred to by the Greek letters α, β, γ, and δ, respectively. CXC, CC, and CX3C chemokines all have four conserved cysteines, whereas C chemokines have only two, corresponding to the second and fourth cysteines in the other groups (Fig. 1). A small subgroup of CC chemokines has six cysteines. CXC and CX3C chemokines are distinguished by the presence of one (CXC) or three (CX3C) aa between the first and second cysteines, whereas the first two cysteines of CC chemokines are adjacent. Both the CC and CXC groups have many known members, whereas human lymphotactin α and β (Kelner et al., 1994) and fractalkine (Bazan et al., 1997) and their equivalents in other species are the only known examples of C and CX3C chemokines, respectively. A cDNA encoding a CXC chemokine-like protein has been discovered in lamprey, suggesting that the origin of the family, and possibly the division into subclasses, is ancient (Najakshin et al., 1999).
Chemokine receptors: nomenclature, pharmacology and biology
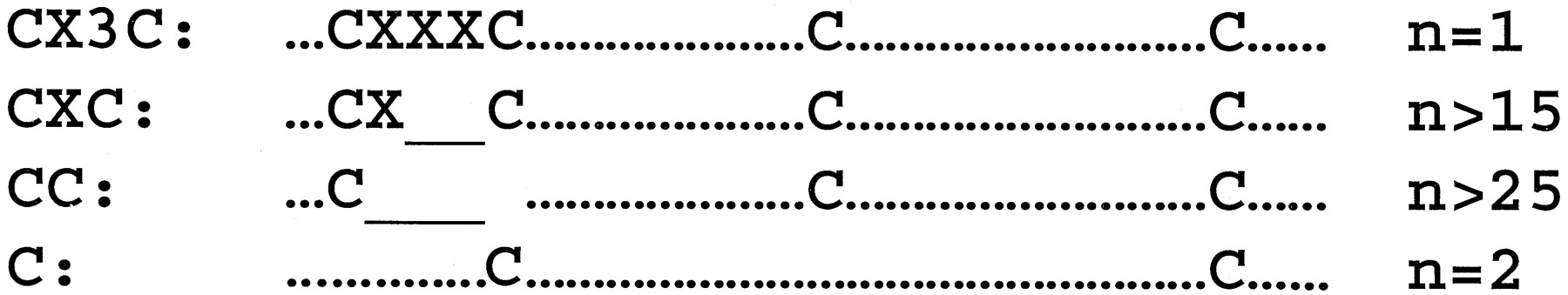
Structural classification of the chemokine family by signature cysteines. The number of members in each subclass is listed at the right of each structure. Underlines indicate gaps in the alignment; X, an amino acid other than cysteine; and dots, other amino acids. Spacing between cysteines is similar in all four groups. The N and C termini can vary in length.
Fractalkine is an interesting model for how chemokines may be presented to target cells. It has a multimodular structure consisting of a chemokine domain fused to a mucin-like stalk plus a transmembrane domain, which anchors the molecule to the plasma membrane, and a cytoplasmic domain. Consistent with this, it functions as an adhesion molecule by binding directly to CX3CR1 (Imai et al., 1997b). Fractalkine also induces cell migration as either a tethered or shed ligand. Although other chemokines lack a transmembrane domain and are secreted, they are able to use glycosaminoglycans for tethering to plasma membrane. This provides a mechanism for gradient formation under conditions of high blood flow. Once “posted” in this manner, chemokines may be “read” by passing leukocytes, which then activate β2-integrins, bind to endothelium and transmigrate from blood to tissue (Tanaka et al., 1993). However, fractalkine is the only chemokine shown to act as a direct cell adhesion molecule.
CXC chemokines can be further subclassified by structure into ELR+ and ELR− molecules based on the presence or absence of the tripeptide motif glutamic acid-leucine-arginine (ELR) N-terminal to the first cysteine. This provides the only strong functional correlate of the structural classification: the specificity of ELR+ CXC chemokines for neutrophils (Hebert et al., 1991). A second classification scheme based on function and expression pattern has also been proposed. It includes an inflammatory/inducible group, which is regulated by proinflammatory stimuli such as lipopolysaccharide and primary cytokines such as IL-1 and tumor necrosis factor, and which together orchestrate innate and adaptive immune responses; a homeostatic/constitutive group, which is important in lymphocyte and dendritic cell trafficking in immune surveillance (Cyster, 1999a,b); and an overlap group. Genes encoding inflammatory chemokines are typically found in two major clusters on human chromosomes 4 (CXC) and 17 (CC), whereas genes for homeostatic chemokines are located alone or in small clusters on chromosomes 1, 2, 5, 7, 9, 10, and 16. Homeostatic receptors include CXCR4, CXCR5, CCR4, CCR7, and CCR9. Inflammatory receptors include CXCR1, CXCR2, CXCR3, CCR1, CCR2, CCR3, CCR5, and CCR6.
C. Chemokine Receptor Classification and Nomenclature
The classification of chemokine receptors is restricted to those defined at the molecular level. Native receptors are more difficult to study specifically because few selective agonists and antagonists are available and because multiple receptor subtypes with overlapping ligand specificities may be expressed in the same cell.
Although most chemokine receptors recognize more than one chemokine, they are almost always restricted to a single subclass (Table4). Thus, the nomenclature system is rooted by the chemokine subclass specificity of the receptor. Human CC and CXC chemokine receptor names consist of the root CCR or CXCR, respectively, followed by a number. The lymphotactin and fractalkine receptors are named XCR1 [“X” to distinguish it from complement receptor 1 (CR1)] and CX3CR1, respectively. The use of the letter “R” in receptor names is nonstandard for pharmacologists but is widely accepted practice for immunologists and was therefore authorized as an exception by NC-IUPHAR. Thus, these receptors are referred to as, for example, “CXCR1,” and not “the CXCR1 receptor,” which would be redundant. Splice variants, if pharmacologically distinct, are designated by a lowercase letter starting from the beginning of the alphabet subscripted in parentheses after the receptor name. Species orthologs are indicated by an appropriate species abbreviation followed by a space before the receptor name (Vanhoutte et al., 1998). By consensus agreement of the conferees at the 1996 Gordon Conference on Chemotactic Cytokines, new names are assigned by a committee composed of Phil Murphy (pmm{at}nih.gov), Craig Gerard (gerard_c{at}gonzo.tch.harvard.edu), and Tom Schall (tschall{at}chemocentryx.com).
Ligand and leukocyte specificities for human chemokine receptors
Like the receptor names, systematic chemokine names, shown in Table 2with their corresponding common names, are also built from cysteine subclass roots, followed by “L” for “ligand” and a number. In general, the numbers correspond to the same number used in the corresponding gene nomenclature, which takes the form “SCY” for “small cytokine,” followed by “A”, “B”, “C,” or “D” for “CC”, “CXC”, “C,” or “CX3C” subclass, respectively, followed by the number.
Analysis of chemokine receptors presents problems not faced with other types of GPCRs. Most imposing is the large number of receptors and endogenous ligands and their overlapping specificities for each other and for leukocyte subtypes. In addition, both chemokines and their receptors may vary markedly in sequence among species, as much as 55% aa divergence in the case of certain chemokines. As a result, even though chemokine orthologs from different species usually cross-activate receptors, the receptors may have markedly different biology and pharmacology. Even the repertoire of chemokine and chemokine receptors may differ in different species. For example, mouse orthologs of IL-8 and CXCR1 have not been found, and there is persuasive evidence in the case of IL-8 that a mouse form does not exist (Modi and Yoshimura, 1999). Why these molecules are evolving so rapidly is unknown, but it is a property shared fairly selectively with the class of genes involved in immunity and inflammation (Murphy, 1993).
D. Chemokine Receptor Structure
The sequences of chemokine receptors have 25 to 80% aa identity (Fig. 2), indicating a common ancestor. However, many other G protein-coupled peptide receptors also have ∼25% aa identity to chemokine receptors, illustrating that the structural boundary is not sharp. Although they lack a single structural signature, there are several features that together are found more frequently among chemokine receptors than other types of GPCRs. These include a length of 340 to 370 aa; an acidic N-terminal segment; the sequence DRYLAIVHA, or a variation of it, in the second intracellular loop; a short basic third intracellular loop; and a cysteine in each of the four extracellular domains. A tyrosine sulfation motif is commonly found in the N terminus of chemokine receptors and has been shown to be critical for HIV coreceptor activity for CCR5 (Farzan et al., 1999).

Structural relationships among human chemokine receptors. The dendrogram was constructed by Marc Rothenberg using default parameters in the PILEUP algorithm of the University of Wisconsin Genetics Computer Group.
The three-dimensional structure of chemokine receptors is unknown, but a reasonable working model can be constructed for the transmembrane domains based on analogy with rhodopsin (Baldwin, 1993; Unger et al., 1997; Lomize et al., 1999). Models of the extracellular and intracellular domains are completely speculative, although in some cases domain-specific antibodies have verified the general location. Evidence has been reported that CCR2, CCR5, and CXCR4 form homodimers (Benkirane et al., 1997; Lapham et al., 1999; Rodriguez-Frade et al., 1999), and in the case of CCR2, a dimer has been implicated as the functional form of the receptor, which may be needed for signaling.
In contrast, many chemokine structures have been determined, including both CC and CXC subtypes, and a common fold is apparent (Clark-Lewis et al., 1995; Clore and Gronenborn, 1995). The N terminus before the first cysteine is structurally disordered, whereas the C terminus after the last cysteine is α-helical. The remainder of the molecule is constrained by disulfide bonding between the first and third and the second and fourth cysteines and contains three β-sheets separated by short loops arranged in the shape of a Greek key. The backbone structures are largely superimposable. Chemokines appear to act as monomers, despite the fact that in most cases they are dimers or higher-order multimers at high concentrations or in crystals (Baggiolini et al., 1997).
The N terminus is not usually important for high-affinity receptor binding but is typically critical for receptor triggering. Native chemokines purified from biological material often exist as families of peptides derived from the same gene that differ in the length of the N- and C-terminal domains, which in some cases has been attributed to the action of specific proteases such as CD26 (a prolylpeptidase) and cathepsin G (Walz and Baggiolini, 1990; Oravecz et al., 1997). The length of the N-terminal segment is important in determing whether a given chemokine binds to receptor, and if so, whether it functions as an agonist or antagonist. Truncation may also cause a switch in receptor specificity as in the cases of NAP-2 and MCP-1.
E. Chemokine Receptor Specificity for Ligands and Leukocytes
Each chemokine receptor has a distinct chemokine and leukocyte specificity (Table 4), but the specificities can overlap considerably, because some chemokines can bind multiple receptor subtypes, and some receptors can bind multiple chemokines. Mutagenesis has indicated that the ligand binding site of chemokine receptors is highly complex, being composed of multiple noncontiguous domains and at least two distinct subsites: one for docking and the other for triggering (Ahuja et al., 1996; Monteclaro and Charo, 1996; Crump et al., 1997). At least for CCR5 and CXCR4, the first two TMDs and associated loops, but not the N-terminal segment, appear to be dispensable for normal receptor expression and function (Ling et al., 1999). Multiple low-affinity interactions together provide the high-affinity binding energy. A conserved HIV gp120 glycoprotein structure has been solved that is involved in chemokine receptor binding (Rizzuto et al., 1998).
Inflammatory chemokines (mainly those encoded by the chromosome 4 and 17 clusters of genes) have highly promiscuous relationships with receptors. There are fewer homeostatic chemokines, but those that map to the same chromosome tend to bind to the same receptor [e.g., MDC and TARC at CCR4; EBI ligand chemokine (ELC) and secondary lymphoid tissue chemokine (SLC) at CCR7]. Recently, the number of monogamous chemokine ligand-receptor relationships, which had previously been regarded as exceptional, has risen substantially (e.g., SDF-1 and CXCR4; TECK and CCR9; BLC and CXCR5; LARC and CCR6; lymphotactin and XCR1; fractalkine and CX3CR1).
Adding to the complexity of the system, distinct receptor subtypes specific for the same chemokine and the same function can be coexpressed on the same cell (Morohashi et al., 1995), distinct chemokines acting at separate receptors coexpressed on the same cell can induce the same cellular response (Zaitseva et al., 1997), and the same receptor can sort signals from different ligands to distinct signaling pathways (Zhang et al., 1999). Also, chemokine receptors are not limited to leukocytes but in specific cases may also be expressed on endothelial cells, neurons, epithelial cells, and microglial cells of the brain (Hadley et al., 1994; He et al., 1997; Horuk et al., 1997;Gupta et al., 1998b; Salcedo et al., 1999). There is intense interest in understanding the biological roles of these receptors in these ectopic sites.
With rare exceptions (Blanpain et al., 1999), functional human chemokines are agonists at leukocyte receptors. In contrast, naturally occurring chemokine antagonists have only been found in viruses (Table5). For example, the viral chemokines MC148R from the poxvirus Molluscum contagiosum virus (Damon et al., 1998) and vMIP-II from human herpesvirus 8 (HHV8) (Kledal et al., 1997) are broad-spectrum chemokine receptor antagonists, suggesting roles in immune evasion and the importance of normal chemokine signaling for antiviral host defense. Various orthopoxviruses (e.g., myxoma, vaccinia) deploy an alternative strategy to block chemokines, through two structurally unique classes of secreted, broadly specific chemokine scavengers, one of which also binds interferon-γ (Graham et al., 1997; Smith et al., 1997; Alcami et al., 1998). Neither has structural homology to chemokines, chemokine receptors, or any other proteins currently in the public databases. They may be good leads for development of novel anti-inflammatory agents, particularly for topical or single-use administration (Table5).
Viral chemokines and chemokine receptors
Recently, a growing number of structurally diverse, naturally occurring, nonchemokine ligands for chemokine receptors has been identified. These include HIV tat at CCR2 (Albini et al., 1998) and CXCR4 (Xiao et al., submitted), HIV gp120 at various HIV coreceptors (Berger et al., 1999), a secreted domain of tyrosyl tRNA synthetase at CXCR1 (Wakasugi et al., 1999), and the human β-defensin HBD2 at CCR6 (Yang et al., 1999).
A major new concept to emerge recently from studies of the leukocyte selectivities of chemokines is that interaction between antigen-loaded dendritic cells and antigen-specific T cells to achieve proper cell positioning in the periphery or in secondary lymphoid tissue for an adaptive immune response is not random but instead results in part from dynamic and coordinated changes in chemokine receptor expression. Moreover, the nature and strength of the immune response may be governed in part by specific chemokine receptor expression patterns. Thus, T lymphocytes and dendritic cells undergo highly dynamic regulation of chemokine receptors depending on whether the T cell is naı̈ve or memory, Th1 or Th2, and resting or activated, and whether the dendritic cell is immature or mature (Sallusto et al., 1999a; Sozzani et al., 1999). For example, when naive T lymphocytes are activated and differentiate into memory/effector cells, they down-regulate receptors for constitutive chemokines such as CXCR4 and CCR7 and acquire receptors for inflammatory chemokines such as CCR3, CCR5, and CXCR3. Also, dendritic cell maturation after antigen loading is accompanied by a transition from expression of inflammatory to homeostatic chemokine receptors. Distinct selectivities for Th1 and Th2 polarized T lymphocytes have been reported for CC chemokine receptors, and actual chemokine receptor markers of these cell types have been claimed and debated (Sallusto et al., 1998; Annunziato et al., 1999). Moreover, homing of memory T cells to specific anatomic sites has been strongly correlated with specific chemokine receptor expression patterns (Campbell et al., 1999).
With this as a general introduction, the next sections are discussions of the molecular pharmacology and biology of individual chemokine receptor subtypes. Note that the voluminous literature correlating the presence of specific chemokines in disease has been extensively reviewed (Baggiolini et al., 1997) and is not repeated here. Instead, we emphasize direct tests of function of specific receptors in disease.
III. CXC Chemokine Receptor Subtypes
A. CXCR1 and CXCR2
CXCR1 and CXCR2 were the first chemokine receptor subtypes to be defined. They are the only known mammalian receptors for ELR+ CXC chemokines, including IL-8, which binds to both receptors with similar high affinity; they do not bind other types of chemokines. They are also the major chemokine receptors expressed on neutrophils and are prototypic receptors for inflammatory/inducible chemokines. They appear to operate mainly in acute inflammation and innate immunity although a role in macrophage accumulation in atherosclerotic plaque has also been demonstrated for CXCR2 (Boisvert et al., 1998). They are considered together because of these shared properties.
CXCR1 cDNA was first cloned from rabbit neutrophils by homology hybridization using a probe based on conserved sequences in TMD 2 of known neuropeptide-specific GPCRs (Thomas et al., 1990). When expressed in frog oocytes, it appeared to be specific for formyl-methionyl-leucyl-phenylalanine, but this could not be reproduced in mammalian cells where IL-8 was a functional ligand (Thomas et al., 1991). Consistent with this, human CXCR1 cDNA was isolated independently from a neutrophil library by expression cloning using an125I-IL-8 binding assay in COS-7 cells (Holmes et al., 1991). CXCR2 cDNA was first cloned by homology hybridization from a dibutyryl cAMP-induced HL-60 cell library using an oligonucleotide probe corresponding to TMD2 of rabbit CXCR1 (Murphy and Tiffany, 1991); later, cDNAs were also isolated from a neutrophil library (Lee et al., 1992). The genes, designated il8ra and il8rb, are located 20 kb apart on human chromosome 2q35, and there is a linked pseudogene of CXCR2 named il8rp (Ahuja et al., 1992; White et al., 1994). The open reading frames (ORFs), which each occupy a single exon, are 350 codons for CXCR1 and either 355 or 360 codons for CXCR2 (both of two in-frame ATG codons are flanked by favorable Kozak sequences). CXCR1 and CXCR2 are 78% identical in aa sequence.
In addition to neutrophils and monocyte/macrophages, CXCR1 and CXCR2 have been detected on cytokine-activated eosinophils, basophils, T lymphocytes, mast cells, and dendritic cells, but important functional roles in vivo have not been clearly demonstrated (Chuntharapai et al., 1994; Hammond et al., 1995; Morohashi et al., 1995; Xu et al., 1995;Heath et al., 1997; Sozzani et al., 1997; Nilsson et al., 1999;Ochensberger et al., 1999; Petering et al., 1999). CXCR2, but not CXCR1, has been identified on brain Purkinje cells by mAb and radioligand binding, but function remains undefined there as well (Horuk et al., 1997).
In calcium flux and chemotaxis assays, CXCR2 is relatively nonselective for IL-8 versus all other ELR+ CXC chemokines studied (<10-fold range in EC50), whereas CXCR1 is highly selective for IL-8 (>50-fold difference in EC50) (Lee et al., 1992; Loetscher et al., 1994; Ahuja and Murphy, 1996); GCP-2 is an equipotent agonist at both CXCR1 and CXCR2 (Wuyts et al., 1997). Thus, GROα, NAP-2, and ENA-78 are selective ligands for CXCR2. Recently, a selective nonchemokine endogenous ligand was identified for CXCR1: the N-terminal cytokine module of human tyrosyl tRNA synthetase, which contains an ELR motif and functions as a neutrophil chemoattractant in vitro. Its biological function is not established but could involve inflammatory signaling by apoptotic cells (Wakasugi and Schimmel, 1999). Consistent with coexpression of CXCR1 and CXCR2 on neutrophils, IL-8 effectively blocks binding of other ELR+ CXC radioligands to human neutrophils and interferes with signaling (calcium flux), but conversely, other ELR+ CXC chemokines can only partially block IL-8 binding to neutrophils and subsequent calcium flux (Moser et al., 1991;Ahuja and Murphy, 1996). The receptors appear to function independently.
Antagonists at CXCR2 include N-terminal truncations of IL-8 and GROα (Hesselgesser et al., 1995), selective neutralizing monoclonal and polyclonal antibodies (Hammond et al., 1995; Green et al., 1996; Jones et al., 1996), a small peptide of undefined selectivity (Hayashi et al., 1995), and SB 225002 [N-(2-hydroxy-4-nitrophenyl)-N′-(2-bromophenyl)urea], a selective small molecule, nonpeptide inhibitor of CXCR2 (White et al., 1998) (Fig. 3). The latter is a potent antagonist of 125I-IL-8 binding with an IC50 value of 22 nM and has >150-fold selectivity over CXCR1. In vitro, SB 225002 potently inhibits human and rabbit neutrophil chemotaxis induced by both IL-8 and GROα, and in vivo it selectively blocks IL-8-induced neutrophil margination in rabbits.

Structures of nonpeptide small molecule antagonists of specific chemokine receptors. Note that a basic nitrogen is a common feature. (Figure courtesy of Kurt Jarnagin.)
In vivo roles of IL-8 and related ligands have been extensively studied, but specific receptor roles are less well defined. The mouse chemokines MIP-2 and KC are human GRO homologs specific for mouse CXCR2 (Bozic et al., 1994; Lee et al., 1995). CXCR2 knockout mice fail to mobilize neutrophils to chemically irritated peritoneum in vivo, and −/− neutrophils do not migrate in vitro in response to KC or MIP-2, indicating that CXCR2 is the dominant neutrophil receptor for these chemokines (Cacalano et al., 1994).
Unexpectedly, CXCR2 −/− mice have massive expansion of neutrophils and B cells throughout the hematopoietic system when derived in specific pathogen-free conditions but not in germ-free conditions (Moore et al., 1995). The explanation may reside in part in the fact that CXCR2 is a negative regulator of hematopoiesis (Broxmeyer et al., 1996). Alternatively, Cacalano et al. (1994) speculated that the inability to properly survey tissues and eliminate external pathogens in the knockouts may result in the release of cytokines that stimulate neutrophil and B cell production. However, the animals have not been reported to have increased susceptibility to infectious disease from either environmental or challenge pathogens.
The defect in neutrophil-mediated inflammation in these mice is consistent with the effects of CXCR2 ligand neutralization in mouse (KC, MIP-2) and rabbit (IL-8) in diverse models of acute inflammation (skin, airway, pleura, glomeruli) (Sekido et al., 1993; e.g., Broaddus et al., 1994). These results suggest indications for IL-8 receptor antagonists in diseases such as psoriasis, coronary artery reperfusion injury, and acute glomerulonephritis. Still, it is important to point out that rodents are poor models of the human IL-8 signaling system: they lack IL-8, a mouse counterpart of CXCR1 has not been identified, and rat CXCR1 is expressed in macrophages not neutrophils (Dunstan et al., 1996). Nevertheless, IL-8 receptor function in the monocyte/macrophage lineage may be more important than was initially appreciated. In particular, IL-8 can trigger firm adhesion of human monocytes to vascular endothelium under flow conditions (Gerszten et al., 1999), and CXCR2 is critical for macrophage accumulation in atherosclerotic lesions of LDL receptor-deficient mice (Boisvert et al., 1998).
CXCR1 and CXCR2 have been reported to carry out different functional roles in human neutrophils in vitro. CXCR1 appears to be dominant for chemotaxis, superoxide production, and phospholipase D activation in response to IL-8 (Hammond et al., 1995; Jones et al., 1996), as well as for chemotaxis to NAP-2 at high concentrations (>1 μM) (Ludwig et al., 1997), whereas CXCR2 appears to mediate neutrophil chemotaxis to NAP-2 (and GROα) at low concentrations. Calcium flux and degranulation are mediated through both receptors. However, cell migration may be more important than cell activation for IL-8 receptor function in vivo, as suggested by the accumulation of unactivated neutrophils and the lack of inflammatory pathology at sites of KC transgene expression in mice; this may be a general property of chemokines (Lira et al., 1994).
Despite abundant evidence that IL-8 is important in acute inflammation, proof of concept is still lacking for differential roles of CXCR1 versus CXCR2 in vivo and in human disease. As suggested earlier, major obstacles include the inadequacy of small animal models and the lack of adequate selective small molecule antagonists. Other major unanswered questions about these receptors include their structure, the relative roles of CXCR1 and CXCR2 in ELR+ CXC chemokine-induced angiogenesis and modulation of myelopoiesis (Broxmeyer et al., 1997), and the putative function of CXCR2 in brain (Horuk et al., 1997).
Two functional viral homologs of CXCR2 have been identified, ECRF3 of Herpesvirus saimiri (Ahuja et al., 1993) and KSHV GPCR of KSHV (HHV8) (Arvanitakis et al., 1997), which are quite different from CXCR2 and are reviewed in a later section.
B. CXCR3
CXCR3 is the first chemokine receptor identified that is highly induced by T cell activation. The ORF was first identified in incomplete form in 1995 on a genomic clone isolated by polymerase chain reaction-based homology hybridization. The gene was namedGPR9 and was originally mapped incorrectly to human chromosome 8p11.2-12 (Marchese et al., 1995) and later mapped correctly to Xq13 (Loetscher et al., 1998a). A full-length cDNA was independently isolated from an IL-2-activated T cell library (Loetscher et al., 1996). The ORF is interrupted by one intron in the region encoding the N-terminal segment and predicts a polypeptide 368 aa in length. The deduced protein sequence of human CXCR3 is ∼30% identical with CXCR1 and CXCR2.
CXCR3 binds three highly potent, inflammatory/inducible, ELR-negative CXC chemokine agonists, I-TAC, Mig, and IP-10 (Loetscher et al., 1998a;Cole et al., 1998; Weng et al., 1998), all of which chemoattract and induce calcium flux in activated T cells, tumor-infiltrating lymphocytes, and CXCR3-transfected cells. The rank order of binding affinity is I-TAC > Mig ∼ IP-10. Curiously, the human CC chemokines eotaxin and MCP-4 also bind to CXCR3-transfected cells but with much lower affinity (K i ∼ 60 nM) and without activating the receptor (Weng et al., 1998). Also, the mouse CC chemokine SLC/6Ckine has been reported to induce calcium flux through mouse CXCR3 (Soto et al., 1998), but this was not observed with human 6Ckine with either human or mouse CXCR3 (87% aa identity) (Jenh et al., 1999). A CXCR3-specific mAb named 1C6 has been reported that blocks human IP-10, but not human Mig, binding to CXCR3 (Qin et al., 1998).
CXCR3 is expressed on a portion of circulating blood T cells, B cells, and natural killer (NK) cells (Qin et al., 1998). Although freshly isolated T cells respond to Mig, curiously they are relatively less responsive to IP-10. Expression and responsiveness are both markedly increased by T cell activation (Rabin et al., 1999), classifying CXCR3 as an inflammatory/inducible type of chemokine receptor. CXCR3 has been detected preferentially on Th1 T cell lines and clones in vitro but could not discriminate between Th1- (Crohn's disease) and Th2- (systemic sclerosis) dominant responses in vivo and therefore may not be a practical marker of Th1 cells, as had been suggested (Bonecchi et al., 1998; Sallusto et al., 1998, 1999b; Annunziato et al., 1999). Blood T cells expressing CXCR3 are mostly CD45RO+ memory cells and express high levels of β1-integrins. Virtually all T cells in rheumatoid arthritis synovial fluid and in various inflamed tissues, such as in ulcerative colitis, chronic vaginitis, and sarcoidosis, express CXCR3, particularly in perivascular regions, whereas fewer T cells within normal lymph nodes are positive (Agostini et al., 1998;Qin et al., 1998). CXCR3 is also consistently detected in functional form on transformed B cells from CLL patients (Trentin et al., 1999).
The biological role of CXCR3 is not yet known, and it has not been established as a disease target, although a role in Th1 dominant diseases have been anticipated. Antagonists and gene knockouts have not been reported. In addition to T cell chemotaxis, CXCR3 ligands are angiostatic factors in vivo, but mechanisms are not defined.
C. CXCR4
CXCR4 is the first chemokine receptor shown to be an HIV-1 coreceptor (Feng et al., 1996) and the only one shown to be essential for life, at least in mice (Ma et al., 1998; Tachibana et al., 1998;Zou et al., 1998). Four groups identified it based on “orphan receptor” cloning strategies, whereas Feng et al. (1996) rediscovered the cDNA by expression cloning of its HIV-1 coreceptor activity and named the protein “fusin.” Specificity for the homeostatic CXC chemokine SDF-1 was established shortly thereafter (Bleul et al., 1996;Oberlin et al., 1996), and fusin was renamed CXCR4.
The ORF is interrupted by one intron in the region encoding the N-terminal segment and predicts a polypeptide 352 aa in length. A splice variant of unclear significance has been found, which affects the length of the N-terminal portion of the molecule upstream of TMD1, but not affinity for ligand (Heesen et al., 1997; Frodl et al., 1998;Gupta and Pillarisetti, 1999).
CXCR4 is unusually widely expressed on most hematopoietic cell types, including neutrophils, monocytes, T lymphocytes, B cells, B cell precursors, CD34+ progenitor cells from blood and bone marrow, blood-derived dendritic cells, Langerhans cells, T cells and macrophages, and both immature and mature T cells in thymus (Bleul et al., 1997; Zaitseva et al., 1997, 1998). It is also expressed at high levels on vascular endothelial cells (Gupta et al., 1998b), neurons from both the central and peripheral nervous systems (Hesselgesser et al., 1997), and microglia and astrocytes (He et al., 1997). In blood-derived T cells, CXCR4 is preferentially expressed on the naive, unactivated CD26low CD45RA+ CD45R0− subset (Bleul et al., 1997), and expression is rapidly up-regulated by phytohemagglutinin and IL-2 (Loetscher et al., 1996) and down-regulated by SDF-1 (Amara et al., 1997).
CXCR4 has also been implicated in platelet formation. Although there is agreement over whether it is expressed throughout platelet development, there is some disagreement about its function (Power et al., 1995a;Hamada et al., 1998; Wang et al., 1998; Kowalska et al., 1999). SDF-1-induced transendothelial migration by mature marrow megakaryocytes and megakaryocyte progenitors has been reported by at least one group but not consistently confirmed. The receptor is on mature platelets but appears to be functionally uncoupled.
The SDF-1 gene is alternately spliced to form SDF-1α and SDF-1β, which differ by a 4-aa extension at the C terminus (Shirozu et al., 1995). These variants, originally isolated from bone marrow stromal cells, are functionally indistinguishable and are the only known endogenous ligands and agonists for CXCR4, inducing calcium flux and chemotaxis in transfected and primary cells in vitro. Genetic disruption of SDF-1 and CXCR4 in the mouse gives the same phenotype (Nagasawa et al., 1996; Ma et al., 1998; Tachibana et al., 1998; Zou et al., 1998), suggesting that they make up a monogamous signaling unit in vivo. The animals die in the perinatal period, the only known chemokine system components for which this is true, and have ventricular septal defects, defective gastric vasculogenesis and cerebellar development, abnormal bone marrow myelopoiesis, and defective B cell, but normal T cell, lymphopoiesis. Functions of CXCR4 in the adult are not defined. In one study, human stem cell engraftment was reported to be regulated by CXCR4 in NOD/SCID mice (Peled et al., 1999). Both SDF-1 and CXCR4 have highly conserved sequences (e.g., 98 and 94% aa identity between human and mouse, respectively), which is highly atypical for chemokines and chemokine receptors, which are among the most rapidly evolving proteins in mammals (Murphy, 1993).
HIV-1 strains able to use CXCR4 for cell entry in vitro are named X4 strains (Berger et al., 1998). They are typically isolated late in the course of infection and correlate more or less with T cell line cytotropism and the syncytium-inducing methods of classification used before the discovery of HIV-1 coreceptors (reviewed in Berger et al., 1999). The importance of CXCR4 in HIV pathogenesis has been suggested but not proved by the detection of X4 HIV in CCR5-deficient HIV-positive individuals (Michael et al., 1998), and the discovery of a single nucleotide polymorphism in the 3′-UTR of SDF-1α (SDF1-3′A) that is associated with slowed progression to AIDS (Winkler et al., 1998). Direct studies of the effect of this polymorphism on SDF-1 production in vivo have not been reported, but any effect could conceivably modulate the extent of X4 HIV interaction with CXCR4.
gp120 from HIV-1 envelope glycoprotein can bind to CXCR4 in the presence of CD4 (Lapham et al., 1999), and X4 virus entry is dependent on CD4 (Feng et al., 1996). However, CD4 independent association of gp120 to CXCR4 has also been demonstrated. Purified X4 gp120 can function as a CXCR4-dependent monocyte chemoattractant, perhaps to recruit more targets, and can induce apoptosis of the human neuronal cell line hNT (Hesselgesser et al., 1998a). Consistent with this, chemokines can regulate hippocampal neuronal signaling and gp120 neurotoxicity (Meucci et al., 1998). These findings may be relevant to the pathogenesis of HIV encephalitis and AIDS dementia. Interaction of gp120 with CXCR4 on macrophages can also induce apoptosis of CD8+ T cells, suggesting a coreceptor mechanism of CTL suppression (Herbein et al., 1998).
Several mAbs have been developed that bind CXCR4, including the prototype 12G5, which blocks HIV infection (Endres et al., 1996). Several small molecules and peptides, including some originally identified in HIV drug discovery programs, have been shown to selectively block chemokine receptor and/or HIV coreceptor activities of CXCR4. They include SDF-1 derived peptides (Loetscher et al., 1998b,Heveker et al., 1998); the synthetic peptide T22 ([Tyr5,12,Lys7]polyphemusin II), which consists of 18 aa residues and an analog of polyphemusin II isolated from the hemocyte debris of American horseshoe crabs (Limulus polyphemus) (Murakami et al., 1997); the related synthetic peptides T134 and T140 (Tamamura et al., 1998; Xu et al., 1999); the polyarginine ALX40-4C (Doranz et al., 1997); the peptoid CGP64222 (Daelemans et al., 2000); and the bicyclam AMD3100 (Schols et al., 1997; Donzella et al., 1998; Bridger et al., 1999) (Fig. 3). The distamycin analog 2,2′-[4,4′-[[aminocarbonyl]amino]bis[N,4′-di[pryrrole-2-carboxamide-1,1′-dimethyl]]-6,8-naphthalenedisulfonic acid] hexasodium salt (NSC 651016) also blocks X4 viral use of CXCR4, but it has a broad specificity for multiple other chemokine receptors (Howard et al., 1998). CXCR4 has also been blocked with intrakines, which are modified forms of SDF-1 delivered by gene therapy that remain in the endoplasmic reticulum and block surface expression of newly synthesized CXCR4 (Chen et al., 1997).
In addition, the HIV protein Tat, which has a highly basic domain but lacks a chemokine fold, can block both SDF-1-induced calcium flux at CXCR4 and X4 HIV entry of target cells (Xiao et al., submitted). The inability of Tat to affect CCR5 function suggests a possible mechanism for restriction of HIV to R5 strains early in infection but cannot explain the appearance of X4 strains late in infection during immune system collapse. Moreover, it conflicts with the reported ability of Tat to up-regulate CXCR4 and serve as a vaccine target in nonhuman primates (Gallo, 1999).
The clinical development of CXCR4 blocking agents in HIV infection will have to confront safety questions of whether the virus will evolve to use other coreceptors and whether one or more of the phenotypes seen in CXCR4 knockout mice will occur. To date, CXCR4 has not been established as a therapeutic target for other diseases.
D. CXCR5
CXCR5 was the first chemokine receptor shown to be involved in lymphocyte homing and development of normal lymphoid tissue (Forster et al., 1996) and the first B cell selective chemokine receptor (Gunn et al., 1998a; Legler et al., 1998). Two cDNAs for CXCR5 were cloned independently by two groups as orphans and named, according to the source, monocyte-derived receptor 15 (MDR15; Barella et al., 1995) and Burkitt's lymphoma receptor 1 (BLR1; Dobner et al., 1992). The ORF of MDR15 has 327 codons and is 45 codons shorter at the N terminus than BLR1 due to alternative splicing of the gene. Distinct pharmacology has not been demonstrated for the two forms. The aa sequence is ∼40% identical with CXCR1 and CXCR2.
Using a mAb directed to the N terminus of BLR1, CXCR5 has been detected on all peripheral blood and tonsillar B cells but only on a fraction of cord blood and bone marrow B cells. It is also present on a small subset of peripheral blood CD4+ (14%) and CD8+ (2%) T cells, which are also CD45R0+, IL-2R−, CD44high, andl-selectinlow, suggesting a memory phenotype. In contrast, in secondary lymphatic tissue, the majority of CD4+ cells are positive, and in cord blood, T cells are negative (Forster et al., 1994). The murine homolog of CXCR5 has been cloned, and specific transcripts found in a pattern similar to the human receptor, including expression on mature B cells and a subpopulation of T helper cells, as well as in secondary lymphatic organs and to a lesser extent in brain, specifically in the granule and Purkinje cell layer of the cerebellum (Kaiser et al., 1993). RNA in situ hybridization localizes transcripts to primary follicles and to the mantle zone of secondary follicles. Like other chemokine receptors, CXCR5 is dynamically regulated on T cells. After T cell receptor (TCR) stimulation, CXCR5 is up-regulated on memory/effector T cells, whereas IL-2 causes down-regulation (Sallusto et al., 1999b). Up-regulation of CXCR5 on antigen-activated T cells implies a role for movement of Th cells to B cell follicles (Ansel et al., 1999).
To date, B cell-attracting chemokine 1 (BCA-1, also known as BLC) is the only known agonist for CXCR5 (Gunn et al., 1998a; Legler et al., 1998). Conversely, CXCR5 is the only known receptor for BCA-1. Signaling includes chemotaxis and Ca2+mobilization. BCA-1, a member of the homeostatic class of chemokines, is B cell selective and constitutively expressed in secondary lymphoid organs. It has weak effects on small numbers of T cells and macrophages. Consistent with this, CXCR5 knockout mice have a severe defect in normal B cell migration and localization (Forster et al., 1996). The animals lack inguinal lymph nodes, have few Peyer's patches, and have abnormal primary lymphoid follicles and no functional germinal centers in spleen. Nevertheless, immunoglobulin levels are normal. Disease phenotypes have not been reported.
Thus, although the biological importance of this receptor is established, evidence is lacking for its significance as a therapeutic target in disease. No CXCR5 antagonists or neutralizing mAbs have been developed yet. Recently, CXCR5 was reported to have coreceptor activity selective for HIV-2 (Kanbe et al., 1999).
IV. CC Chemokine Receptor Subtypes
A. CCR1
CCR1 was the first CC chemokine receptor identified and the first shown to have a functional viral homolog, US28 of human cytomegalovirus (Gao et al., 1993; Neote et al., 1993; Gao and Murphy, 1994). The gene is on human chromosome 3p21 in a cluster with CCR2, CCR3, CCR4, CCR5, CCR8, CCR9, XCR1, CX3CR1, and several orphans (Samson et al., 1996c). The ORF is on a single exon, and the predicted polypeptide is 355 aa in length.
Using a polyclonal rabbit antibody, Su et al. (1996) identified CCR1 on human peripheral blood lymphocytes and monocytes. A majority of CD3+, CD4+, CD8+, and CD16+ lymphocytes were positive. Among CD4+ peripheral blood T cells, CD45RO+ cells expressed greater amounts of CCR1 than CD45RO− cells, suggesting selective expression on the memory subtype. Expression studies using an anti-CCR1 mAb have not been reported.
CCR1 binds multiple inflammatory/inducible CC chemokines with similar high affinity, including MIP-1α, RANTES, MCP-2, MCP-3, leukotactin-1/MIP-5, MPIF-1 and HCC-1 (Neote et al., 1993; Gao et al., 1993; Youn et al., 1997; Gong et al., 1997; Tsou et al., 1998; Zhang et al., 1999; Nardelli et al., 1999). MIP-1β and MCP-1 bind with much lower affinity and are poor agonists (Neote et al., 1993). HCC-1 may be selective. Mouse CCR1 (80% aa identity) binds human and mouse MIP-1α with high affinity; agonists include mouse and human MIP-1α and human leukotactin-1/MIP-5 (Gao and Murphy, 1995; Post et al., 1995; Zhang et al., 1999). A closely related mouse orphan named MIP-1α-RL1 (65% aa identity) has also been cloned, but it has no human counterpart (Gao and Murphy, 1995).
CCR1 signaling includes calcium flux, inhibition of adenylyl cyclase, and chemotaxis (Myers et al., 1995; Pease et al., 1998). Coupling to both Gi and G14, but not Gq/11 or G16, has been reported in transfected COS cells (Kuang et al., 1996). Signaling can be blocked efficiently by RANTES variants that have been modified at the N terminus, including truncated forms (Arenzana-Seisdedos et al., 1996; Struyi et al., 1998), Met-RANTES (Proudfoot et al., 1996), and amino-oxypentane (AOP)-RANTES (Simmons et al., 1997); however, none of these is selective for CCR1 over the other RANTES receptors, CCR3 and CCR5. High CCR1 selectivity has been reported by Berlex Biosciences for 4-hydroxypiperidines (K i = 40–4000 nM) (Hesselgesser et al., 1998b; Ng et al., 1999) (Fig. 3), particularly 2-2-diphenyl-5-(4-chlorophenyl)piperidin-lyl)valeronitrite, which inhibits MIP-1α binding to CCR1 (K i∼ 40 nM) and blocks MIP-1α-induced extracellular acidification, Ca2+ mobilization, and chemotaxis of peripheral blood mononuclear cells; effects in disease have not been reported yet. Other small molecule CCR1 antagonists have also been disclosed but have either lower potency or selectivity than the Berlex Biosciences compound.
Clear disease indications have not yet been identified for CCR1. Nevertheless, there is a fair amount now known about its biology from the phenotype of CCR1 knockout mice. The receptor is dispensable for growth, development, and reproduction, and the mice do not acquire spontaneous infections from environmental pathogens. It is the dominant receptor used by MIP-1α for induction of mouse neutrophil chemotaxis and calcium flux in vitro, mobilization of neutrophils and hematopoietic progenitor cells in vivo, and regulation of hematopoietic progenitor cell proliferation (Gao et al., 1997; Broxmeyer et al., 1999). Consistent with this, MIP-1α functions as a negative regulator of hematopoiesis (reviewed in Broxmeyer et al., 1997), and in vitro anti-CCR1 antibodies block MIP-1α inhibition of colony formation by burst-forming unit-erythroid from purified human CD34+ bone marrow cells (Su et al., 1997). In this regard, BB10010, an agonistic variant of MIP-1α (British Biotech, Inc.), has been tested in phase I and II clinical trials as a stem cell protective agent in patients undergoing chemotherapy. The agent was safe in the doses tested, but only small therapeutic effects were noted on myelopoiesis, perhaps because of insufficient stress on the bone marrow by the chemotherapy regimens tested (Clemons et al., 1998; Marshall et al., 1998). Another CCR1 agonist, MPIF-1 (Human Genome Sciences, Rockville, MD), has recently undergone phase I trial for the same indication.
Consistent with a role in neutrophils, CCR1 −/− mice have reduced alveolitis in a pancreatitis-alveolitis mouse model (Gerard et al., 1997), as well as increased lethality when infected withAspergillus fumigatus, an organism controlled primarily by neutrophils (Gao et al., 1997). However, this is an example where mouse and human orthologs may differ in biological function, because the major CCR1 agonists MIP-1α and RANTES are poor agonists for human neutrophils (Coulin et al., 1997; Youn et al., 1997; Zhang et al., 1999). CCR1 also regulates granuloma formation and Th1/Th2 cytokine balance in response to Schistosome eggs deposited in mouse lung, but it is not a dominant receptor for MIP-1α-induced macrophage chemotaxis in vitro (Gao et al., 1997). Nevertheless, CCR1 deficiency did not reduce neutrophil accumulation in a nephrotoxic nephritis mouse model; disease was actually exacerbated with increased accumulation of macrophages and CD4+ and CD8+ T cells, as well as enhanced effector immune responses (Topham et al., 1999). However, CCR1 deficiency suppressed development of acute and chronic cardiac allograft rejection in several mouse models (Gao et al., 2000). Thus CCR1 can modulate inflammatory responses either positively or negatively, depending on the context, through effects on multiple leukocyte subtypes. The phenotype of MIP-1α knockout mice includes protection from coxsackievirus myocarditis, influenza A alveolitis, and acute experimental allergic encephalomyelitis; however, specific roles for CCR1 are not defined (Cook et al., 1995; Kennedy et al., 1998).
B. CCR2
CCR2 is the only leukocyte MCP-1 receptor identified so far, and it is important in inflammation, including atherosclerosis. The ORF is on two alternatively spliced exons that encode two distinct polypeptides 360 (CCR2(a)) and 374 (CCR2(b)) aa in length (Charo et al., 1994; Wong et al., 1997). The two have an identical sequence until aa 313, which is located in the C-terminal cytoplasmic region, and similar functional properties. Both RNAs are detectable in monocytes, blood-derived DC and NK cells and T lymphocytes but not in resting neutrophils or eosinophils. CCR2(b) appears to be the predominant form. mAbs have identified functional CCR2 in monocytes, activated memory T cells, B cells, and basophils (Frade et al., 1997;Rabin et al., 1999). In vivo, chronic inflammation may potentiate neutrophil migration to MCP-1 (Johnston et al., 1999).
Signaling through CCR2 in transfected cells includes calcium mobilization, inhibition of adenylyl cyclase, and chemotaxis (Myers et al., 1995). Receptor triggering may require receptor dimerization (Rodriguez-Frade et al., 1999). CCR2 binds multiple inflammatory/inducible ligands with similar high affinity, including MCP-1, MCP-2, MCP-3, MCP-4, and mouse MCP-5 (Charo et al., 1994;Ben-Baruch et al., 1995; Garcia-Zepeda et al., 1996; Gong et al., 1997;Sarafi et al., 1997). Only MCP-1 is selective versus other chemokine receptors, although it also binds to D6 and Duffy (see later). The HIV Tat protein is also an agonist at CCR2, which has suggested a possible mechanism for recruitment of target cells to sites of HIV infection (Albini et al., 1998).
mAb MCP-1 R02 directed to the CCR2 N terminus is also an agonist, whereas mAbs directed to the third extracellular domain (MCP-1R04 and MCP-1 R05) are antagonists (Rodriguez-Frade et al., 1999). Small molecule CCR2 antagonists have been reported in the patent literature by Roche Biosciences (Fig. 3).
Mouse CCR2 has 80% aa identity to human CCR2, is expressed in peritoneal macrophages, and is specific for the mouse chemokines JE and FIC, which have highest sequence homology to MCP-1 and MCP-3, respectively (Boring et al., 1996; Kurihara and Bravo, 1996). Using chemokine neutralization in mice, Karpus's group found that acute and relapsing forms of experimental autoimmune encephalomyelitis are regulated by differential expression of MIP-1α and JE/MCP-1, respectively, implicating CCR2 in relapsing forms of EAE (Kennedy et al., 1998). Correlative studies have also implicated chemokines and chemokine receptors in the pathogenesis of multiple sclerosis (Ransohoff, 1999).
Mice lacking CCR2 develop normally but do not recruit macrophages in an experimental peritoneal inflammation model, fail to clearListeria monocytogenes, have smaller granulomas after i.v. injection with yeast β-glucan, and have smaller granulomas and lower production of interferon-γ in draining lymph nodes when challenged with immobilized PPD by embolization to lung (Boring et al., 1997; Kurihara et al., 1997; Kuziel et al., 1997). As for CCR1 knockouts, this suggests a role in immunomodulation as well as in direct recruitment of monocytes/macrophages to sites of inflammation. They also have defective cockroach allergen-induced bronchial hyperreactivity (Campbell et al., 1999). Consistent with a pathogenetic role for MCP-1 and macrophages in human atherosclerotic plaques, CCR2 −/− mice have a sustained ∼50% reduction in size of atherosclerostic lesions when challenged with a Western diet on an apolipoprotein E −/− genetic background, which normally produces severe atherosclerosis (Boring et al., 1998). This is consistent with results from similar studies of JE/low-density lipoprotein receptor double knockout mice and JE deficiency in mice overexpressing human apolipoprotein B (Gu et al., 1998; Gosling et al., 1999). The protective effects are not mediated by changes in lipid levels and occur at both high and low levels of plasma lipids. The effect of blocking CCR2 in established atherosclerosis has not been reported yet.
CCR2(b) has HIV-1 coreceptor activity in vitro, but the activity and strain specificity are both low (compared with CCR5 and CXCR4; Doranz et al., 1996; Zhang et al., 1998a). Nevertheless, a role in disease has been suggested by discovery of a common variant CCR2 allele named CCR2-64I that is associated with a 2- to 4-year delay in progression to AIDS in some HIV-1 seroconvertor cohorts (Smith et al., 1997). To date, the mechanism of action of this mutation has not been defined. In particular, it does not appear to directly affect either the chemokine receptor or HIV coreceptor activities of CCR2 or other coreceptors (Lee et al., 1998). Interestingly, CCR2-64I but not wild-type receptor has been observed to heterodimerize with native CXCR4 when transfected in human embryonic kidney 293 cells; however, functional correlates have not been defined (Mellado et al., 1999). CCR2-64I could also potentially be linked to a disease-modifying mutation in CCR5, because the two genes are located within 10 kb on chromosome 3.
C. CCR3
CCR3 is an eosinophil chemoattractant receptor for multiple inflammatory/inducible CC chemokines (Heath et al., 1997) and may be important in allergic inflammation, including asthma, and antihelminthic host defense where eosinophils greatly outnumber other leukocytes; it is also an HIV-1 coreceptor (Doranz et al., 1996; Choe et al., 1996). A human CCR3 cDNA highly expressed in eosinophils was first reported by Combadiere et al. (1995a). Its ligands were originally reported incorrectly as MIP-1α, MIP-1β, and RANTES and later corrected to eotaxin (Kitaura et al., 1996). Two other groups independently characterized CCR3 as an eotaxin receptor and identified additional agonists (Daugherty et al., 1996; Ponath et al., 1996). The ORF is on a single exon and predicts a polypeptide 355 aa in length. CCR3 is most similar to CCR1 in sequence (62% aa identity) and ligands.
CCR3 is difficult to express in foreign cells, and ligand binding is highly dependent on pH and salt concentration (Dairaghi et al., 1997) Ligands and agonists for human CCR3 include eotaxin, eotaxin-2, eotaxin-3, RANTES, MCP-3, MCP-4, MIP-5/leukotactin-1, and HIV Tat (Daugherty et al., 1996; Garcia-Zepeda et al., 1996; Kitaura et al., 1996, 1999; Ponath et al., 1996; Forssmann et al., 1997; Gong et al., 1997; Heath et al., 1997; Youn et al., 1997; Albini et al., 1998;Shinkai et al., 1999). Eotaxin, eotaxin-2, and eotaxin-3 appear to be the most potent and are selective.
Human CCR3 distribution on eosinophils (Heath et al., 1997), basophils (Uguccioni et al., 1997), mast cells (Ochi et al., 1999), and a subset of Th2 T lymphocytes (Gerber et al., 1997; Sallusto et al., 1997) is compatible with a role in allergic inflammation. The receptor is also found on dendritic cells (Rubbert et al., 1998) and microglial cells of the brain (He et al., 1997). CCR3 activities on human cells in vitro include eosinophil arrest under flow conditions (Kitayama et al., 1998), eosinophil and Th2 cell chemotaxis (Heath et al., 1997; Sallusto et al., 1997), degranulation of eosinophils and basophils (Uguccioni et al., 1997), HIV-1 entry of microglial cells (He et al., 1996), and HIV-specific T cell cytotoxicity mediated by RANTES (Hadida et al., 1998).
A fully antagonistic mAb specific for human CCR3 named 7B11 was developed and used to show that eosinophil and basophil responses to eotaxin, RANTES, MCP-2, MCP-3, and MCP-4 are mediated entirely by CCR3 in most donors (Heath et al., 1997; Uguccioni et al., 1997). The chemokine derivative Met-chemokine β7 is a potent and specific antagonist (Nibbs et al., 2000). Less specific antagonists include Met-RANTES, AOP-RANTES, the distamycin analog previously mentioned in the section on CXCR4, and vMIP-II (Kledal et al., 1997). It has also been reported that vMIP-II chemoattracts human eosinophils in a CCR3-dependent manner, a discrepancy that has not been reconciled (Boshoff et al., 1997). Most of these blocking agents also block HIV interaction with CCR3. A small molecule antagonist has been reported by Takeda in the patent literature (Fig. 3).
The discovery of eotaxin and CCR3 raised hopes that this would be a dominant signaling system in allergic inflammation and a major new drug target; however, so far, the evidence from animal models is inconclusive. Eotaxin knockout mice have been generated by two groups, but reported effects on airway eosinophilia after ovalbumin challenge have conflicted: Rothenberg et al. (1997) reported a ∼40% reduction, whereas Yang et al. (1998) saw no effect. Eotaxin is required for the baseline level of tissue eosinophils (Matthews et al., 1998). A CCR3 knockout mouse has not yet been reported. Mouse may be a poor host for modeling eotaxin and CCR3, because CCR1 is highly expressed in mouse versus human eosinophils (Gao et al., 1996); mouse CCR3, unlike human CCR3, binds MIP-1α in addition to eotaxin (Post et al., 1995); and mouse CCR3 is expressed only on eosinophils (Grimaldi et al., 1999). The guinea pig has been proposed as a superior model, which can now be studied with a neutralizing mAb recently developed against guinea pig CCR3 (Sabroe et al., 1998). Meanwhile, previous work on the neutralization of guinea pig eotaxin revealed partial blockade of airway eosinophilia on allergen challenge (Humbles et al., 1997) and partial suppression of eosinophil mobilization from bone marrow (Palframan et al., 1998).
CCR3 has broad specificity for R5-, X4-, and dual-tropic HIV envelope glycoproteins and is used by HIV for entry of microglial cells in vitro (Choe et al., 1996; He et al., 1997; Bazan et al., 1998). However, use in vivo is not established.
D. CCR4
CCR4 has been reported to be a selective marker for Th2 T lymphocytes and is up-regulated by T cell receptor activation (Bonecchi et al., 1998; Sallusto et al., 1998, 1999b). Current concepts of CCR4 function include dendritic cell trafficking, T cell recirculation from tissue to draining lymph node, T cell transmigration through thymus during T cell maturation, T cell migration to ectopic lymphoid tissue (Sozzani et al., 1999), and homing of memory T cells to inflamed skin but not to gut (Campbell et al., 1999).
The cDNA was originally cloned from a human basophilic leukemia cell line library (Power et al., 1995). The ORF is on a single exon and predicts a polypeptide 360 aa in length. High-affinity ligands and high-potency agonists include MDC and TARC (Imai et al., 1997a, 1998), which are constitutively made and selective for CCR4. Activities include calcium flux and chemotaxis. MIP-1α, RANTES, and MCP-1 are also agonists when CCR4 is expressed in frog oocytes and transmembrane currents are measured, but potencies have not been reported (Power et al., 1995). A cDNA encoding a mouse counterpart of CCR4 has been isolated that has 80% aa identity (Hoogewerf et al., 1996). Knockout mice, neutralizing mAbs, and small molecule antagonists have not been reported yet.
At present, potential roles for CCR4 in disease have been inferred based on analysis of its known ligands in mouse disease models. In one study, it was implicated in the pathogenesis of bacteria-induced fulminant hepatic failure in mice, based on a protective effect of injecting anti-TARC mAbs (Yoneyama et al., 1998). In a second study, neutralization of mouse MDC was protective in a model of airway hyperreactivity and eosinophilic inflammation (Gonzalo et al., 1999).
Purified native human MDC has been reported to be a broad-spectrum HIV-1 suppressive agent in vitro, but its mechanism of action is unlikely to involve CCR4, because it has not been observed to function as an HIV coreceptor and because TARC lacks this activity (Pal et al., 1997). Several laboratories have been unable to reproduce this effect with recombinant and synthetic MDC.
E. CCR5
CCR5 is a major HIV-1 coreceptor that controls susceptibility to HIV-1 infection and disease. The first report of the sequence and ligands for human CCR5 was published on March 19, 1996 (Samson et al., 1996a), followed on March 29 and June 14 by mouse CCR5 (Boring et al., 1996; Meyer et al., 1996) and in July 1996 by two additional independent reports of human CCR5 (Combadiere et al., 1996; Raport et al., 1996). The HIV coreceptor activity was described in five reports by five independent groups published within 1 week in June 1996 (Alkhatib et al., 1996; Choe et al., 1996; Deng et al., 1996; Dragic et al., 1996; Doranz et al., 1996), and 2 months later the first of a series of reports describing the defective CCR5Δ32 allele, which established the pivotal in vivo role of CCR5 in HIV pathogenesis, was reported (Dean et al., 1996; Huang et al., 1996; Liu et al., 1996;Samson et al., 1996b; Zimmerman et al., 1997).
The human CCR5 ORF is on a single exon and predicts a polypeptide 355 aa in length. It is expressed on peripheral blood-derived dendritic cells (Granelli-Piperno et al., 1996; Rubbert et al., 1998), CD34+ hematopoietic progenitor cells (Ruiz et al., 1998), and activated/memory CD26highCD45RAlow CD45R0+ Th1 lymphocytes (Bleul et al., 1997; Loetscher et al., 1998c). Fresh monocytes express low levels that can be increased by culture in vitro (Alkhatib et al., 1996). Likewise, freshly isolated T cells express low amounts that increase with prolonged stimulation by IL-2, mitogens, and other activating protocols ex vivo (Bleul et al., 1997) or by Th1 type inflammation in vivo (e.g., in synovial fluid from patients with rheumatoid arthritis;Qin et al., 1998). CCR5 is also expressed on CD4+and CD8+ thymocytes (Zaitseva et al., 1998) and Langerhans cells (Zaitseva et al., 1997). Reports of CCR5 on neurons, astrocytes, capillary endothelial cells, epithelium, vascular smooth muscle, and fibroblasts have also been published, but functional roles are not established.
In vivo, T lymphocytes and macrophages in both lymphoid and nonlymphoid tissues express CCR5 and CXCR4, but follicular dendritic cells in lymph node express neither, suggesting that trapping of HIV by these cells does not involve the major HIV coreceptors. CCR5-positive cells are more frequently identified in the colon than in the rectum and more frequently identified in the cervix than in the vagina, suggesting that the expression levels of coreceptors are differentially regulated at different anatomic sites (Zhang et al., 1998b).
High-affinity ligands and high-potency agonists include MIP-1α (P form is more potent than S form; Nibbs et al., 1999), RANTES, MIP-1β, and MCP-2, but none is selective (Combadiere et al., 1996; Raport et al., 1996; Samson et al., 1996a; Gong et al., 1998). Additional ligands include MCP-3, MCP-4, MCP-1, and eotaxin. MCP-3 appears to be an antagonist (Blanpain et al., 1999). gp120 from R5 HIV strains are also ligands and induce receptor triggering and chemotaxis in a CD4-dependent manner (Weissman et al., 1997). CCR5 has been found to associate constitutively with CD4 (Xiao et al., 1999), but the physiological role is unknown. Chemokine ligands block CCR5 use by R5 HIV-1 strains, and vice versa (Cocchi et al., 1995; Alkhatib et al., 1996; Trkola et al., 1996).
CCR5 blocking agents include mAbs, some of which selectively block HIV coreceptor activity but not chemokine binding (Wu et al., 1997; Olson et al., 1999), and chemokine derivatives, such as truncated versions of RANTES, Met-RANTES, and AOP-RANTES and the viral chemokine KSHV vMIP-II, all of which block both chemokine and HIV interaction with CCR5 but are not selective (Arenzana-Seisdedos, 1996; Kledal et al., 1997; Simmons et al., 1997). Met-RANTES and AOP-RANTES are pure and partial antagonists at CCR5, respectively. AOP-RANTES induces calcium flux but not chemotaxis and is the most potent RANTES derivative in blocking R5 HIV entry. Moreover, it blocks HIV entry of all target cells tested, whereas wild-type RANTES, for unclear reasons, fails to efficiently block R5 HIV entry of macrophages.
CCR5 can be blocked selectively by mAbs; by intrakines, which are chemokines delivered by gene therapy and targeted for endoplasmic reticulum retention that trap CCR5 intracellularly (Yang et al., 1997); by hammerhead ribozymes (Goila and Banerjea, 1998); and by the small molecule TAK-779 (N,N-dimethyl-N-[4-[[[2-(4-methylphenyl)-6,7-dihydro-5H-benzocyclohepten-8-yl]carbonyl]amino]benzyl]tetrahydro-2H-pyran-4-aminium chloride) (Baba et al., 1999) (Fig. 3). Moreover, a novel fusion-competent HIV vaccine strategy has been discovered in which CCR5, CD4, and a cross-linking agent are used to trap neutralizing epitopes of gp120 for presentation to the immune system (LaCasse et al., 1999). Immunization of mice led to the production of neutralizing antisera for diverse primary HIV isolates.
CCR5 is blocked naturally by inheritance of CCR5Δ32, a mutant allele common in whites that encodes a truncated, inactive receptor due to a 32-bp deletion in the ORF (Dean et al., 1996; Huang et al., 1996; Liu et al., 1996; Samson et al., 1996b; Zimmerman et al., 1997). CCR5Δ32 homozygotes, which represent ∼1% of North American whites, exhibit high resistance to HIV infection and appear otherwise healthy. This suggests that normal CCR5 function is well compensated or redundant. CCR5Δ32 heterozygotes have reduced normal CCR5 expression on cells, due in part to a dominant negative effect of the CCR5Δ32 protein (Benkirane et al., 1997) and, if infected with HIV, progress less rapidly to AIDS in some but not all cohorts that have been studied. An even stronger effect on disease progression has been associated with a single nucleotide polymorphism located in the CCR5 promoter (P1 or 59029 G/A alleles), which affects gene transcription (McDermott et al., 1998; Martin et al., 1998). Why the CCR5Δ32 allele is so common in whites is unknown, but a reasonable mechanism is selection through an earlier epidemic within the past two millennia (Libert et al., 1998). In this regard, it is interesting to note that myxoma, a rabbit poxvirus, can use CCR5 (as well as several other chemokine receptors) as a cell entry factor (Lalani et al., 1999). Perhaps variola could also use CCR5 but not CCR5Δ32, which was enriched in populations by smallpox epidemics.
Mouse CCR5 is similar in ligand selectivity to the human receptor, and CCR5 −/− mice, like CCR5-deficient humans, appear healthy (Zhou et al., 1998). Subtle defects have been identified in stressed mice, including reduced efficiency in the clearance of Listeriainfection, relative resistance to lipopolysaccharide-induced endotoxemia, increased susceptibility to Cryptococcus infection (Huffnagle et al., 1999), enhanced delayed-type hypersensitivity reaction, and increased humoral responses to T cell-dependent antigenic challenge, indicating a role for CCR5 in down-modulating T cell-dependent immune responses.
To date, CCR5 is the only chemokine receptor (excluding Duffy for the moment) for which proof of concept is available for a role in human disease. The development of therapeutic and preventive strategies that mimic the near-perfect safety and efficacy of CCR5Δ32 in the prevention of infection of HIV-exposed populations is therefore of substantial interest. However, blocking CCR5 alone in the setting of established infection is unlikely to be effective because viruses are likely to emerge that can use CXCR4 or other coreceptors. To date, one phase I clinical trial has addressed this question, using the MIP-1α variant BB10010 as a CCR5 blocking agent in HIV-positive individuals. No significant effect was observed on viral burden or CD4 counts, but the maximal administered dose was insufficient to achieve blocking levels in vivo (L. Czaplewski, personal communication).
F. CCR6
CCR6 is the only known receptor for LARC (also known as MIP-3α, exodus and ck-β4) (Baba et al., 1997; Greaves et al., 1997; Liao et al., 1997b; Power et al., 1997), and it mediates responsiveness of diverse subsets of memory T cells to LARC (Liao et al., 1999). Additional unusual features include its location on human chromosome 6q27 outside of the main CCR cluster on 3p (Liao et al., 1997a), its functional expression on nonactivated memory T cells (Campbell et al., 1998; Liao et al., 1999), and down-regulation during dendritic cell maturation (Dieu et al., 1998). Although the biology and pharmacology of CCR6 are not yet developed, it is predicted to be important in memory T cell and dendritic cell trafficking to secondary lymphoid organs. The CCR6 ligand MIP-3α appears to specifically regulate constitutive homing of Langerhans-type dendritic cells to the epidermis, whereas other types of dendritic cells respond to multiple other chemokines (Charbonnier et al., 1999).
The ORF is on two exons and predicts a polypeptide 368 or 374 aa in length, depending on which of two deduced methionines is considered to initiate translation. The aa sequence is 76% identical with mouse CCR6 (Varona et al., 1998). CCR6 mRNA is present constitutively in secondary lymphoid tissue (spleen, lymph nodes, appendix) and fetal liver, in peripheral blood CD4+ and CD8+ T cells with a memory phenotype, and in B cells but not NK cells, monocytes, or granulocytes. It is also selectively expressed in human dendritic cells derived from CD34+ cord blood precursors and in dendritic cells derived from peripheral blood monocytes (Yang et al., 1999a). TCR activation of T cells causes down-regulation of CCR6 (Sallusto et al., 1999b).
LARC, which is produced by activated macrophages, dendritic cells, and endothelial cells, is the only high-affinity ligand (K d = 0.1–12 nM on transfected cells, 0.4 nM on lymphocytes) and high-potency chemokine agonist for CCR6. Signaling includes calcium flux and chemotaxis. Recently, the human β-defensin HBD2 has been identified as a nonchemokine functional ligand for CCR6. HBD2 is produced by epithelial cells during infection and functions as a direct antimicrobial factor but also chemoattracts CCR6+ dendritic cells and memory T cells, suggesting a chemokine receptor link between innate and adaptive immunity (Yang et al., 1999b). The activity of other defensin family members at chemokine receptors has not been reported.
G. CCR7
CCR7 is a major homing receptor of the immune system, critical not only for trafficking of B lymphocytes, T lymphocytes, and dendritic cells across high endothelial venules but also for their correct positioning in T cell zones of secondary lymphoid organs (Cyster et al., 1999; Forster et al., 1999; Sozzani et al., 1999). A favored model views CCR7 as a homing switch that is turned on during the activation of resting T cells and maturing dendritic cells (Gunn et al., 1998b; Sallusto et al., 1998, 1999b; Sozzani et al., 1998a;Yanagihara et al., 1998; Kellerman et al., 1999; Forster et al., 1999;Saeki et al., 1999). Its importance is revealed by the profound disorganization of secondary lymphoid tissue in CCR7 −/− mice and the failure of these mice to mount rapid antibody responses as well as contact sensitivity and delayed-type hypersensitivity responses to T-dependent antigens (Forster et al., 1999).
CCR7 cDNAs were cloned first from B cells by two groups working independently: one using an Epstein-Barr virus-infected versus uninfected B lymphocyte subtraction cDNA library (Birkenbach et al., 1993), and the other using a Burkitt's lymphoma cell library. Thus, the receptor was originally named EBI-1, for Epstein-Barr virus-induced cDNA #1, and BLR-2, for Burkitt's lymphoma receptor-2 (Burgstahler et al., 1995). The gene is on human chromosome 17q12-21.2 outside the main CCR cluster on 3p21 (Schweickart et al., 1994). The ORF is on two exons separated by an intron in the N-terminal domain and predicts a polypeptide 378 aa in length.
ELC and SLC are highly specific functional ligands for CCR7 that bind with similar affinity and induce chemotaxis of CCR7-positive cells (Yoshida et al., 1997, 1998a; Campbell et al., 1998). Both ELC and SLC are expressed constitutively within the T cell zone of secondary lymphoid tissue and mucosa-associated lymphoid tissue but not in B cell areas or sinuses or blood-derived leukocytes (Willimann et al., 1998). SLC expression has been finely mapped to interdigitating dendritic cells and high endothelial venules of secondary lymphoid tissue and in lymphatic endothelium of various organs, where it promotes adhesion and chemotaxis of naı̈ve T lymphocytes (Gunn et al., 1998a). This is consistent with the phenotype of mice homozygous for the spontaneous mutation plt (paucity of lymph node T cells), which maps to the SLC locus and in which SLC is not produced. The exactplt mutation has not been identified, but it is not within SLC exons or introns (Gunn et al., 1999). In these mice, naive T cells fail to home to lymph nodes or lymphoid regions of spleen, and skin dendritic cells fail to traffic to and accumulate in spleen and lymph node T cell zones. The defect is associated with markedly increased sensitivity to infection with murine hepatitis virus. The phenotypes of CCR7 −/− mice and plt mice are very similar (Forster et al., 1999). Defective B and T cell homing in CCR7 −/− mice has been shown by adoptive transfer experiments to be due to defective cell migration.
CCR7 is also dynamically regulated during thymocyte maturation; however, a defect in T cell development was not found in CCR7 −/−/ mice (Forster et al., 1999; Suzuki et al., 1999). Evidence for chemoattraction of activated T lymphocytes by maturing dendritic cells via up-regulation of CCR7 ligands has also been reported (Tang and Cyster, 1999).
Using an anti-CCR7 mAb, CCR7 phenotype has been used to define subsets of T cells that mediate two functionally distinct aspects of immunological memory (Sallusto et al., 1999). CCR7 negative memory cells, designated as TEM (effector memory T cells), express receptors for migration to inflamed tissue and have immediate effector function. CCR7+ memory cells or TCM (central memory T cells) express lymph node homing receptors, efficiently stimulate dendritic cells, and differentiate into TEM on secondary stimulation but lack immediate effector function.
H. CCR8
CCR8 is notable for high expression in thymus (Napolitano et al., 1996) and association with Th2 lymphocytes (Zingoni et al., 1998). It was molecularly defined as a previously cloned orphan receptor (Roos et al., 1997; Tiffany et al., 1997; Goya et al., 1998; Horuk et al., 1998). The ORF is on a single exon and predicts a polypeptide 355 aa in length, 32 to 45% identical with other CCR subtypes. Mouse CCR8 has 71% aa identity to human CCR8 (Goya et al., 1998).
In addition to thymus and Th2 lymphocytes, CCR8 mRNA is found in brain, spleen, lymph node, and monocytes (Napolitano et al., 1996; Tiffany et al., 1997). I-309 is a selective human ligand; however, interestingly, it also binds three viral chemokines: vMIP-I and vMIP-II from KSHV and MC148R (also called vMCC-I) from M. contagiosum virus (Damon et al., 1998; Dairaghi et al., 1999; Endres et al., 1999). In calcium flux and chemotaxis assays, I-309 and vMIP-I are agonists, whereas MC148R is an antagonist; vMIP-II has been reported as an antagonist by one group (Dairaghi et al., 1999) and as a chemotactic agonist by another (Sozzani et al., 1998b). TARC and MIP-1β have also been reported as chemotactic agonists (Bernardini et al., 1998), but this has not been confirmed (Dairaghi et al., 1999; H. L. Tiffany and P. M. Murphy, unpublished data). Mouse CCR8 is also expressed in thymus. Both I-309 and its mouse homolog TCA-3 are high-affinity ligands and potent agonists at mouse CCR8 (Goya et al., 1998).
The biological roles played by CCR8 and its ligands are speculative at this point. Knockout mice, mAbs, and antagonists are currently unavailable. Expression of CCR8 in Th2 cells correlates well with the ability of I-309 to chemoattract these cells and suggests a possible role in allergic inflammation. Selective action of viral chemokines at CCR8 suggests a role in Kaposi's sarcoma (KS) and M. contagiosum, possibly through the modulation of Th2 function. Of note, Th2 cells are found in KS lesions, whereas M. contagiosum lesions are notable for the paucity of leukocytes. Apart from migration, the ability of I-309 to inhibit apoptosis of dexamethasone-treated mouse thymic lymphoma cells has suggested that CCR8 may be involved in this and possibly other apoptotic processes (Van Snick et al., 1996).
In transfected cells, CCR8 can function as an HIV-1 coreceptor for diverse T-cell tropic, dual-tropic, neutrotropic, and macrophage-tropic HIV-1 strains, and I-309 can inhibit this activity (Horuk et al., 1998). However, the use of CCR8 by HIV-1 in primary cells and in pathogenesis is undefined.
I. CCR9
CCR9 is the previously cloned orphan GPR 9-6 (Youn et al., 1999;Yu et al., 2000; Zaballos et al., 1999; Zabel et al., 1999). The chemokine binding protein D6 had previously been inappropriately named CCR9 (see later). Two splice variants, designated CCR9(a) and CCR9(b), have been identified. CCR9(a) is predicted to have 12 additional aa at its N terminus (compared with CCR9(b)) and is the predominant form in all cell types examined (Yu et al., 2000). TECK, a homeostatic/constitutive type chemoattractant for dendritic cells, thymocytes, intestinal homing T lymphocytes, mucosal lymphocytes, and activated macrophages, is the only known agonist, and it acts with slightly higher potency at CCR9(a) than CCR9(b). Constitutive expression of human and mouse CCR9 is very high in thymus and low in lymph nodes and spleen. Immature and mature thymic T cells express CCR9, suggesting a role in T cell development in the thymus.
J. CCR10
CCR10 is the former orphan receptor GPR2 (Marchese et al., 1995). The only ligand identified to date is the placenta and skin-associated CC chemokine CCL27 (also known as ESkine, skinkine, and CTACK), which attracts skin-homing memory T cells (Baird et al., 1999; Morales et al., 1999; Homey et al., 2000; C. Gerard et al., manuscript submitted).
K. CCR11
CCR11 is the second MCP-1 receptor identified. However, it has not yet been detected on leukocytes. The gene maps to 3p22 separate from the major CCR cluster at 3p21. Chemotactic ligands include MCP-1, MCP-2, and MCP-4, and it is expressed in heart, small intestine, and lung (Schweickart et al., 2000).
V. CX3C Chemokine Receptor Subtypes
A. CX3CR1
CX3CR1 is unique among chemokine receptors in functioning directly as a cell-cell adhesin, including under physiological shear (Imai et al., 1997b; Fong et al., 1998; Haskell et al., 1999). Its powerful adhesive action may be important for leukocyte extravasation in the setting of high blood flow. CX3CR1 has highest similarity to CC chemokine receptors (30–42%); consistent with this, the gene is located on human chromosome 3p21 in the major CCR cluster (Combadiere et al., 1995b).
As revealed first in rat and later in human and mouse, CX3CR1 mRNA is expressed at highest levels in brain, but it is found in all organs examined (Harrison et al., 1994, Combadiere et al., 1995b, 1998a). The only known ligand is fractalkine, which is also referred to as neurotactin in the mouse, a multimodular protein containing a chemokine domain, a mucin-like stalk, a transmembrane domain, and a cytoplasmic domain, as well as the CX3C signature (Bazan et al., 1997; Imai et al., 1997b; Pan et al., 1997). Fractalkine is found in a 95-kDa shed form as well as a membrane-tethered form expressed on endothelial cells and neurons (Bazan et al., 1997; Imai et al., 1997b; Pan et al., 1997;Harrison et al., 1998). Chemotactic signaling by CX3CR1 is G protein-mediated (pertussis toxin-sensitive), whereas direct adhesion to tethered fractalkine is not (Imai et al., 1997b). Blood-derived neutrophils, monocytes, NK cells, and T lymphocytes are positive for CX3CR1 and respond chemotactically to fractalkine (Bazan et al., 1997;Imai et al., 1997b). The recombinant chemokine domain of neurotactin is chemotactic for neutrophils both in vitro and in vivo (Pan et al., 1997). Membrane-bound fractalkine, in addition to being proadhesive, can also trigger receptors to signal and induces chemotaxis.
CX3CR1 appears to mediate cell adhesion to both endothelial cells and neurons. In rat brain, fractalkine has been detected on neurons, where it is up-regulated by axonal injury and mediates direct interactions with CX3CR1-expressing microglia, possibly to mediate nerve repair (Harrison et al., 1998). In the Wistar-Kyoto rat crescentic glomerulonephritis model, a setting of high blood flow, fractalkine is markedly induced in glomerular endothelium and CX3CR1 is increased on infiltrating leukocytes. Moreover, anti-CX3CR1 antibody treatment dramatically inhibits leukocyte infiltration in the glomeruli, prevents crescent formation, and improves renal function (Feng et al., 1999). Effects on established disease are not defined.
In transfected cells, CX3CR1 can function as an HIV-1 coreceptor for a limited number of HIV-1 strains, and fractalkine can block this activity (Combadiere et al., 1998b). However, the use of CX3CR1 in primary cells by HIV-1 and in pathogenesis is undefined.
VI. Chemokine Receptor Subtypes
A. XCR1
XCR1 is the former orphan receptor GPR5 and the only C chemokine receptor, specific for the T lymphocyte directed molecule lymphotactin (Yoshida et al., 1998b). Biology and pharmacology have not yet been developed. However, a potential role in cancer therapy was suggested by the ability of lymphotactin to synergize with IL-2 in producing an antitumor immune response and tumor regression in mice (Dilloo et al., 1996).
VII. Chemokine Binding Proteins
A. Duffy
Duffy is the red cell receptor for the malaria-causing protozoanPlasmodium vivax and a highly promiscuous chemokine binding protein, specific for some but not all CC and CXC chemokines, such as ELR+ (IL-8, GROα) but not ELR− CXC chemokines and basic (RANTES, MCP-1) but not acidic (MIP-1α) CC chemokines (Miller et al., 1975,1976; Darbonne et al., 1991; Chaudhuri et al., 1993, 1994; Horuk et al., 1993). Despite having a 7TMD architecture (338 aa) and ∼25% aa identity to chemokine receptors, no signaling function has been observed on chemokine ligation (Neote et al., 1994). Because of its long history in the malaria and blood group literature (Horuk, 1994), the nomenclature committee has decided that the name of this molecule may remain Duffy even if signaling is demonstrated at some future date.
Normally, Duffy RNA is present in multiple organs, including brain, spleen, bone marrow, and lung, and in addition to red cells, Duffy antigen can be detected on endothelial cells of postcapillary venules (Hadley et al., 1994; Chaudhuri et al., 1997) and Purkinje cells of the cerebellum (Horuk et al., 1997). However, Duffy is selectively missing from red cells of individuals from equatorial and southern Africa (Chaudhuri et al., 1995; Peiper et al., 1995), the result of fixation of a variant allele bearing a single nucleotide mutation named −46C in an erythroid-specific GATA-1 site in the Duffy promoter (Tournamille et al., 1995). Accordingly, red cells from these individuals do not bind chemokine or plasmodium ligands for Duffy (Horuk et al., 1993; Chitnis and Miller, 1994); the individuals are resistant to P. vivax; and P. vivax is virtually absent from this region of Africa. Analogous to CCR5 and HIV, these individuals have no obvious health problems attributable specifically to Duffy deficiency on red cells. Furthermore, an apparently healthy white individual has been described with complete Duffy deficiency, the result of a 14-bp deletion in the ORF (Mallinson et al., 1995). Together these experiments of nature suggest that Duffy is a dispensable gene. This presents a challenge to hypotheses regarding the normal physiological function of Duffy, which include a potential role on red cells as a chemokine clearance factor to maintain chemokine gradients from tissue to blood (Horuk, 1994), and a role on endothelial cells in the presentation of chemokines to leukocytes (Hadley et al., 1994).
Presumably, fixation of the mutant Duffy allele in Africa was caused by selective pressure of P. vivax malaria. An opportunity to test this hypothesis prospectively may exist in Papua New Guinea, whereP. vivax infection is endemic and where the identical promoter mutation has occurred independently on a different Duffy haplotype but is currently present at only a low frequency in the population (Zimmerman et al., 1999).
Chemokine and Plasmodium ligands have no significant sequence homology but cross-inhibit the binding of each other to Duffy. GROα variants have been identified that block red cell invasion byP. knowlesi without activating CXCR1 or CXCR2 (Hesselgesser et al., 1995). A glutamic acid for alanine-substituted mutant named E6A is the most potent variant identified, but clinical development has not been reported. The Duffy-specific mAb Fy6 blocks chemokine binding (Horuk et al., 1993).
A murine ortholog of Duffy has been identified, and the gene is namedDfy (Luo et al., 1997; Tang et al., 1998). Features common with human Duffy include 63% aa identity; localization on mouse chromosome 1; expression in skeletal muscle, spleen, bone marrow, and brain; presence of an intron between codons 7 and 8; and a common chemokine binding profile. One difference is expression of the mouse gene in liver. Duffy-deficient mice have not yet been reported.
B. D6
The nomenclature for D6 has changed several times due to confusion about its signaling properties. Mouse D6 was originally identified as a bone marrow cDNA expressed in hematopoietic progenitor cells and placenta (Nibbs et al., 1997a). Ligands include multiple CC chemokines, including MIP-1α, MCP-1, RANTES, and MCP-3, which were originally reported to induce calcium flux in transfected cells. Two human cDNAs were then reported that encode proteins 70% identical with mouse D6. Their sequences differ by 4 aa, which is thought to be due to allelic variation of the same gene. One variant was originally named “CCR9” in the GenBank database, but “D6” in the article describing it (Nibbs et al., 1997b). The other variant was named “CCR10” (Bonini et al., 1997). Both variants were shown to bind similar CC chemokines as mouse D6, with the exception of MIP-1α (see later), but signaling could not be demonstrated. Moreover, signaling for mouse D6 could not be reproduced (Nibbs et al., 1999). Therefore, the committee has not proposed a CCR designation for this molecule, and the groups originally responsible for its identification have agreed to use of D6 pending further research (Nibbs et al., 1999; J. Bonini, personal communication).
The difference in specificity of human and mouse D6 for MIP-1α has been reconciled recently through the appreciation that there are two distinct but highly related MIP-1α genes in human, named LD78α and LD78β, but only one in mouse. The LD78α and LD78β products have been tentatively named MIP-1αS and MIP-1αP, respectively, because of signature serine and proline residues that distinguish their structure and specificity for human D6 (Nibbs et al., 1999) In particular, MIP-1αP is a selective agonist versus MIP-1αS at human D6 in calcium flux assays. In vivo roles of D6 are undefined.
VIII. Virus-Encoded Chemokine Receptors
A. ECRF3
ECRF3 is another name for ORF 74 of Herpesvirus saimiri, a T lymphotropic γ-herpesvirus that is not pathogenic in its natural host, the squirrel monkey, but causes T cell transformation and acute leukemias and lymphomas in other primate hosts. The ECRF3 sequence was discovered as part of the saimiri genome sequencing project, and its relationship to chemokine receptors was identified through bioinformatics (Albrecht et al., 1992). When it is expressed inXenopus oocytes, agonists include GROα > IL-8 > NAP-2, which is the same specificity but a different hierarchy as for CXCR2, its closest known mammalian homolog (Ahuja and Murphy, 1993). Signaling includes calcium mobilization. Functional expression has not been successfully accomplished in mammalian cell lines, and there is no information about its biological role or expression in the context of virally infected cells.
B. US28
Human cytomegalovirus (HCMV) is a β-herpesvirus that causes life-threatening systemic infections in immunocompromised hosts, including patients with AIDS. UL33, US27, and US28 are ORFs in the unique long and unique short genomic regions of HCMV, respectively, that encode homologs of GPCRs (Chee et al., 1990). UL33 and US27 remain orphans, whereas US28 (30% aa identity to CCR1) has been demonstrated to bind the CC chemokines MIP-1α, MIP-1β, RANTES, MCP-1, and MCP-3 with similar affinity in transfected cells, as well as in the context of HCMV infection of human fibroblasts (Neote et al., 1993; Gao and Murphy, 1994; Bodaghi et al., 1998; Vieira et al., 1998). These chemokines are also US28 agonists, inducing calcium flux in both transfected and HCMV-infected cells, with RANTES having the highest potency (Gao et al., 1994; Vieira et al., 1998).
HCMV-infected fibroblasts are able to deplete endogenous RANTES and MCP-1 from media in a US28-dependent manner, suggesting a novel antichemokine mechanism for immune evasion (Bodaghi et al., 1998;Vieira et al., 1998). The CC chemokine specificity of US28 has recently been broadened to include fractalkine, which has suggested a possible adhesive role of US28, analogous to that of the cellular fractalkine receptor CX3CR1, that could mediate cell-cell spread of virus (Kledal et al., 1998). Consistent with this, US28 enhances cell-cell fusion by different viral proteins (Pleskoff et al., 1998). In this regard, US28 also has HIV coreceptor activity (Pleskoff et al., 1997), which has suggested a potential symbiotic relationship between the two viruses, with HIV providing immunosuppression needed for maximal HCMV replication and HCMV providing an additional mechanism for cell entry by HIV.
Recently, US28 was shown to transduce smooth muscle cell migration in response to RANTES and MCP-1 (Streblow et al., 1999). This provides a potential molecular mechanism to explain the link between smooth muscle cell infection by HCMV and vascular disease.
C. KSHV GPCR
KSHV GPCR is the product of ORF 74 of KS-associated herpesvirus or HHV8, a γ-herpesvirus and purported causal factor in KS, and two rare B cell neoplasms found in AIDS patients: primary effusion lymphoma and multicentric Castleman's disease (Cesarman and Knowles, 1999). KSHV GPCR is syntenic with ECRF3, the CXC chemokine receptor of Herpesvirus saimiri, and was discovered through bioinformatics as part of the HHV8 Genome Sequencing Project. It has been detected at the mRNA level in KS tissue and in B cell lymphomas (Cesarman and Knowles, 1999). Ligands include both CC and CXC chemokines, with highest apparent affinity for IL-8 (Arvanitakis et al., 1997). The receptor is unique among chemokine receptors in being constitutively active. Several chemokine ligands, most potently GROα, can increase the activity in transfected mammalian cells and are therefore agonists (Gershengorn et al., 1998), whereas other chemokines, such as IP-10, vMIP-II, and SDF-1, decrease the activity and are inverse agonists (Geras-Raaka et al., 1998a,b;Rosenkilde et al., 1999). Signaling includes pertussis toxin-resistant phospholipase C activation in transfected COS cells and cell transformation. Activation of protein kinase C and cotransfection of GRK-5 have also caused down-regulation of activity (Geras-Raaka et al., 1998c). The transduction mechanism is not defined but does not appear to be Gi as for other chemokine receptors. Biological function includes angiogenesis and oncogenesis in transfected cells (Bais et al., 1998). KSHV GPCR may be responsible in part for KS pathogenesis. If so, the development of inverse agonists may be therapeutically useful.
D. UL12
The T lymphotropic β-herpesvirus HHV- 6 causes acute and latent infections and encodes two GPCR homologs, U12 and U51. The U12 gene is expressed late in infection from a spliced mRNA, and when expressed in transfected cells, the encoded protein functions as a calcium-mobilizing CC chemokine receptor specific for RANTES, MIP-1α, MIP-1β, and MCP-1 (Isegawa et al., 1998). Function in the context of HHV6 infected cells and biological roles in vivo have not been delineated.
E. E1
ORF E1 of equine herpesvirus 2 has high aa sequence similarity to CC chemokine receptors (Telford et al., 1995) and was recently demonstrated to mediate calcium flux and chemotaxis in response to eotaxin (Camarda et al., 1999).
The subject of viral chemokine and orphan chemokine receptor-like proteins is quite large and interesting, but unfortunately is beyond the scope of this review, which is limited to 7TMD chemokine binding proteins and receptors. Information about putative viral chemokine receptors, viral chemokines, and non-7TMD viral chemokine binding proteins, some of which have already been demonstrated to be virulence factors, is summarized in Table 5 and in recent reviews (Pease and Murphy, 1998; Lalani and McFadden, 1999).
IX. Conclusions
The attention paid to chemokine receptors has greatly increased as AIDS has merged with inflammation in a common area of pharmacological opportunity. Nevertheless, notably absent from most receptor descriptions given here are lists of potent, selective antagonists typically found for other types of GPCRs and well-defined indications in disease. This is in part because the field is young and has had an inverted history relative to classic receptors, which began with antagonists and even approved drugs before the molecular basis of action was known. In the future this will no doubt change for chemokine receptors, but the molecular information reviewed here already indicates reason to anticipate significant, idiosyncratic hurdles, including major cross-species differences in both structure and repertoires of receptors and ligands; unanticipated difficulties in identifying lead compounds, which has already been encountered for CXCR1; and extensive redundancy in the system, with regard to both endogenous ligands, nonchemokine classic chemoattractant receptors (Murphy, 1994), and biology. As these complexities are sorted out, it is the sincere hope of the committee that the nomenclature system described herein will help to prevent any ambiguity in communication about specific receptor targets.
Acknowledgments
We thank Josh Farber for critical reading of the manuscript and excellent suggestions.
Footnotes
-
↵1 Address for correspondence: Dr. Philip M. Murphy, Laboratory of Host Defenses, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 10, Room 11N113, Bethesda, MD 20892. E-mail: pmm{at}nih.gov
Abbreviations
- NC-IUPHAR
- Nomenclature Committee of the International Union of Pharmacology
- 7TMD
- seven-transmembrane domain
- HHV8
- human herpesvirus 8
- KS
- Kaposi's sarcoma
- NK
- natural killer
- GPCR
- G protein-coupled receptor
- HIV
- human immunodeficiency virus
- AIDS
- acquired immune deficiency syndrome
- mAb
- monoclonal antibody
- IL
- interleukin
- ORF
- open reading frame
- AOP
- amino-oxypentane
- aa
- amino acid(s)
- HCMV
- human cytomegalovirus
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
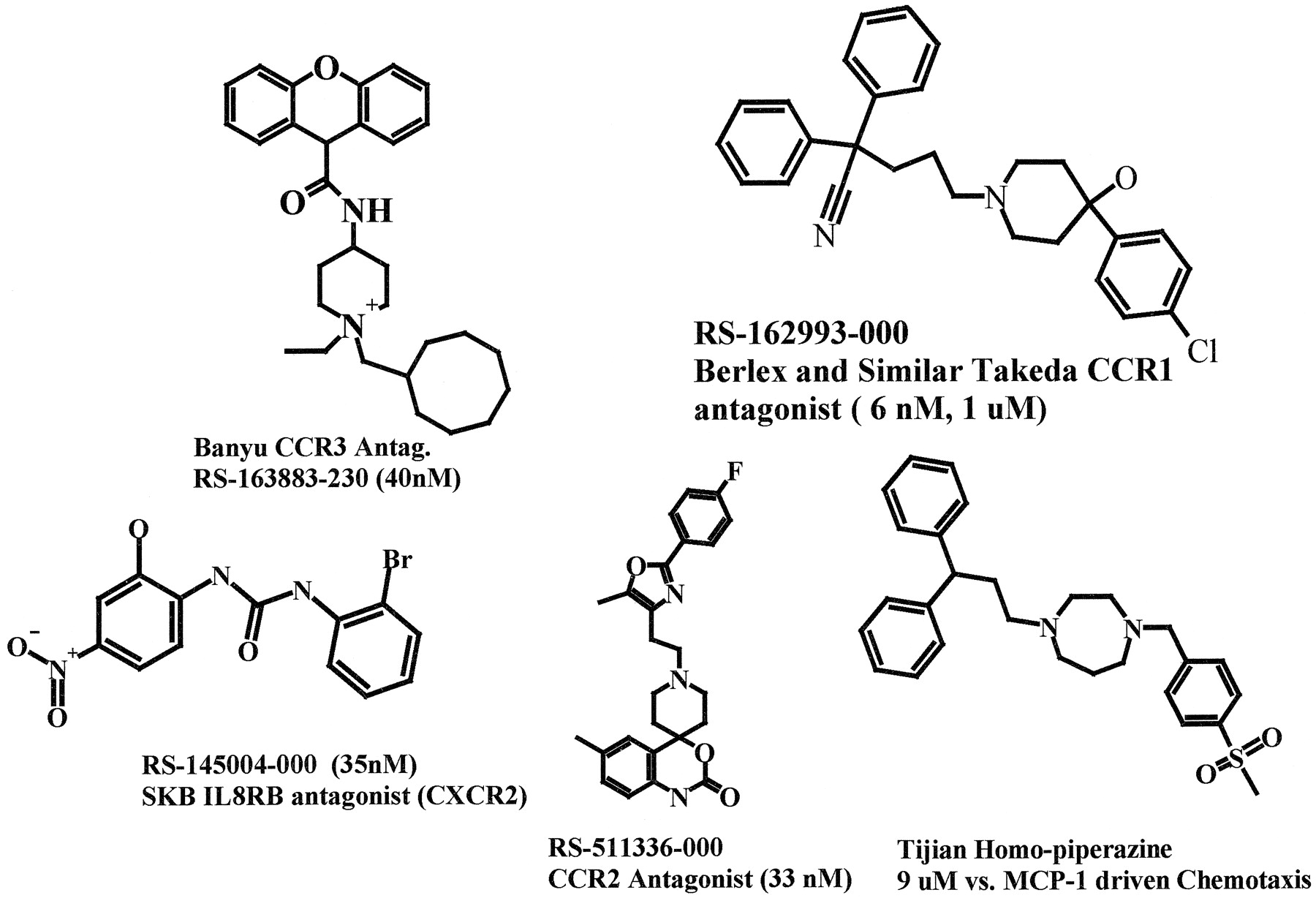{kind=link}
Jump to section
- Article
- Abstract
- I. Overview
- II. Introduction
- III. CXC Chemokine Receptor Subtypes
- IV. CC Chemokine Receptor Subtypes
- V. CX3C Chemokine Receptor Subtypes
- VI. Chemokine Receptor Subtypes
- VII. Chemokine Binding Proteins
- VIII. Virus-Encoded Chemokine Receptors
- IX. Conclusions
- Acknowledgments
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters