


Abstract
The type-1 parathyroid hormone receptor (PTHR1) is a family B G protein–coupled receptor (GPCR) that mediates the actions of two polypeptide ligands; parathyroid hormone (PTH), an endocrine hormone that regulates the levels of calcium and inorganic phosphate in the blood by acting on bone and kidney, and PTH-related protein (PTHrP), a paracrine-factor that regulates cell differentiation and proliferation programs in developing bone and other tissues. The type-2 parathyroid hormone receptor (PTHR2) binds a peptide ligand, called tuberoinfundibular peptide-39 (TIP39), and while the biologic role of the PTHR2/TIP39 system is not as defined as that of the PTHR1, it likely plays a role in the central nervous system as well as in spermatogenesis. Mechanisms of action at these receptors have been explored through a variety of pharmacological and biochemical approaches, and the data obtained support a basic “two-site” mode of ligand binding now thought to be used by each of the family B peptide hormone GPCRs. Recent crystallographic studies on the family B GPCRs are providing new insights that help to further refine the specifics of the overall receptor architecture and modes of ligand docking. One intriguing pharmacological finding for the PTHR1 is that it can form surprisingly stable complexes with certain PTH/PTHrP ligand analogs and thereby mediate markedly prolonged cell signaling responses that persist even when the bulk of the complexes are found in internalized vesicles. The PTHR1 thus appears to be able to activate the Gαs/cAMP pathway not only from the plasma membrane but also from the endosomal domain. The cumulative findings could have an impact on efforts to develop new drug therapies for the PTH receptors.
I. Introduction
The type-1 parathyroid hormone receptor (PTHR1) mediates the biologic actions of two distinct polypeptide ligands—the endocrine-acting parathyroid hormone (PTH) and the paracrine-acting PTH-related protein (PTHrP) (Fig. 1). The receptor, also referred to as the PTH/PTHrP receptor, is a member of the “family B” class of G protein–coupled receptors (GPCRs), which is comprised of 15 different receptors, each of which recognizes a moderately sized peptide ligand (Alexander et al., 2013). The PTHR1 is expressed in bone and kidney cells, where it mediates the calcium- and phosphate-regulating actions of PTH, and in a number of developing tissues, including the skeleton, where it mediates the morphogenetic actions of PTHrP. The PTHR1 couples primarily to the Gαs/adenylyl cyclase/cAMP/protein kinase A (PKA) signaling pathway; however, it can also couple to at least several other signaling cascades including the Gαq/phospholipase C (PLC)β/inositol trisphosphate (IP3)/intracellular Ca/protein kinase C (PKC) pathway, the Gα12/13-phospholipase D/RhoA pathway (Singh et al., 2005), and the mitogen-activated protein kinase (extracellular signal-regulated kinase [ERK1/2]) signaling cascade (Syme et al., 2005; Gesty-Palmer et al., 2006; Rey et al., 2006; Sneddon et al., 2007). Both ligands, PTH and PTHrP, show similar agonist response profiles in regard to activation of the different coupling pathways. The so-called type-2 parathyroid hormone receptor (PTHR2), discovered via a PTHR1 homology-based screening approach, binds a peptide ligand, called tuberoinfundibular peptide-39 (TIP39), and although the biologic role of the PTHR2/TIP39 system is not as defined as that of the PTHR1, it likely functions in the neuroendocrine system and is as well required for spermatogenesis (Table 1).

Ligand recognition and second-messenger signaling at PTHR1. The PTHR1 is a family B GPCR that mediates the actions of two peptide ligands—parathyroid hormone and PTH-related protein. In response to either ligand, the PTHR1 can couple to a variety of signal transduction pathways, including most prominently, the Gαs/cAMP/PKA pathway but also the Gαq/PLC/PKC pathway, the Gα12/13/RhoA/PLD pathway, and the ERK-1/2–MAP-kinase pathway, the latter via G protein–dependent and G protein–independent/β-arrestin–dependent mechanisms.
Properties of PTHR1 and PTHR2
Understanding the mechanisms by which PTH ligands mediate their actions has long been a focus of experimental study, beginning at least with the demonstration by Collip in the 1920s that an acid extract of oxen parathyroid gland could correct the tetany in parathyroidectomized dogs and concomitantly raise their blood calcium levels (Collip, 1925). The eventual purification and Edman degradation analysis of the active substance in the 1970s revealed the single-chain PTH(1–84) polypeptide sequence (Brewer and Ronan, 1970; Niall et al., 1970). This led soon thereafter to the chemical synthesis of the PTH(1–34) peptide fragment, which proved to be equipotent to the intact hormone (Potts et al., 1971), and indeed the PTH(1–34) peptide has since proven to be the mainstay scaffold for structure-activity relationship studies performed on the hormone and its receptor. Moreover, recombinant PTH(1–34) has been developed as the first pharmaceutical targeted to the PTHR1 and is thus in use for the treatment of osteoporosis and has since been joined in this regard only by intact PTH(1–84) (Bilezikian et al., 2005; Baron and Hesse, 2012). The current pharmaceutical application of PTH peptides for osteoporosis is based on their capacity to stimulate new bone formation, but clinical trials are also underway to use the peptides to treat the smaller-scale population of patients with hypoparathyroidism (Winer et al., 2012; Mannstadt et al., 2013). For either treatment group, frequent (e.g., daily) dosing is required, and via, because of the peptidic nature of the drug, a nonparenteral (i.e., subcutaneous injection) route of delivery.
Given the proven efficacy of such PTH ligand administration to stimulate bone anabolism in patients with osteoporosis and to normalize serum calcium in patients with hypoparathyroidism, there stands considerable interest in the development of an orally active, nonpeptidic compound that acts as a potent agonist at the PTHR1, yet despite extensive drug discovery effort, no such compound has been reported. The reason for the apparent failure to find such a mimetic agonist for the PTHR1 remains unresolved, but the problem can be seen as a common thread within the family B peptide hormone GPCRs, which, as a class, have yielded only very few small molecule agonist ligands and likely relates to their basic molecular modes of action (Hoare, 2007; Koole et al., 2013). Defining these modes of action could thus provide insights not only into the basic mechanism by which these receptors function but also into how they might be better targeted for the development of more effective therapeutic agents. (For more information see: http://www.iuphar-db.org/DATABASE/FamilyMenuForward?familyId=53 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=53&familyType=GPCR.)
II. Biologic Actions at the Type-1 Parathyroid Hormone Receptor In Vivo
A. Parathyroid Hormone
Ionized calcium (Ca2+) plays an essential role in numerous biologic processes and cell functions and thus must be available in ready and reliable supply. Parathyroid hormone plays a key role in maintaining the levels of Ca2+ in the blood and extracellular fluids and thus acts to control the minute-by-minute fluctuations that occur in these levels throughout the day. The hormone is secreted from the parathyroid gland as blood Ca2+ levels fall below a set point level, which in humans is ∼1.2 ± 0.1 mM, and then acts on specific bone and kidney cells to increase their rates of calcium mobilization. In bone, PTH acts on surface osteoblasts as well as on the bone-embedded osteocytes, and these cells respond by producing the receptor activator of nuclear factor-κB ligand, which, in turn, acts on osteoclasts to stimulate their rates of bone matrix resorbtion and mineral release (Silva et al., 2011; Boyce et al., 2012; O'Brien et al., 2013; Saini et al., 2013). In the kidney, PTH acts on cells of the distal tubules to increase their rates of Ca reabsorption via effects on calcium transport proteins, including the transient receptor potential vanilloid type V channel active on the lumenal cell surface (van Abel et al., 2005; de Groot et al., 2009). In renal proximal tubule cells, PTH acts to decrease the rate at which inorganic phosphate (Pi) is reabsorbed, thus reducing the blood levels of this counter-ion and ensuring the increased Ca in the blood remains in free ionized form. The hypophosphatemic/hyperphosphaturic effect of PTH occurs via reductions in the levels of expression of the sodium-dependent phosphate transporter type 2A on the lumenal brush border membrane of the renal proximal tubule cells (Biber et al., 2009; Picard et al., 2010; Nagai et al., 2011). In addition to these effects of PTH on Ca and Pi mobilization processes, which are rapid responses occurring within minutes of hormone exposure, PTH further acts in renal proximal tubule cells to increase the levels of the 25-hydroxyvitamin D3 1-α-hydroxylase, in turn resulting in increased blood levels of active vitamin D3, which has independent but slower-acting effects on blood Ca levels, thus providing for a longer-term period (hours to days) of defense against hypocalcemic risk. PTH also plays a role in the bone-remodeling process that goes on continuously and involves coordinated actions of osteoblasts and osteoclast (Miao et al., 2004; Martin et al., 2008).
B. Parathyroid Hormone–Related Protein
PTH-related protein was discovered in the 1980s as the long-sought tumor-released factor responsible for the hypercalcemia of malignancy seen frequently in cancer patients. The factor, which had long been suspected of being a PTH-like entity, was indeed found to be a polypeptide that not only induced the same effects on bone and kidney cells as PTH did, but shared amino acid sequence homology with PTH, at least within the N-terminal region that was known to be critical for activity (Fig. 2) (Suva et al., 1987; Nissenson et al., 1988; McCauley and Martin, 2012). A true biologic role for PTHrP was then established in 1994 by the gene “knockout” approach, because the PTHrP-null mice died at birth with developmental abnormalities, particularly affecting the skeleton (Karaplis et al., 1994). The growth plates of the developing long bones of these mice thus showed perturbed patterns of cell differentiation, resulting in abnormal bone elongation and mineralization. PTHrP was thus found to be produced by cells at the peripheries of the developing bone template and to act in a paracrine fashion on the chondrocytes within the template to promote their proliferation and slow their terminal differentiation toward the hypertrophic state (Kronenberg, 2006). It is now clear that in addition to bone, PTHrP plays a role in the development of several other tissues, including the mammary glands (Wysolmerski et al., 1998) and teeth (Philbrick et al., 1998), and is widely expressed among various organ systems, suggesting a generally broad functional role as a paracrine-acting regulatory factor (Trivett et al., 2005; McCauley and Martin, 2012).

Sequences of PTH, PTHrP TIP39. Sequences of the bioactive 1–34 portions of PTH and PTHrP and the intact TIP39 sequence are shown with residues that are identical in PTH and PTHrP and retained in TIP39 shown red fill. The bars represent peptide fragment lengths that correspond to approximate minimum-length functional domains of the bioactive peptides.
III. Structure-Activity Relationships in Parathyroid Hormone and Parathyroid Hormone–Related Protein Ligands
A. Parathyroid Hormone
The chemical synthesis of PTH(1–34) in 1970 by Potts and colleagues and their demonstration that the N-terminal fragment was as potent as the intact native hormone, PTH(1–84), for inducing biologic responses in cells and in animals (Potts et al., 1971) launched a broad program of studies aimed at determining the structural features in the hormone that determine its function. Thus, peptide truncation and substitution approaches revealed that the key determinants of hormone signaling and receptor binding reside within the N-terminal and C-terminal portions of the PTH(1–34) fragment, respectively. One key early observation was that the PTH(3–34) fragment exhibited markedly reduced biologic activity (Tregear et al., 1973; Goltzman et al., 1975), a finding that, in turn, led to the development by Rosenblatt and colleagues, of the first and still currently most effective PTH antagonist peptides, based largely on the PTH(7–34) scaffold fragment (Fig. 2) (Rosenblatt et al., 1977; Segre et al., 1979; Horiuchi et al., 1983). PTH(1–34)-based peptides that have the highly conserved valine-2 replaced by bulkier amino acids, such as tryptophan, benzoyl-phenylalanine (BPA), or arginine, also exhibit antagonist properties, although typically with more residual partial agonism than found in the PTH(7–34) peptides (Gardella et al., 1991, 1994; Carter et al., 1999a; Behar et al., 2000).
Further structure-activity relationship studies revealed that the PTH(15–34) sequence comprised the hormone's principal receptor-binding domain, because the PTH(15–34) peptide was the smallest fragment that retained at least some capacity to inhibit the binding of [125I]PTH(1–34) radioligand to the receptor, albeit this binding was quite weak, with a Ki value of ∼1 × 10−6 M, compared with that of PTH(1–34), which exhibits a Ki value of around 1 × 10−9 M (Nussbaum et al., 1980; Rosenblatt et al., 1980). Amino-terminal PTH fragments, such as PTH(1–14), could be inferred from the antagonist studies to contain the principal determinants of receptor signaling, but initially such peptides were found to be inert. Eventually, peptide optimization studies revealed that such N-terminal PTH peptides did in fact possess the expected intrinsic signaling activity (see below).
B. Parathyroid Hormone–Related Protein
As for PTH, the (1–34) fragment of PTHrP, which in total is 141 amino acids in length, is fully active on the PTHR1 (Kemp et al., 1987; Nissenson et al., 1988), and the principal determinants of receptor-signaling potency and binding affinity reside within the N-terminal and C-terminal portions of that peptide, respectively. Consistent with a key role in receptor activation, the N-terminal portion of PTHrP exhibits the highest homology with PTH, because 8 of the first 13 amino acids are identical. The (15–34) region of PTHrP shares only two identities with PTH—arginine-20 and leucine-24 (Fig. 2); however, similarity is preserved, suggesting a common mode of action, and indeed, the (15–34) fragment of PTHrP was found to inhibit the binding of intact [125I]PTH(1–34) radioligand to the receptor as effectively as PTH(15–34) fragment (Caulfield et al., 1990). The early supposition that the principal binding domains of PTH and PTHrP adopt similar conformations when binding to the receptor has now largely been confirmed by the recent crystallographic analyses of each of ligand fragment in complex with the same binding portion of the receptor, as discussed further below.
The biologic roles, if any, of the portions of PTH and PTHrP that extend C-terminally from position 34 are unknown. These regions of the two ligands share little or no amino acid sequence homology, and they do not appear to contribute importantly to interactions with the PTHR1. This is demonstrated by the finding that chemically synthesized PTH(1–84) and PTHrP(1–141) polypeptides each exhibit binding affinities and signaling potencies on the PTHR1 that are indistinguishable from those of the counterpart N-terminal peptide fragment (Dong et al., 2012; Li et al., 2012). It nevertheless remains possible that the C-terminal regions of the ligands modulate, directly or indirectly, interaction with the PTHR1 in target tissues in vivo. It is also interesting to consider that the differences in the C-terminal regions of the two ligands account, to some extent, for the distinct modalities by which the two ligands carry out their biologic functions—i.e., the endocrine modality by which PTH controls Ca and Pi homeostasis versus the paracrine modality by which PTHrP controls cell differentiation programs. For PTH, it has been shown that C-terminal peptide fragments derived from the secreted hormone can be detected in the circulation (Bringhurst et al., 1982; D'Amour et al., 2005), and synthetic C-terminal fragments can bind with moderate affinities to some non-PTHR1, but as yet unidentified binding sites in cultured cells (Divieti et al., 2005). There is thus possibility that the C-terminal domains of PTH and PTHrP can function autonomously and independently of the PTHR1.
A large number of solution-phase NMR studies have been conducted on various PTH and PTHrP peptides in efforts to determine their most stable and potentially bioactive three-dimensional conformation. Although a good deal of variation is seen in the structures obtained from these studies, depending on the specific experimental conditions, e.g., peptide sequence, solvent systems used, a generally consistent finding is the presence of a relatively stable α-helical segment that extend for several turns within the approximate (15–34) region, with the N-terminal portion tending to be more random or to contain a short segment of weak α-helix (Barden and Kemp, 1994, 1996; Marx et al., 1998, 2000; Chen et al., 2000). TIP39 has also revealed a bihelical domain structure (Piserchio et al., 2000b) (Fig. 3).
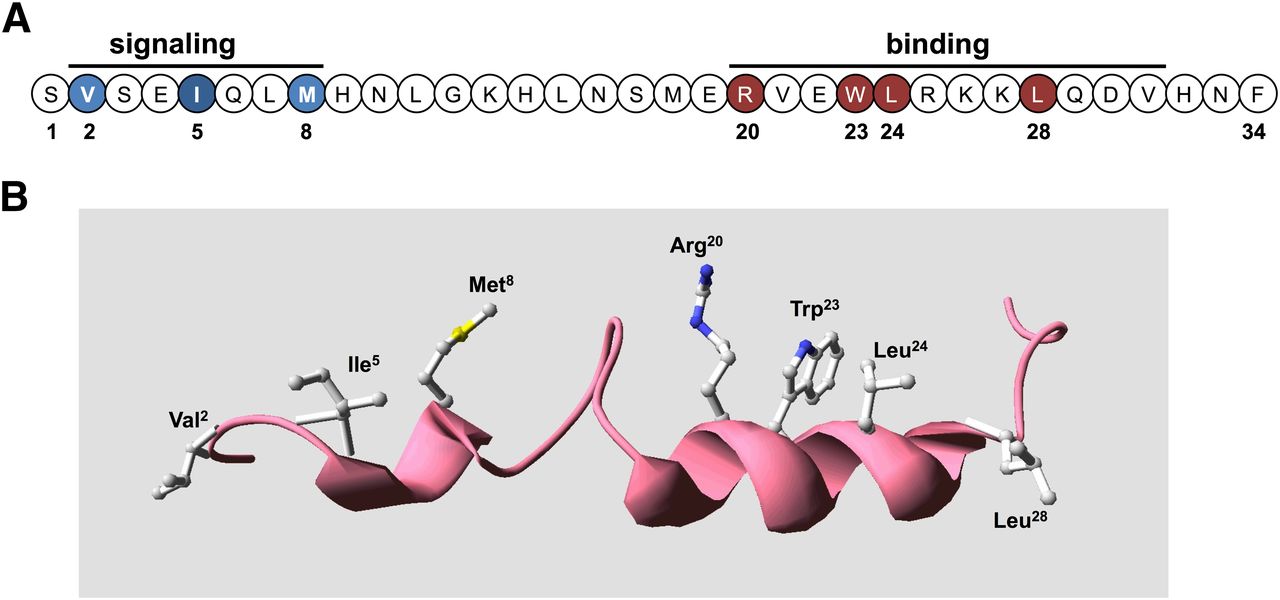
Bioactive domains and key functional determinants of PTH(1–34) receptor ligands. (A) Shown is the sequence of the bioactive (1–34) segment of parathyroid hormone in linear format with residues in the amino-terminal portion that are critical for signaling shown in blue and those in the C-terminal domain that are critical for binding shown in red. (B) The PTH(1–34) ligand is presented in three-dimensional ribbon format to display the two-helical-domain structure typically observed in solution-phase NMR studies, with the side chains shown of the key binding and signaling residues. The structure was generated using the solution-phase NMR-derived coordinate file PDB ID 1HPH deposited in the Protein Data Bank (Marx et al., 1995).
The presence of backbone flexibility or even a β-turn motif between the amino and C-terminal domain has raised the possibility that the bioactive PTH(1–34) peptide ligand adopts a folded or U-shaped structure (Marx et al., 1998, 2000; Chen et al., 2000). One of the NMR studies, however, specifically addressed this question for PTH(1–34) and failed to find evidence for interdomain tertiary interactions and thus argued against such a U-shaped structure (Pellegrini et al., 1998). A crystallographic analysis of PTH(1–34) revealed a single continuous α-helix that extended from the N terminus to the C terminus.
In both PTH and PTHrP, the C-terminal α-helix in the (15–34) domain is seen to be amphipathic in nature, with opposing hydrophobic and hydrophilic faces, and mutational analyses of this region in PTH revealed the importance of residues on the hydrophobic face of the helix, particularly Trp23, Leu24, and Leu28, for binding to the receptor (Gardella et al., 1993; Oldenburg et al., 1996). In PTHrP the corresponding residues are Phe23, Leu24, and Ile28. It is now clear from these considerations and especially the recent X-ray crystallographic studies discussed below that the (15–34) domains of both PTH and PTHrP do indeed adopt similar conformations, an amphipathic α-helix, when binding to the receptor and in fact occupy nearly the same contact surface within the receptor's binding domain.
At least two observations suggest that the N-terminal portion of the ligand adopts at least some α-helical structure in the bioactive state. First, the deletion of residues 1 and 2 in PTH peptides has been seen in solution phase NMR studies to result in a marked loss in the N-terminal α-helix (Marx et al., 1998). Valine-2, one of the most conserved residues in PTH and PTHrP ligands and a critical determinant of receptor activation, may thus not only form direct contacts with the receptor but help stabilize the bioactive ligand conformation. Second, as assay systems became available that used the cloned PTHR1 overexpressed in heterologous cells, weak cAMP agonist activity was revealed for short N-terminal PTH fragment peptides, such as PTH(1–14), and through a series of structure-activity relationship studies aimed at optimizing such N-terminal peptides, a number of modifications were identified that in aggregate not only greatly improved binding and cAMP signaling potency in the so-called “M” modified M-PTH(1–14) analogs (Luck et al., 1999; Shimizu et al., 2000a,b, 2001a,b) but also markedly increased their peptide α-helical structure, as assessed by solution-phase NMR spectroscopy analyses (Tsomaia et al., 2004) (Fig. 4). The N-terminal modifications, which typically include conformationally constraining amino acid analogs, such as α-amino isobutyric acid substituted at positions 1 and/or 3, also conferred detectable cAMP-signaling potency to the otherwise inactive PTH(1–11) peptide (Shimizu et al., 2001a,b, 2004), along with at least some α-helicity (Barazza et al., 2005; Fiori et al., 2007; Caporale et al., 2009a,b, 2010; Cupp et al., 2013b). It thus may be that for the native ligand, the N-terminal domain becomes structurally more organized as an α-helix upon binding to the receptor (Shimizu et al., 2001b, 2004), as has been suggested for other peptide ligands that bind to a family B GPCR (Parthier et al., 2009).

Optimized PTH(1–14) domain. Shown is the sequence of the human PTH(1–14) peptide and the six amino acid substitutions that comprise the “M” set of modifications that together increase cAMP signaling potency of the PTH(1–14) fragment by five orders of magnitude and stabilize α-helical structure in the otherwise disordered PTH(1–14) peptide (Tsomaia et al., 2004). The graphs depict dose-response curves for cAMP generation obtained for PTH(1–34), M-PTH(1–14), and native PTH(1–14) in COS-7 cells transiently transfected to express either the intact wild-type PTHR1 (left) or the PTHR1-delNT construct (right), which is deleted for most of the ECD. These data reveal that whereas potency of PTH(1–34) is about 100-fold weaker on PTHR1-delNT than on the wild-type PTHR1, the PTH(1–14) fragments exhibit the same potency on the two receptors. The data thus demonstrate that whereas PTH(1–34) requires both the ECD and the TMD regions to obtain full potency, the PTH(1–14) portion of the ligand only interacts with the TMD region of the receptor (Shimizu et al., 2001b).
The collective body of research on PTH peptides and analogs and their biologic actions that originated with the first synthesis of PTH(1–34) by Potts et al. (1971) and expanded deeply and broadly to include the studies highlighted above as well as numerous others not mentioned here, has provided for a solid framework on which to approach the higher-order problem of elucidating the specific molecular mechanisms by which the ligands specifically bind to the PTH receptor and trigger signal transduction processes. This next level of study is the focus of the sections that follow.
IV. The Type-1 Hormone Receptor Gene Identification, Classification, and Structure
The PTH receptor was initially identified in molecular terms in 1991 with the cloning of cDNAs encoding a protein with the capacity to bind [125I]PTH(1–34) from libraries of mRNA prepared from kidney and bone cell lines (Jüppner et al., 1991; Abou-Samra et al., 1992). Although the receptor sequence exhibited the seven hydrophobic transmembrane domain motif typical of the GPCR class, it otherwise lacked any direct homology with most of the GPCRs identified at the time, such as the β2-adrenergic receptor. Instead the receptor showed similarity to the newly identified receptors for secretin and calcitonin, and it was thus realized that these receptors formed a distinct GPCR subgroup, thenceforth called the family B GPCRs (Barwell et al., 2012; Couvineau and Laburthe, 2012). The family B GPCR subgroup is comprised of 15 distinct receptors, each of which binds a peptide hormone ligand, which, in addition to PTH, calcitonin, and secretin, also include glucagon, glucagon-like peptide-1, corticotrophin-releasing factor (CRF), and several others (Hollenstein et al., 2014). The sequence homologies within the related family B GPCRs predict at least some similarity in their modes of action, and indeed each binds a ligand that is a moderately sized, single-chain polypeptide of 30 to 40 amino acids in length; the obvious, if not peculiar, exception to this is the PTHR1, which binds the longer-length PTH (84 amino acids) and PTHrP (141 amino acids), yet for which the (1–34) portions retain full binding and signaling activity.
There was little doubt early on that the receptor identified through the cloning efforts was the true biologic receptor for PTH, in that the clones were obtained from cDNA libraries derived bone and kidney cells, and subsequent tissue distribution analyses revealed the robust expression patterns in the expected bone and kidney target sites (Urena et al., 1993). That the same receptor was also the true biologic mediator of the actions of PTHrP, however, was not firmly established until somewhat later when gene knockout studies showed that PTH receptor-null mice died at birth with nearly the same skeletal and developmental defects as the PTHrP-null mice (Lanske et al., 1996; Vortkamp et al., 1996). It is thus now clear that, in a somewhat unusual biologic arrangement, the one GPCR identified mediates the distinct biologic actions of two different endogenous peptide ligands, PTH and PTHrP.
In humans, the gene encoding the PTHR1 is located on chromosome 3p and consists of 14 coding exons. The mature mRNA encodes a protein that of 593 amino acids, including a 22-amino acid N-terminal signal peptide that is removed during intracellular processing. The basic protein topology is defined by a relatively large amino-terminal extracellular domain (ECD) of ∼165 amino acids, a transmembrane domain (TMD) region containing the seven membranes spanning helices and interconnecting loops, and a carboxy-terminal tail of about 130 amino acids (Fig. 5). The ECD of the PTHR1 contains a 45 amino acid segment that is encoded by a separate exon, called E2, that is not found in the other family B receptors, including the PTHR2, and, in fact, can be deleted without affecting hormone binding or signaling potency (Lee et al., 1994).

The PTH receptor type 1. This “snake” diagram of the human the PTHR1 illustrates the receptor's 593 amino acids in a topological arrangement typical of the family B GPCRs. The receptor thus has a relatively large amino ECD of about 160 amino acids (minus the 23 amino acids of the N-terminal signal sequence) that are involved in initial ligand binding, the seven helical transmembrane domains and connecting loops that mediate agonist-induced receptor activation and signal transduction events, and a C-terminal tail of about 130 amino acids that contains sites involved in mediating ligand-induced receptor internalization, trafficking, and signal termination events. Key specific amino acids identified include the four pairs of extracellular cysteine (C) residues that form a disulfide bond network that is conserved in the family B GPCRs and maintains receptor structure and function (Lee et al., 1994; Pioszak and Xu, 2008; Pioszak et al., 2009); four glycosylated asparagine (N) residues in the ECD (Zhou et al., 2000); Thr33 and Gln37, which modulate interaction with tryptophan-23 in the ligand (Mannstadt et al., 1998; Mann et al., 2008); Phe184 and Arg186, which mediate interactions involving ligand residues at or near lysine-13 (Adams et al., 1998; Carter et al., 1999a); Ser370, Ile371, Met425, Trp437, and Gln440, which contribute interactions involving ligand residues at or near valine-2 and likely play a role in receptor activation (Gardella et al., 1994; Lee et al., 1995; Bisello et al., 1998; Behar et al., 1999; Gensure et al., 2001a); Arg233 and Gln451, which participate in an interhelical interaction network (dashed connectors) that likely helps modulate PTHR activation (Gardella et al., 1996a) and is conserved in the family B GPCRs (Hollenstein et al., 2013); conserved Pro132 in the ECD, which is the site of an inactivating mutation (Leu) in Blomstrand’s chondrodysplasia (Zhang et al., 1998); His223, Thr410, and Ile458, at which mutations result in constitutive signaling activity and in patients result in Jansen’s chondrodysplasia (Schipani et al., 1999); Lys319, at which mutations impair Gαq signaling (Iida-Klein et al., 1997); Lys388, at which mutations impair Gαq and Gαs signaling (Huang et al., 1996). Key residues in the C-tail include the seven serine (S) residues that are phosphorylated upon agonist activation and mediate recruitment of β-arrestins (Malecz et al., 1998; Qian et al., 1998; Tawfeek et al., 2002; Vilardaga et al., 2002; Rey et al., 2006) and the C-terminal ETVM sequence that mediates interaction with the NHERF family of proteins (Mahon et al., 2002, 2003; Ardura et al., 2011; Mamonova et al., 2012). In each transmembrane domain, the residue identified as the most conserved residue among the family B GPCRs is enclosed in a hexagon (Wootten et al., 2013).
V. The Parathyroid Hormone-2 Receptor and Tuberoinfundibular Peptide-39
A. The Type-2 Parathyroid Hormone Receptor
PTHR2 was identified in 1995 through a homology-based cDNA cloning strategy aimed at identifying paralogs of the previously identified PTH/PTHrP receptor (PTHR1) (Usdin et al., 1995). The cDNA library was derived from human brain tissue, and the sequence revealed an amino acid coding region that shared 51% identity with the hPTHR1. The tissue distribution profile of the PTHR2, assessed by mRNA expression analysis in the rat, was also distinct from that of the PTHR1, because the PTHR2 was detected at various loci in the brain, including the hypothalamus, in pancreatic islet somatostatin cells, thyroid parafollicular cells, gastrointestinal secretory cells; as well as in the heart and vasculature (Usdin et al., 1996).
Initial functional studies on the PTHR2 expressed heterologously in HEK-293 cells or COS-7 cells revealed a ligand recognition profile distinct from that of the PTHR1 in that the human PTHR2 variant responded potently in cAMP and intracellular Ca signaling assays to PTH(1–34) but not to PTHrP(1–34) (Usdin et al., 1995; Behar et al., 1996). Subsequent studies, however, revealed that the PTHR2 isolated from the rat responded poorly not only to PTHrP but also to PTH (Hoare et al., 1999). This finding supported the view that the endogenous ligand for the PTHR2 was not PTH or PTHrP, but rather some other distinct ligand. Evidence for such a ligand was indeed provided by the finding that extracts of bovine hypothalamus tissue contained a protein factor that could stimulate the rat or human PTHR2 more effectively than either PTH or PTHrP (Usdin, 1997; Hoare et al., 1999). The active factor was purified and sequenced and thus shown to be a polypeptide of 39 amino acids, called TIP39 (Usdin et al., 1999; Usdin, 2000). The identified peptide sequence shares only modest homology with PTH and PTHrP and includes a two-amino acid N-terminal extension (Fig. 2). The sequence maintains the predicted C-terminal amphipathic α-helix, and in solution phase NMR studies, TIP39 exhibits the bihelical structure fairly typical for the PTH and PTHrP ligand family (Piserchio et al., 2000b).
B. Tuberoinfundibular Peptide-39; Functional Properties
TIP39 exhibits strong receptor selectivity as it potently activates the PTHR2 but is inactive on the PTHR1 and binds to the PTHR2 with approximately 20-fold higher affinity than it does to the PTHR1 (Hoare et al., 2000a). As with PTH ligands acting on the PTHR1, the deletion of the N-terminal residues of TIP39 markedly reduces the signaling potency of the ligand on the PTHR2, while having a relatively modest impact on PTHR2 binding affinity, such that N-terminal fragments, such as TIP(7–39) and TIP(9–39) exhibit antagonist properties on the PTHR2 (Hoare et al., 2000a; John et al., 2002). Interestingly, such N-terminal truncations tend to reverse the binding selectivity seen for the TIP(1–39) ligand, in that they lower binding affinity for the P2R but improve affinity for the P1R, such that TIP(7–39) binds to the PTHR1 with a 20-fold greater affinity than it does to the PTHR2 (Hoare et al., 2000a; Hoare and Usdin, 2000). This increase in PTHR1 binding affinity is not accompanied by any gain in signaling activity, and as a result, TIP(9–39) and TIP(7–39) function as potent antagonists on the PTHR1 and are at least as effective as the classic PTHR1 antagonists based on the PTH(7–34) scaffold (Hoare et al., 2000a; Hoare and Usdin, 2000, 2002; Jonsson et al., 2001).
In a search for novel ligands that would act as selective antagonists for the PTHR2, and thus ultimately might be useful in defining the biologic role of this receptor in vivo, Kuo and Usdin (2007) performed a site-directed mutagenesis-based analysis of the TIP39 sequence, searching the mutants for absence of cAMP signaling activity on the PTHR2. The study yielded the TIP(1–39) analog having the sequence Leu-Ala-Asp-Asp at positions 4–7 replaced by the sequence His-Tyr-Trp-His. In cell-based assays, the new analog, called HYWH-TIP39, was shown to be 15-fold more effective for inhibiting agonist-induced signaling at the PTHR2 than at the PTHR1 (Kuo and Usdin, 2007). Thus, HYWH-TIP39, indeed shows the opposite receptor antagonist selectivity profile as TIP(7–39). Although the new analog has not been assessed in vivo, it could potentially be useful as a pharmacological reagent with which to explore the biologic role of TIP39 and the PTHR2 in normal physiology.
Clues about the potential biologic roles of TIP39 come from several lines of investigations that focus on anatomic gene expression patterns as well as functional responses to ligand or receptor modulation. Thus in situ hybridization studies in mice reveal generally wide tissue distribution patterns for TIP39 mRNA, but high levels are seen in several nuclei of the brain, including the hypothalamus and brain stem areas (subparafascicularis thalmi) as well as in the testis (Dobolyi et al., 2002; John et al., 2002). In functional tests of neuronal actions, administration of TIP39 into rats either peripherally via subcutaneous injection or centrally via intracerebroventricular cannulation, was found to increase blood levels of adrenocorticotropic hormone as well as luteinizing hormone, results that suggest a neuroendocrine role of the ligand in the hypothalamo-pituitary-adrenal and hypothalamo-pituitary-gonadal axes (Ward et al., 2001). In other studies performed in mice, microinjection of TIP39 locally into the plantar region of the paw was found to induce paw-withdrawal responses, a result consistent with a potential facilitatory role of TIP39 in nociception via activation of the PTHR2 on sensory neurons (Dobolyi et al., 2002). A clear role of TIP39 in the testis has been established by genetic knockout studies in mice. Thus, ablation of the TIP39 gene was found to result in sterility in males, and the testes of the mutant mice were devoid of spermatids (Usdin et al., 2008). Thus TIP39 is essential for normal spermatogenesis. The mechanism has not been fully elucidated, but the ligand likely acts in an autocrine or paracrine fashion to promote sperm cell development, as the phenotype could be rescued by genetic expression of TIP39 locally within the spermatids (Usdin et al., 2008). There is no evidence to suggest that endogenous TIP39 has any action upon the PTHR1 or that either PTHR2 or TIP39 plays a role in mineral ion metabolism or have actions on bone.
VI. Evolution of Parathyroid Hormone Receptors and Their Ligands
In evolutionary terms, the PTH receptor system appears to be ancient. Orthologs of the PTHR1 can be detected in species as distant as fish and birds (Cardoso et al., 2006; Bhattacharya et al., 2011; Suzuki et al., 2011; Pinheiro et al., 2012), and a sequence with some 50% amino acid identity to the human PTHR1 is apparent in the genome of the sea squirt Ciona intestinalis (Kamesh et al., 2008) (Fig. 6A). In addition, PTH family ligands have been identified in representatives of the early vertebrate groups of fish and birds (Fig. 6B), providing evidence for the expected parallel coevolution of ligand-receptor pairs. A recent evaluation of family B GPCRs in the genomes of various invertebrate species revealed putative receptors in insects and nematodes that align with a subgroup of human family B GPCRs comprised of the PTHR1 along with the receptors for secretin and glucagon; the findings would thus suggest that PTH receptors originated before the protostome-deuterostome divergence that is estimated to have occurred about 1000 million years ago (Cardoso et al., 2014).

Phylogenetic relationships among PTH receptors and ligands from different species. (A) The amino acid sequences of PTH receptors from humans, zebrafish (Danio rerio) and chicken were aligned after removal of the predicted signal peptides and the segment of the human PTH1R encoded by exon E2 using the ClustalW(2.012) program (gap penalties: opening, 10; extending, 0.2 multiple, 0.1 pairwise) and an unrooted tree in which branch distances indicate amino acid sequence divergence was generated using the Phylip(3.67) DrawTree program. The diagram illustrates the three subtypes of PTH receptors, for which the PTH1R is present in all vertebrates, the PTH2R is present in humans and fish (Danio) but absent in birds, and the PTH3R is present in birds and fish but not in higher vertebrates. The tree includes a PTHR-like sequence that was identified in the genome of the tunicate Ciona intestinalis and which shows ∼35% overall identity to the human PTHR1. The Protein database accession numbers for the sequences used are as follows: human PTH1R, Q03431; chicken PTH1R, 418507; Danio PTH1R, Q9PVD3; chicken PTH3R, Danio PTH3R, Q9PVD2; Ciona PTHR-L, ci0100139945; human PTH2R, P49190; Danio PTH2R, Q9PWB7. (B) Also shown is an alignment of the (1–37) portions of PTH and PTHrP ligands and full-length TIP39 from human, zebrafish (D. rerio), and chicken species. Key residues involved in signaling, aligning with Val2, Ile5, and Met8 in the human PTH sequence, and binding, aligning with Arg20, Trp23, Leu24, and Leu28, are colored green and blue, respectively. Database accession numbers are shown for each ligand along with the specifies-ligand identifying label.
Apparent gene-duplication events occurring during evolution, and the speciation process have given rise to paralogs of the PTH receptor, which may be present in certain species but not others. The so-called PTH-3 receptor (PTHR3) is thus found in birds and fish but not in mammals; whereas the PTHR2 is found in fish but it is absent in birds. The PTHR3 is more closely related in amino acid sequence to the PTHR1 than the PTHR2 (Fig. 6A), and functional studies performed on the zebrafish (z)PTHR3 show that it binds and responds to both PTH and PTHrP peptides but not the PTHR2-selective ligand TIP39 (Rubin and Jüppner, 1999; Hoare et al., 2000b; Bhattacharya et al., 2011). Although initial studies on the zPTHR3 indicated that it bound PTHrP but not PTH, subsequent analyses revealed that this apparent selectivity was limited to the human-derived PTH ligand, because rat PTH(1–34) was found to bind and activate the zPTHR3 at least as well as PTHrP (Hoare et al., 2000b). As of yet, no biologic function or intended ligand has been identified for the PTHR3 in birds and fish, and so whether it plays a role in adapting to the unique demands on calcium regulation associated with life in aquatic environments or egg shell calcification is unknown. In parallel with the receptors, apparent homologs of the known PTHR ligands, PTH, PTHrP, and TIP39, have each been detected in the teleost fish, in which the genes are typically duplicated to give rise to two isoforms of each ligand (Power et al., 2000; Danks et al., 2003; Gensure et al., 2004; Papasani et al., 2004; Hogan et al., 2005; Guerreiro et al., 2007). Immuno-staining methods have also detected PTHrP in the lamprey, a jawless fish that is thought to represent a time in the evolutionary past of some 540 million years ago (Trivett et al., 2005).
VII. Structural Features of the Type-1 Parathyroid Hormone Receptor and Mode of Ligand Binding
A. Two-Site Model of Binding
The mode of ligand binding and activation used by the PTHR1 has been approached using biochemical and mutational methods that employ various mutant receptors and altered ligand analogs. These studies have yielded considerable mechanistic insights and have together led to the so-called “two-site” model of binding for the PTH/PTHR interaction. Thus, by this model, the C-terminal portion of the PTH(1–34) fragment, representing the principal binding domain, contacts the N-terminal ECD region of the receptor to establish initial docking interactions, and then the N-terminal portion of the ligand, containing the principal determinants of signaling, interacts with the TMD portion of the receptor to induce the conformational changes involved in receptor activation and G protein coupling (Fig. 7).
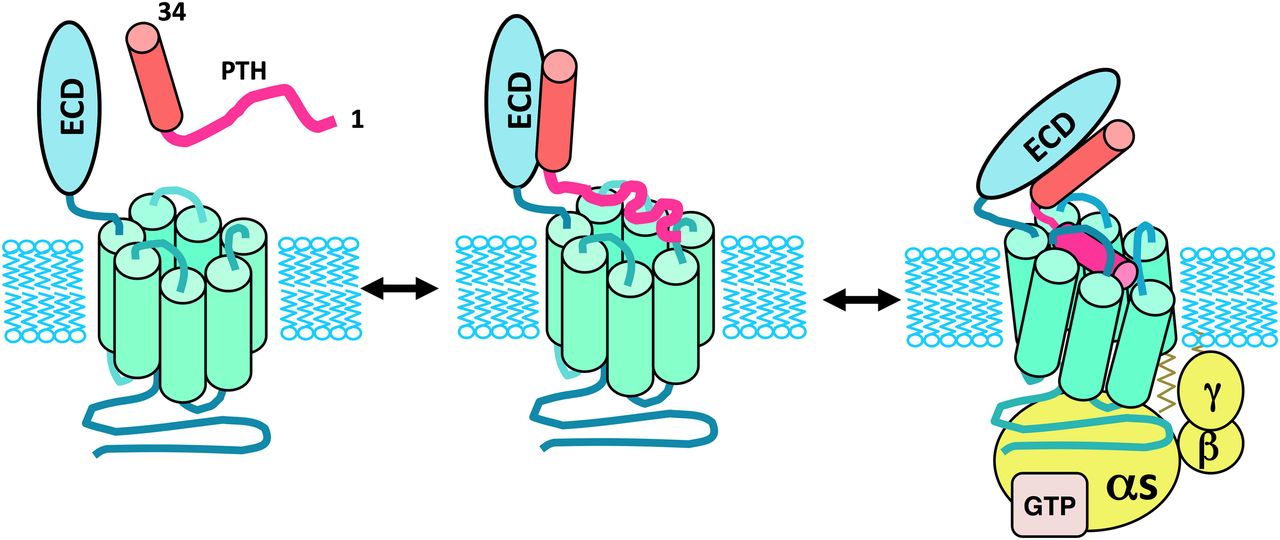
Two-site model of the PTH/PTHR1 interaction mechanism. Illustrated is the two-site mechanism of PTH-PTHR1 interaction, according to which the C-terminal portion of PTH(1–34), in α-helical conformation, first interacts with the amino-terminal extracellular domain (ECD) of the PTHR1, and then the N-terminal portion of the ligand binds to the transmembrane domain (TMD) region of the receptor, leading to conformational changes involved in receptor activation and coupling to heterotrimeric G proteins. Whereas the C-terminal portion of PTH(1–34) binds as a preformed α-helix, the N-terminal portion of the ligand is shown to undergo a coil-helix transition during the binding process, as suggested by structure-activity studies on PTH(1–14) peptide analogs (Shimizu et al., 2001b). The resulting folding cooperativity could contribute to the overall affinity of binding, as also suggested for other family B GPCRs (Parthier et al., 2009).
Some of the early findings that suggested this mode of binding were based on the differences in the binding affinities that C-terminal PTH fragments, as represented by the PTH(7–34) fragment, exhibited for the rat and human receptors and the demonstration using receptor-chimeras that the differences were determined by the ECD region of the receptors (Jüppner et al., 1994). Similar chimera and mutation-based studies revealed that the altered agonist/antagonist properties that the ligand analog Arg2-PTH(1–34) exhibited on the opossum and rat receptors could be assigned to the TMD region and specifically to residues at the extracellular end of TMD helix 5 (Gardella et al., 1994).
The two-site model emerging from such mutational studies then gained further support from the use of photoaffinity cross-linking approaches, which identified by direct physical means, specific contacts between sites in the ligand and in the receptor. These approaches used ligand probes modified at specific amino acid positions with photoreactive functional groups; thus incorporated as the amino acid analog, para-benzoyl-l-phenylalanine (BPA), or with the photoreactive benzophenone group attached to the epsilon amino group of the lysine-13 side chain (Nakamoto et al., 1995). Thus, for example, PTH and PTHrP ligand analogs modified with BPA at position 23, normally tryptophan in PTH and phenylalanine in PTHrP, cross-linked to a site near the amino-terminal end of the receptor's ECD (Mannstadt et al., 1998), whereas analogs modified with BPA at position 1 (serine or alanine) or 2 (valine) cross-linked to Met425 at the extracellular end of helix 6 (Bisello et al., 1998; Behar et al., 2000; Gensure et al., 2001a; Monaghan et al., 2008).
Although the overall cross-linking data were generally supportive of the basic two-site model of ligand-receptor interaction, they also suggested a higher level of complexity, as for example ligands modified with benzophenone at position-27 cross-linked to the first extracellular loop of the TMD region (Greenberg et al., 2000) and ligands modified at position 21 interacted with the mid-region of the ECD (Wittelsberger et al., 2006). Nevertheless, the basic model, although no doubt lacking in important specifics of the interaction not yet revealed, stands largely supported by the aggregate data provided by numerous mutational and biochemical studies that have focused on this problem for the PTHR1 (Lee et al., 1995; Bergwitz et al., 1996, 1997; Turner et al., 1996; Carter et al., 1999a; Shimizu et al., 2000b; Hoare and Usdin, 2001; Hoare et al., 2001; Shimizu et al., 2001b; Gensure et al., 2005; Wittelsberger et al., 2006). It is also now clear that the same general mode of interaction is used by most of the other family B GPCRs (Fortin et al., 2009; Parthier et al., 2009; Pal et al., 2012; Dong et al., 2014). Ultimately, any such model will need to be consistent with direct physical analyses of ligand-receptor complexes. Recent breakthroughs in the field are providing important steps in this direction and are indeed revealing high-resolution views of at least some of specific sites of contact between the ligand and receptor.
B. The Parathyroid Hormone Receptor-1's Amino-Terminal Extracellular Domain
Although the complete three-dimensional structure of the intact PTHR1 has not yet been solved, nor has it been for any family B GPCR, important advances were recently made in obtaining structural information on the PTHR1 and how it interacts with PTH and PTHrP peptide ligands. A key breakthrough was the determination in 2008 by Pioszak and Xu (2008) of the X-ray crystal structure of the PTHR1 ECD in complex with the PTH(12–34) peptide, which contains the ligand's principal binding domain (Fig. 8). The ECD was produced in Escherichia coli and purified under conditions that promoted disulfide bond formation; this measure was taken because it was clear from prior mutational studies, as well from the sequence conservation patterns, that the six conserved cysteines in the ECD play a critical role in receptor function and were thought to likely form an intramolecular disulfide bond network that stabilizes the bioactive fold (Lee et al., 1994). In addition to the PTHR1, X-ray crystallography or NMR approaches have been used to obtain three-dimensional structures for the isolated ECD regions of several other family B GPCRs, including the gastric-inhibitory polypeptide receptor (Parthier et al., 2007), the CRFR1 (Grace et al., 2004, 2007; Pioszak et al., 2008), the CRFR2 (Perrin et al., 2006; Pal et al., 2010), and the pituitary adenylate cyclase-activating polypeptide receptor (Kumar et al., 2011). A common protein fold for the ECD is observed for each of these ECDs, and the six conserved cysteines indeed form an intramolecular disulfide bond network that maintains the overall structural fold. The general protein folding pattern seen in each of these structures follows that of the so-called “sushi” domain, which is a structural scaffold unit present in many extracellular proteins, including proteins of the complement system, as well as in the extracellular portions of some transmembrane proteins (Lehtinen et al., 2004). The ECD of the PTHR1, as seen in the other family B GPCRs, presents an oblong shape that is rather flat, at ∼10-Å thick, such that there is no deep buried core to the domain. The core shape consists of two pairs of antiparallel β-strands forming the center surfaces with a β-hairpin between β-strands-1 and -2 and a more extended loop between β-strands-3 and -4 extending inward toward the center of the core; the core is then flanked by a long N-terminal α-helical segment on one side and a shorter C-terminal helical segment on the other. The three disulfide bonds interconnect these elements of secondary structure and thus maintain the overall shape (Figs. 5 and 8).
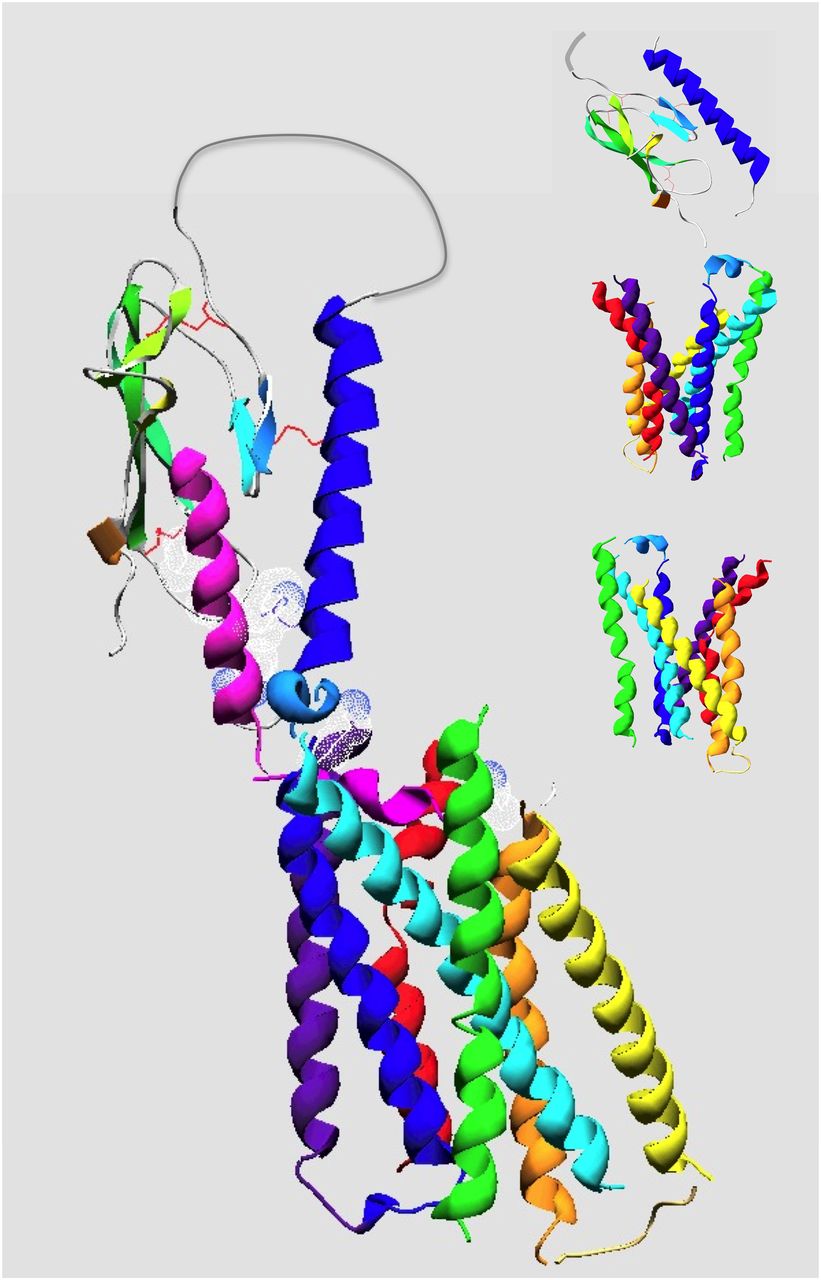
Molecular model of the PTH(1–34)•PTHR1 complex. Shown is a plausible model of PTH(1–34) bound to the PTHR1. The protein backbone chains are shown in ribbon format with the ligand colored magenta and the receptor colored by segment with the amino-terminal extracellular domain (ECD) colored blue-green and the transmembrane domain (TMD) colored by transmembrane helices as follows: purple (TM1), blue (TM2), cyan (TM3), green (TM4), yellow (TM5), orange (TM6), and red (TM7). The structure of the PTH(15–34) in complex with the ECD is according to the structural coordinates reported by Pioszak and Xu (2008) (PDB ID 3C4M). The TMD region of the PTHR1, spanning residues Thre175 to Ser491, was modeled by homology using the crystal structure coordinates of the CRFR1 (Hollenstein et al., 2013) (PDB ID 4K5Y) as a template. The PTH(1–14) segment, modeled as a partial α-helix, was docked to the TMD region manually, placing Val2 and Lys13 of the ligand near the extracellular ends of TMs 6 and 1, respectively. The ECD•PTH(15–34) component was positioned onto the TMD•PTH(1–14) component, manually, allowing for a bend in the ligand between the amino-terminal and carboxyl-terminal domain, as suggested by NMR structural studies on PTH peptide ligands and photoaffinity cross-linking and mutational data on PTH•PTHR1 complexes (Adams et al., 1998; Behar et al., 2000; Marx et al., 2000; Gensure et al., 2001a, 2003; Peggion et al., 2002; Wittelsberger et al., 2006). The side chains of ligand residues Val2, Lys13, Arg20, Trp23, Leu24, and Leu28 are shown in dotted surface format, and the three disulfide bonds in the ECD are in red. The insets show the unliganded ECD and two views of the TMD, rotated 180° relative to each other, which highlight the V-shaped putative ligand-binding groove that forms between the extracellular ends of the TM helices.
Several of the hallmark residues that are highly conserved in the ECD regions of family B GPCRs are seen to participate in intramolecular packing interactions. Among these are Asp113 in the central hairpin loop between β-strands-1 and -2 and Pro132 at the end of β-strand-3. The latter residue is the site of a PTHR1 point mutation, P132L, that is associated with Blomstrand’s chondrodysplasia, a homozygous neonatal lethal condition of exceedingly high fetal bone mass; studies in vitro show that the P132L is mutation is incompatible with PTHR1 function, likely due to effects on protein folding and hence expression, thus explaining the lethal phenotype (Zhang et al., 1998).
In the cocrystal, the bound PTH ligand domain, residues (16–34), is situated in a groove that runs along the center of the ECD. The ligand domain is bound as an α-helix, and the majority of the key intermolecular contact interactions is hydrophobic in nature. Thus, Trp23, Leu24, and Leu28, which form the hydrophobic surface of the ligand helix, make extensive contacts with hydrophobic residues lining the floor and walls of the central groove. The structure is thus fully consistent with the prior structure-activity relationship studies that predicted that the PTH(15–34) domain binds to the receptor as an amphipathic α-helix (Epand et al., 1985; Gardella et al., 1993; Oldenburg et al., 1996). The Trp23 side-chain is also seen to be in close proximity to residues in the N-terminal α-helix, including Gln37 (Fig. 8), which is in agreement with prior cross-linking data with Bpa23-labeled PTH analogs as well as mutagenesis data that indicated proximity between these two sites (Mannstadt et al., 1998; Mann et al., 2008).
Soon after the PTH•PTHR1-ECD structure was solved, the same group reported the crystal structure of the PTHR1 ECD in complex with the binding domain fragment of PTHrP (Pioszak et al., 2009). The mode of binding observed for PTHrP was found to be highly similar that for PTH, such that the new structural data combined were fully consistent with and indeed confirmed the prior functional studies that indicated that, despite the glaring sequence variation, the (15–34) domains of PTH and PTHrP bind to the same or at least largely overlapping sites in the receptor (Jüppner et al., 1988; Caulfield et al., 1990). The cocrystal structure thus shows that the PTHrP domain binds as an amphipathic α-helix with ligand residues Phe23, Leu24, and Ile28 along one helical surface forming extensive hydrophobic interactions with receptor residues lining the groove of the ECD (Pioszak et al., 2009). A slight bend, however, is present in the PTHrP helix at position-27, which leads to a modest divergence in the receptor contacts made by the C-terminal portions of the two ligands. Whether such a divergence in binding modes correlates with some of the differences in functional responses that have been observed for the two ligands in cell-based kinetic binding and signaling assays (Dean et al., 2008) is unclear. For both ligands, the guanidinium side-chain makes extensive, mostly polar contacts with a cluster of receptor residues that form a small pocket at the proximal end of the groove (Pioszak and Xu, 2008; Pioszak et al., 2009). The elaborate set of interactions made by the Arg20 side chain is fully consistent with the high level of evolutionary conservation seen for this residue, as arginine is present at this position in all PTH family ligands studied to date (Fig. 6B). Moreover, structure-activity relationship studies focused on this position have shown that even structurally related amino acid substitutions have significant and deleterious impacts on receptor binding affinity (Barbier et al., 2001; Dean et al., 2006a). Thus the contacts identified in the crystal structures between the Arg20 side chain and the receptor must play a key and critical role in docking the ligand to the ECD and/or maintaining the stability of the ligand/receptor complex once it is formed.
The three-dimensional structures obtained for the PTHR1 ECD, and mirrored by those obtained for several other family B GPCR ECDs, represent only incomplete views of the overall bimolecular interaction, which of course also involves interaction of the ligand's N-terminal residues with the TMD portion of the receptor. Nevertheless the structure begins to suggest a possible mechanism by which the binding of the C-terminal ligand helix to the ECD occurs in such a way that the N-terminal portion of the ligand extends from the ECD and is thus in optimal position to engage the extracellular loops and/or extracellular ends of the membrane spanning helices, and to thus induce the molecular events leading to receptor activation and transmembrane signaling. The next goal is thus to define this TMD component of the ligand-receptor interaction, and recent studies are providing some important clues.
C. The Type-1 Parathyroid Hormone Receptor Transmembrane Domain Region; Structural Properties
At present, the ligand interactions that occur within the receptor's TMD region containing the seven transmembrane helices (TMs) and connecting loops are known with much less certainty than those occurring within the ECD, given the absence of a crystal structure for this region of the PTHR1. Recently, however, a major breakthrough was made by the determination of the X-ray crystal structures of the TMD regions of two other family B GPCRs, the CRF receptor-1 (Hollenstein et al., 2013) and the glucagon receptor (GCGR) (Siu et al., 2013). As with the recent cyrstallographic studies on the β2-adrenergic receptor and several other family A GPCRs (Kobilka, 2011; Granier and Kobilka, 2012), the crystallization of the two family B GPCRs was made feasible by incorporating thermostabilizing protein modifications, such as insertion of a stabilized T4 lysozyme into the second intracellular loop (ICL2) and adding 12 stabilizing point mutations at various positions in the helices and loop regions (Hollenstein et al., 2013). These new family B TMD structures can be used to generate molecular models of the PTHR1 TMD region (Fig. 8) that are likely to be more accurate than any of those constructed previously using structural coordinates derived from a family A GPCR.
Although the seven helical domain architecture revealed by the crystal structures of the CRFR1 and GCGR TMD regions superimpose fairly well with that of the β2-adrenergic receptor and other family A GPCRs, there are readily discernable and important differences. Most prominently is that the opening at the extracellular surface of the helical bundle is wider and deeper in the family B receptors than those in most family A receptors; this larger size opening may well reflect the larger size of the ligand that is bound by the family B GPCRs versus the small-molecule type ligands that are bound by most family A receptors and hence the greater contact surface required in the former versus the latter case. Indeed, a recent genetic and cross-linking analysis of the putative ligand binding surface in the TMD of the CRFR1 found no fewer than 35 of 145 targeted receptor residues formed cross-linking contacts with the peptide ligand, with strong contacts occurring in extracellular loop (ECL)2, ECL3, and the extracellular ends of each TM helix, with exception of TM4 (Coin et al., 2013). Several putative ligand contact sites in the extracellular portions of the PTHR1 TMD region have been identified by mutagenesis and/or cross-linking methods as discussed in a subsequent section.
In the CRFR1 and GCGR TMD crystal structures, the intracellular ends of the TM helices of the family B receptors align more closely with those of the family A GPCRs, likely reflecting an overlap in the repertoire of G proteins, β-arrestins, and other signal modulating proteins that interact with the cytoplasmic surfaces of the receptors (Siu et al., 2013). Although the intracellular and extracellular connecting loops mostly lacked structure, an α-helical segment was observed in ECL1, which is consistent with prior NMR studies by Piserchio et al. (2000a) of an isolated ECL1 peptide of the PTHR1 in solution. Another helical segment was observed at the extracellular end of TMD-1. This so-called “stalk” segment extends for about three helical turns and would serve to connect the TMD portion to the ECD and could conceivably serve a hinge function to enable movement of the ECD toward to the TMD region during the ligand binding process (Coin et al., 2013; Siu et al., 2013). Perhaps consistent with such a scenario, cross-linking and mutational studies of the PTHR1 suggest that residues within this stalk region, comprised of residues Val171–Glu182, interact with residues in the midportion of PTH(1–34) and play important roles in receptor activation (Adams et al., 1998; Carter et al., 1999b; Wittelsberger et al., 2006).
The crystal structures of the two family B TMD regions, the GCGR and CRFR1, each obtained in an antagonist-bound, inactive state, revealed a number of interhelical interactions within the body of the TMD region that likely help modulate the relative positioning and movements of the helical domains, and thus the isomerization of the receptor between inactive and active conformations. At least some of these interactions, which are generally comprised of weak hydrogen bonds and electrostatic interactions, could be functionally akin to those forming the “ionic lock” in the family A GPCRs, because these are comprised principally of electrostatic interactions between a conserved Arg in TM3 and an Asp or Glu in TM6 and serve to control the movements of these two helices during the activation process (Granier and Kobilka, 2012; Bortolato et al., 2014). Although residues directly homologous to those forming the ionic lock are not seen in the family B GPCRs, evidence for such a dynamic relationship between TM3 and TM6 of the PTHR1 is provided by mutational studies in which a zinc-chelation strategy was used to map interhelical interactions and movements required for agonist-induced activation (Vilardaga et al., 2001). Another interhelical interaction identified by mutational methods and potentially involved in receptor activation occurs between Arg233 in TM2 and Gln451 in TM7, at which mutations modulate the binding of PTH(1–34) agonist ligands but not PTH(3–34) antagonists (Gardella et al., 1996a) (Fig. 5). Direct evidence for this Arg233–Gln451 interaction is observed for the homologous residues in the crystal structure of the CRFR1 TMD region (Hollenstein et al., 2013). Other residues in the PTHR1 TMD region likely to be involved in the PTHR1 activation process include the highly conserved His223 in TM2, Thr410 in TM6, and Arg458 in TM7, at which mutations have been identified in patients with Jansen's chondrodysplasia and shown to result in ligand-independent (constitutive) PTHR1 signaling (Calvi and Schipani, 2000) (Fig. 5).
D. The Type-1 Parathyroid Hormone Receptor Transmembrane Domain Region; Ligand Interactions
Although direct three-dimensional structural information on the complex formed between the PTH ligand and the TMD region of the receptor has not yet been obtained, the interaction has been extensively analyzed using both mutational and photoaffinity cross-linking approaches. As mentioned above, cross-linking methods have thus established a proximity between Lys13 in the ligand and Arg186 in the stalk segment at the extracellular end of TMD1 (Adams et al., 1998) as well as between residues 1 and 2 in the ligand and Met425 located at the extracellular end of TM6 (Bisello et al., 1998; Behar et al., 2000; Gensure et al., 2001b). Additionally, substitution of Leu for Met425 converted the functional response to Bpa2-containing PTH ligands used for the cross-linking studies from antagonism to partial agonism (Gensure et al., 2001a). Similarly, earlier mutational studies identified Ser370 and Ile371 located near the extracellular end of TMD5 as key determinants of the antagonist versus agonist properties exhibited by PTH analogs containing arginine at position-2 on PTH1 receptors derived from different species (rat versus opossum) (Gardella et al., 1994). It would thus appear that the highly conserved valine-2 in the ligand interacts with receptor residues at or near the extracellular ends of TMD helices 5 and 6 and that these interactions play key roles in inducing the molecular changes in the TMD helices that lead to receptor activation and G protein coupling. It is highly likely that other ligand residues also participate in such receptor activation responses, and key candidates for this are Ile5 and Met8. Key data in support of this suggestion come from studies on a PTHR1-delNT derivative that has the PTH(1–9) segment tethered, via a four-residue glycine linker, to the extracellular end of TMD1. This PTHR1-tethered PTH(1–9) construct exhibits high basal signaling activity when transfected into a naive host cells (e.g., COS-7 or HEK-293 cells), and alanine-scanning analyses show that this activity is strongly diminished by alanine replacement at either Val2, Ile5, or Met8 (Shimizu et al., 2000a). The three residues of Val2, Ile5, and Met8 would thus appear to comprise the principal agonist pharmacophore of the PTH ligand.
As mentioned above, modified N-terminal PTH peptides as short as M-PTH(1–14) and even M-PTH(1–11) exhibit the same high cAMP signaling potency in cells expressing the intact PTHR1 as they do on the PTHR1-delNT construct (Carter et al., 1999a; Shimizu et al., 2000a,b, 2001a,b, 2005) (Fig. 4). Because these minimized N-terminal PTH analogs adopt stable helical structures in solution, whereas native PTH(1–14) is largely unstructured (Tsomaia et al., 2004), the combined data seem consistent with the possibility that the approximate N-terminal (1–14) portion of PTH adopts at least some α-helical structure as it binds to the orthosteric ligand-binding site within the TMD region of the receptor. This TMD binding site is presumably a pocket or groove formed by the extracellular loops and extracellular ends of the TMD helices, such that the key pharmacophoric determinants in the ligand—Val2, Ile5 and Met8—can make the key intermolecular contacts that induce receptor activation (Shimizu et al., 2000b, 2002; Bisello et al., 2002; Monticelli et al., 2002; Piserchio et al., 2002; Gensure et al., 2003, 2005; Vilardaga et al., 2003). Whether the binding and activation process involves a coil-helix transition in the N-terminal portion of the ligand, which would presumably be mediated by recognition determinants in the receptor and has been suggested by prior studies (Shimizu et al., 2001b), remains unknown. Such a mode of induced N-terminal ligand helix formation during the TMD binding process has also been suggested to apply to other family B GPCRs (Parthier et al., 2009; Coin et al., 2013).
Two key features about the overall interaction that remain to be elucidated are 1) the specific mode of binding and contact sites used by the N-terminal portion of the PTH ligand and the TMD region of the receptor and 2) the spatial relationship between the ECD and TMD components of the complex and the relative movements of these two components during binding and activation. Presumably, in the holo-ligand-receptor complex, the ECD component is oriented in such a way that the N-terminal ligand domain has facile access to the binding pocket located in the TMD portion of the receptor, as generally suggested by the prior cross-linking and mutational studies (Greenberg et al., 2000; Shimizu et al., 2002; Gensure et al., 2003; Wittelsberger et al., 2006). Given the relative flattened and oblong shape of the ECD, it is tempting to imagine that the ECD might fold over and cover the opening of the extracellular surface of the TMD, as would a lid covering a pot, and thus trap the ligand in the TMD active site (Sun et al., 2007). It must be recognized, however, that the overall process of ligand binding and activation at the PTHR1 is still far from being well understood, and it is likely much more complex than ascribed by any such mechanism, including the simple “two-site” model outlined above. Further studies using new approaches and new tools are thus needed to elucidate more clearly the key steps that define the ligand binding and activation mechanisms operating at the PTHR1.
VIII. Mechanisms of Signal Transduction
A. G Protein Coupling and Signal Regulation
There are currently only a few clues from experimental studies that help in understanding how the PTHR mediates transmembrane signal transduction, couples to G proteins, and then reverses the process for signal termination. Several residues in the portions of the receptor predicted to be exposed to the cytoplasmic domain have been identified that when mutated result in alterations to G protein signaling (Fig. 5). Mutation of Lys388 in ICL3 to Ala diminishes signaling via both the Gαs/cAMP/PKA and Gαq/PLC/IP3 pathways, a phenotype that suggests that this lysine is general determinant of G protein coupling (Huang et al., 1996). Mutation of Lys319 in ICL2 to Glu results in a specific defect in Gαq-mediated PLC/IP3 signaling, as Gαs-mediated cAMP/PKA signaling is preserved, suggesting that this lysine interacts with a determinant in Gαq that is not present in Gαs (Iida-Klein et al., 1997). There are likely many other residues in the intracellular loops and at the intracellular ends of the TMD helices that contribute to G protein coupling, but these remain largely undefined.
As far as mechanisms of signal termination at the PTHR1, a number of studies have shown that this process, as for most GPCRs, is mediated largely by receptor phosphorylation via G protein receptor kinases, subsequent recruitment of β-arrestin proteins, and then receptor internalization (Malecz et al., 1998; Qian et al., 1998; Ferrari et al., 1999; Bisello et al., 2002; Chauvin et al., 2002; Tawfeek et al., 2002; Vilardaga et al., 2002; Bouxsein et al., 2005; Rey et al., 2006; Maeda et al., 2013). A cluster of seven serine residues located in the mid-region of the cytoplasmic tail are the sites of ligand-induced receptor phosphorylation and serve to regulate the interaction of activated PTHR1 with arrestin proteins and hence receptor internalization and desensitization of the Gq signaling pathway (Malecz et al., 1998; Qian et al., 1998; Tawfeek et al., 2002; Vilardaga et al., 2002; Rey et al., 2006; Maeda et al., 2013). Recent studies, however, are now indicating that interaction of the PTHR1 with β-arrestins and the internalization process itself can lead to nonclassic modes of Gs/cAMP signaling.
B. Signaling Bias at the Type-1 Parathyroid Hormone Receptor
1. Extracellular Signal-Regulated Kinase-1/2/Arrestin-Mediated Biased Agonism.
The concept of biased agonism, by which a certain modified analog ligand for a given receptor activates a signaling pathway cascade that is distinct from that activated by the nonmodified ligand is drawing considerable attention in the GPCR field, in part because it potentially offers a means to develop novel therapeutics that selectively induce only desirable responses in the target system (Reiter et al., 2012; Luttrell, 2014; Tehan et al., 2014). Moreover, because the PTHR1 can signal through multiple pathways, ligands that selectively activate one pathway but not the others could help in determining which signaling pathways contribute to the biologic responses induced by PTH ligands in bone and kidney target cells. The general concept of biased agonism is based on the notion that GPCRs can adopt a multitude of conformations and thus have a pleuridimensional capacity to mediate altered modes of ligand recognition and signal transmission (Clarke, 2005; Reiter et al., 2012; Luttrell, 2014). Most often discussed in the literature are biased agonists that act on certain Gαs-coupled GPCRs, such as the β2-adrenergic receptor and angiotensin receptor, and selectively activate the ERK-1/2 pathway via β-arrestin, with little or no activation of the Gαs/cAMP/PKA (Shenoy et al., 2006; Shukla et al., 2011).
For the PTHR1, the analog, [d-Trp12,Tyr34]bovine PTH(7–34)NH2, was shown to induce an increase in ERK-1/2 phosphorylation via a G protein–independent, β-arrestin–mediated pathway (Gesty-Palmer et al., 2006). This analog elicits little or no cAMP agonist activity and, indeed, was developed to be a pure PTHR1 antagonist at least for cAMP signaling (Goldman et al., 1988). The analog furthermore acts as an inverse agonist for cAMP signaling at constitutively active PTHR1 mutants (Gardella et al., 1996b; Carter et al., 2001). The new studies revealing ERK-1/2 signaling for the analog used PTHR1-transfected HEK-293 cells; however, the same ligand was also found to induce an increase in bone formation when injected into wild-type mice, and the effects were not observed when it was injected into β-arrestin-2–null mice (Gesty-Palmer et al., 2009). These results are clearly intriguing and suggest that further studies on biased agonism at the PTHR1 are needed. In a previous study, a PTH(7–34) peptide failed to stimulate bone formation when tested in animals (Sebastian et al., 2008). More recent studies using plate reader technologies to assess PTHR1 signaling via multiple pathways including ERK-1/2 and β-arrestin recruitment did not confirm that [d-Trp12,Tyr34]bovine PTH(7–34)NH2 behaves as an ERK-1/2–selective biased agonist in several types including HEK-293, CHO, and the human osteoblastic cell line U2OS (Cupp et al., 2013a; van der Lee et al., 2013). Whether the differences in the findings involve differences in the assay formats or animal or cell systems used remain to be sorted out and thus seem uncertain whether or not the PTH(7–34) scaffold will be a key to the development of therapeutically useful biased agonists for the PTHR1.
2. Gαq/Phospholipase C/Inositol Trisphosphate versus Gαs/cAMP Biased Agonism.
In as much as the PTHR1 couples to both the Gαs/cAMP/PKA pathway and the Gαq/PLC/IP3/PKC pathway, the concept of biased agonism would extend to ligands that selectively activate one of these signaling cascades but not the other. Most PTH and PTHrP agonist ligands couple to both pathways, but several analogs have indeed been identified that are selectively defective for activating Gαq signaling while maintaining good potency for Gαs/cAMP signaling. These analogs are modified at position 1 in the ligand, which is serine in human PTH and alanine in human PTHrP. In some cases the analogs are modified with other substitutions to enhance receptor-binding affinity. One such analog is Gly1,Arg19-PTH(1–28) reported by Takasu et al. (1999a). The PTH(1–28) scaffold was chosen for this analog because the goal was to generate a purely cAMP-based agonist ligand, and the (29–32) region of PTH had been reported to contain determinants of PKC activation (Jouishomme et al., 1994). However, Gly1-PTH(1–34)–based analogs were also found to be selectively PLC defective (Takasu et al., 1999a), whereas PTH(1–31) and PTH(1–30), each containing Ser1 but missing at least some of the (29–32) segment, showed equal effects on signaling via the cAMP and PLC pathways (Takasu and Bringhurst, 1998). Moreover, PTH(3–34) was found to be inactive for PLC/IP3 signaling in hPTHR1-transfected LLC-PK1 cells (Takasu et al., 1999b). The (29–32) region of PTH thus does not appear to be essential for activating the Gαq/PLC/IP3 signaling cascade per se.
Consistent with an important role of residue 1 in activating PLC signaling via the PTHR1, Bisello and colleagues (2002) reported a PTHrP(1–36) analog that had alanine-1 replaced by benzoyl-phenylalanine and that was thus strongly defective for PLC/IP3 signaling yet retained full cAMP potency. Subsequent studies reported that PTHrP(1–36) and an M-PTH(1–28) analog that contained tryptophan, which is structurally similar to BPA, at position 1 were also selectively defective for PLC signaling and hence functioned as biased agonists for the cAMP/PKA pathway (Gesty-Palmer et al., 2006; Nagai et al., 2011). The Gly1,Arg19-PTH(1–28) and Trp1-M-PTH(1–28) analogs have been used in studies in mice to probe the relative importance of the Gαs/cAMP/PKA versus Gαq/PLC/PKC signaling pathways in vivo, and the findings led to the conclusion that the /cAMP pathway plays the major role in mediating both the bone anabolic response (Yang et al., 2007) and the renal phosphaturic response to PTH ligands (Nagai et al., 2011).
In a recent comprehensive analysis of the activities of a wide collection of previously reported PTH and PTHrP analogs in a multimodal array of signaling reporter assays using cells expressing the PTHR1, Cupp and colleagues (2013a) largely confirmed that position 1–modified analogs described above maintain good potency for activating the Gαs/cAMP pathway but are relatively deficient for activating the Gαq/PLC/IP3 signaling. No PTH analog has yet been found that exhibits the reverse selectivity and thus potently activates PLC/IP3/PKC signaling but not cAMP/PKA signaling. Whether this reflects a more efficient coupling of the PTHR1 to Gαs than to Gαq (Iida-Klein et al., 1997), a difference in the pharmacophoric determinants in the ligand and receptor that mediate activation of the two pathways, the more amplified nature of the cellular cAMP response, or a combination of such factors is unclear. The experimental results nevertheless tend to support the notion that although the PLC/IP3/PKC and potentially other pathways may help regulate responses induced by PTH and PTHrP ligands in bone and kidney target cells, it is the cAMP/PKA pathway that plays the dominant role in mediating the biologic actions of these ligands in vivo (Guo et al., 2002, 2010; Yang et al., 2007).
C. Novel Modes of cAMP Signaling at the Type-1 Parathyroid Hormone Receptor
1. Prolonged Signaling via Binding to the R0 Receptor Conformation.
Consistent with current GPCR theory, the PTHR1 is thought to be conformationally dynamic and to thus have the capacity to shift through a variety of receptor conformations, any of which may be bound and stabilized by a certain ligand analog, and thence activate a specific program of cytoplasmic signaling events. Recent studies using pharmacological and/or biophysical approaches provide support for this concept and thus some clues as to how ligand stabilization of distinct conformational states of the PTHR1 may lead different types of biologic responses. Whereas most GPCRs such as the β2-adrenergic receptor have in general been thought to exist in a low affinity ligand-binding state when uncoupled from G protein and to shift to high affinity state only upon G protein coupling (De Lean et al., 1980), recent studies suggest that the PTHR1 can form high affinity complexes with certain ligands, including PTH(1–34), even in the absence of G protein coupling, because the complexes remain stable in the presence of GTPγS, which induces receptor–G protein dissociation (Dean et al., 2006b). In contrast, other agonist ligands, such as PTHrP(1–36) or M-PTH(1–14), form complexes that are more like those formed with β2-adrenergic receptor and dissociate rapidly in the presence of GTPγS. These observations have led to the hypothesis that the PTHR1 has the capacity to form a unique high affinity conformation, called R0 in accordance to similar observations made on the CRFR1 by Hoare et al. (2003), and this R0 conformation is independent of G protein coupling. Thus, whereas PTH(1–34) can form stable complexes with R0, other ligands such as M-PTH(1–14) and PTHrP(1–36) bind with high affinity only to the G protein–coupled receptor conformation, called RG, and thus are more consistent with classic mechanisms of ligand binding at GPCRs (Dean et al., 2006b, 2008).
Although the data suggest that while stable binding to R0 does not appear to require direct G protein coupling, a functional consequence of such binding is that the cAMP signaling response mediated by such ligands is prolonged. One way to explain the observed correlations between stable binding to R0 and the prolonged cAMP signaling responses is that the R0 complex can isomerize to the biologically active RG conformation and perhaps do so repeatedly without a release of the bound agonist ligand after each G protein–uncoupling event. In this regard, the ligand-PTHR1 (R0) complex engages and activates G protein heterotrimers via a catalytic-like mechanism (Rodbell, 1997; Dean et al., 2008).
Other modified PTH analogs, including M-PTH(1–28) and M-PTH(1–34), have also been shown to exhibit high affinity binding to R0 as well as markedly prolonged cAMP signaling responses when applied to PTHR1-expressing cells, and moreover, these analogs mediate markedly prolonged hypercalcemic and hypophosphatemic responses when injected into mice (Okazaki et al., 2008; Maeda et al., 2013). One analog of particular interest, called Long-Acting PTH (LA-PTH), is a unique M-PTH(1–14)/PTHrP(15–36) hybrid peptide. This analog binds to the RG conformation of the PTHR1 with an affinity similar to that seen with PTH(1–34), but it binds to the R0 conformation with an affinity that is severalfold higher than that observed for the unmodified peptide, and although the potencies exhibited by the two peptides in conventional dose-response assays of cAMP formation performed in PTHR1-transfected HEK-293 cells are nearly equivalent, consistent with their equivalent RG binding affinities, the cAMP response induced by LA-PTH persists for a far greater time period after washout than does the response induced by PTH(1–34) (Fig. 9, A–C). Moreover, when injected into mice, LA-PTH induces elevations of serum calcium that last for nearly 24 hours and are thus markedly prolonged, compared with those induced by PTH(1–34), which last for only 4 hours or less (Maeda et al., 2013) (Fig. 9D). The overall findings so far suggest that the prolonged responses to LA-PTH and related analogs observed in vivo are not due to simply a prolonged half-life of the peptides in the circulation but rather to the persistent binding of the ligands to the PTHR1 in bone and kidney target cells. Because of their prolonged actions in vivo, such R0-selective analogs are now being investigated as potential new therapies for patients with hypoparathyroidism (Shimizu et al., 2008; Mannstadt et al., 2013). The extent to which the capacity to bind to the R0 PTHR1 conformation, and hence mediate prolonged signaling, might play in a role in determining the overall biologic response profile induced by endogenous PTH or PTHrP is unclear at present, in part because the key studies so far have been performed largely with truncated and modified variants of PTH and PTHrP and also because there remains some uncertainty about the exact forms of the endogenous PTH and PTHrP ligands that act on the PTHR1 in target tissues.

Properties of a long-acting PTH analog. Shown is the capacity of the long-acting analog, LA-PTH, a hybrid peptide consisting of a modified M-PTH(1–14) segment joined to a modified PTHrP(15–36) segment, to bind with high affinity to the R0 PTHR1 conformation and thus mediate prolonged signaling responses in cells and in animals. The analog is compared with PTH(1–34) for (A) binding to the PTHR1 in the G protein–independent conformation, R0; (B) inducing dose-dependent cAMP signaling responses in HEK-293 cells expressing the rat PTHR1 and the luciferase-based glosensor cAMP reporter; (C) capacity to maintain cAMP responses in the same cells after an initial application of ligand (0.3 nM/15 minutes) and then a washout (at t = 0') of unbound ligand; and (D) stimulating blood ionized calcium responses in mice [after injections with either vehicle, PTH(1–34) at 50 nmol/kg or LA-PTH at 10 nmol/kg]. Not shown is that LA-PTH bound to the G protein–coupled PTHR1 conformation, RG, with an affinity only threefold higher than that of PTH(1–34); this similar affinity for RG likely explains the similar potencies observed in the cAMP dose-response assays, whereas the greater difference in binding to the R0 PTHR1 conformation likely explains the more prolonged cAMP responses in cells and calcemic responses in animals. Adapted from Maeda et al. (2013).
2. Correlation of Prolonged Signaling and Receptor Internalization.
The cellular and molecular mechanisms that underlie the differences in the signaling response profiles of the different PTH and PTHrP analogs are yet to be fully elucidated, but the problem has been approached using optical approaches [fluorescence confocal microscopy, Forster Resonance Energy Transfer (FRET), Total Internal Reflection Fluorescence] as applied to the PTHR1, in some cases with green fluorescent protein tags attached to an innocuous site (E2) in the ECD and expressed in transfected HEK-293 cells, along with fluorescently labeled PTH analogs, typically PTH(1–34) containing TMR attached to lysine-13. Ferrandon et al. (2009) thus performed studies using FRET-based biosensors to assess potential differences in the molecular and cellular mechanisms activated by PTH and PTHrP ligands in live HEK-293 cells. Of the two ligands, PTH(1–34) was found to bind to the R0 conformation with the higher affinity and to mediate longer-lasting cAMP responses (Dean et al., 2008). These single-cell cAMP analyses using the FRET biosensor approach revealed only transient increases in cAMP formation by PTHrP(1–36) and markedly more prolonged responses by PTH(1–34). Moreover, the sustained cAMP response observed at the later times after the brief (∼20 seconds) exposure to PTH(1–34) coincided with the internalization of most if not all of the detectable PTH(1–34)-PTHR complexes into early endosomes, and the complexes were colocalized with Gαs as well as adenylyl cyclases and β-arrestins. Prolonged cAMP signaling associated with concomitant ligand-receptor internalization has also been observed with several other R0-selective and longer-acting PTH analogs, including M-PTH(1–34) (Okazaki et al., 2008) and LA-PTH (Maeda et al., 2013).
D. Evidence for Noncanonical cAMP Signaling at the Type-1 Parathyroid Hormone Receptor via Endosomes
1. Canonical versus Noncanonical G Protein–Coupled Receptor Signaling.
Whereas the transient actions of the RG-selective ligands, such as PTHrP(1–36) and M-PTH(1–14), on the PTHR1 appear compatible with the “canonical” model of G protein signal regulation, which describes mechanisms to guard against prolonged signaling and thus to prevent overstimulation (Lohse et al., 1992), the functional properties of these long-acting PTH analogs appear strikingly at odds with such models (Fig. 10). By the canonical model, GPCR signaling is attenuated rapidly after onset via a series of steps that begin soon after receptor activation with phosphorylation of the receptor on its cytoplasmic tail by G protein–coupled receptor kinases, and this phosphorylation, in turn, promotes the binding of a β-arrestins, which then dislodges the bound G protein by steric interference and prevents binding of additional G proteins (Pitcher et al., 1992; Pippig et al., 1993), and also by recruiting cAMP-specific phosphodiesterase 4 (PDE4) to the plasma membrane to degrade the second messenger cAMP (Perry et al., 2002). Arrestin binding further promotes receptor internalization, a process that relies upon the interaction of β-arrestins with clathrin, a major component of the clathrin-based endocytic machinery (Ferguson et al., 1996), which starts the endocytosis process wherein the ligand-receptor complexes are dissociated and the signal terminates. The internalized receptor is dephosphorylated and either recycled back to the cell surface or else directed to lysozomes for degradation.

Canonical and noncanonical G protein signaling at the PTHR1. The cAMP signaling responses induced by PTH and PTHrP peptides are typically of similar potency and efficacy when measured acutely; however, recent kinetic signaling and fluorescent imaging studies revealed differences in the response mechanisms used. Thus studies performed in HEK-293 cells transfected with green fluorescent protein–tagged PTHR1 thus shows that whereas PTHrP forms complexes with Gαs that are active and thus signal through cAMP only at the plasma membrane and then dissociate, PTH forms complexes that not only signal at the cell surface but remain associated as the complexes internalize. Moreover, as shown in the graphs depicting FRET-based cAMP biosensor responses, the response induced by PTH(1–34) persists for at least 30 minutes after initial binding (the ligand is applied during the time indicated by the bar at the top of the graph and then washed out), whereas the response induced by PTHrP(1–36) is transient and decays after ligand wash out. Prolonged cAMP signaling responses that correlate temporally with bulk internalization of ligand-receptor complexes also correlate positively with the capacity of a given ligand to bind with high affinity to the R0 PTHR1 conformation. The overall results lead to the hypothesis the PTHR1 can mediate cAMP signaling responses both by canonical mechanisms operating at the plasma membrane as well as by noncanonical mechanisms that operate from within the internalized endosomal domain (Dean et al., 2006b, 2008; Okazaki et al., 2008; Ferrandon et al., 2009; Feinstein et al., 2011).
Many studies have indeed verified mechanisms of rapid signal regulation and desensitization for the PTHR1 that involve receptor phosphorylation and arrestin-dependent internalization (Ferrari and Bisello, 2001; Vilardaga et al., 2001; Bisello et al., 2002; Chauvin et al., 2002; Tawfeek et al., 2002; Tawfeek and Abou-Samra, 2004; Rey et al., 2006; Sneddon and Friedman, 2007). However, the sustained cAMP signaling mediated by internalized PTHR1 bound to R0-selective ligands suggested a distinct mode of action and indeed have prompted the hypothesis that sustained signaling at the PTHR1, as induced particularly by ligands that bind preferentially to the R0 PTHR1 conformation, is mediated by ligand-receptor complexes that have moved to the early endosomal domain (Ferrandon et al., 2009; Vilardaga et al., 2012, 2014). One key result in support of this model is the finding that coexpression of an inhibitory (dominant negative) form of dynamin, a protein that promotes vesicle internalization by acting to close the neck of maturing clatherin-coated pits at the plasma membrane, or a dominant negative mutant of Rab5 (Rab5-S34N) that prevents the formation of endosomes led to a reduction in the duration of PTH(1–34)-induced cAMP signaling at the PTHR1 (Ferrandon et al., 2009; Gidon et al., 2014).
Studies carried out on the thyroid-stimulating hormone receptor (Calebiro et al., 2009) and the vasopressin receptor type 2 (Feinstein et al., 2013) also using fluorescent cell imaging and FRET biosensor methods similar to those used for the PTHR1 found that these receptors, which are family A GPCRs, cannot only mediate sustained cAMP signaling in response to binding their cognate peptide ligand but can do so at times when the ligand-receptor complexes are found mainly in endosomes. It thus appears possible that prolonged G protein–mediated signaling via internalized complexes is a signaling mechanism that is used more broadly across the wider GPCR super family (Irannejad and von Zastrow, 2014; Vilardaga et al., 2014).
2. Role of β-Arrestin in Type-1 Parathyroid Hormone Receptor Signal Regulation.
Whereas β-arrestin proteins are typically involved in GPCR signal termination, as described above, additional biochemical, cell-based FRET and imaging studies on the PTHR1 have suggested that coupling of the receptor to β-arrestin can help to prolong Gs-mediated cAMP signaling (Vilardaga et al., 2012; Wehbi et al., 2013). Thus, overexpression of a high affinity β-arrestin mutant (β-arrestin-IVAA) prolonged cAMP signaling responses induced by PTH(1–34) at the PTHR1 (Feinstein et al., 2011). The associated data obtained in this study provide some clues as to a mechanism by which this might occur, which is based on the well known capacity of β-arrestins to assemble signaling complexes that permit internalized GPCRs to activate ERK1/2 (Sorkin and von Zastrow, 2009). The data for the PTHR1 thus suggest that β-arrestins assembled with the PTHR1 in endosomes activates MAP kinases, which in turn phosphorylate and thereby inhibit PDE4, leading to prolongation of the cAMP signaling response (Feinstein et al., 2011). Consistent with such a mechanism, the PTH-dependent increases in cAMP were prolonged when the cells were treated with a PDE4 inhibitor but were damped when treated with inhibitors of an ERK-1/2. The findings thus suggest a positive feedback system for cAMP signaling at the PTHR1, by which endosomal ERK-1/2 signaling, mediated by PTH-bound PTHR-arrestin complexes, promotes cAMP formation from endosomes via inhibition of PDE4.
There are additional studies, however, that suggest another level of involvement of β-arrestins in promoting cAMP signaling by internalized PTHR1 complexes. In this case, β-arrestin appears to promote prolonged signaling by stabilizing interaction of the PTHR1 with Gβγ subunits. Ternary PTH-PTH1R-Gβγ complexes then assemble with Gαs subunits to form GαsGβγ heterotrimers, which, in turn, can then mediate subsequent rounds of Gαs activation (Wehbi et al., 2013). Such a mechanism could conceivably operate in parallel or in a concerted fashion with that by which β-arrestins mediate PDE4D inactivation via ERK-1/2 activation.
The combined results raise the question as to how arrestin can promote Gs signaling at the PTHR1 when it has been well established for a number of GPCRs that the binding of arrestin and G proteins is mutually exclusive. The answer is not clear, but it is worth pointing out that a candidate docking site for Gβγ has been located on the proximal portion of the C-terminal tail of the PTHR1 (Mahon et al., 2006), and this site does not overlap with the cluster of seven G protein–coupled receptor kinase-phosphorylated serine residues that are located in the midportion of the C-tail and known to mediate binding with β-arrestins (Malecz et al., 1998; Qian et al., 1998). There may therefore be two distinct mechanisms by which the PTHR1 can associate with β-arrestins—one involving a direct interaction with the phosphorylated serines on the C-terminal tail and the other involving indirect binding via interaction with Gβγ subunits docked to the upstream portion of the C-terminal tail.
3. Signal Termination and the Retromer Complex.
A separate question that arises involves the mechanism by which the cAMP signal responses induced by the long-acting PTH ligands are eventually terminated. Again, the answer is not clear, but additional recent studies suggest that as the PTHR1-containing internalized vesicles mature through the endosomal pathway, they eventually engage the cargo-sorting retromer complex, and this engagement brings about, or at least coincides with, signal termination (Feinstein et al., 2011; Vilardaga et al., 2012). The retromer complex is a peripheral membrane protein assembly that regulates the sorting cargo proteins through the endosomal system and mediates retrograde flow to the trans-Golgi network (Collins, 2008). The core retromer assembly consists of two membrane-associated sorting nexins (Snx1 and Snx2) and a heterotrimer composed of the vesicle protein sorting subunits: Vps26, Vps29, and Vps35 (Hierro et al., 2007). Feinstein et al. (2011) found that overexpression of Vps subunits in PTHR1-expressing HEK-293 cells shortens the time course of PTH(1–34)-induced cAMP generation, whereas inhibition of Vps function via expression of silencing RNAs prolonged the cAMP signaling response (Feinstein et al., 2011). Moreover, high-resolution imaging analyses showed that the PTHR1 colocalized with the retromer complex within late-stage trafficking vesicles targeted for the Golgi apparatus, and the timing of this colocalization correlated with the termination of cAMP signaling. Whether the PTHR1 directly binds to the retromer complex cannot be readily determined from the current microscopic imaging studies. It is intriguing to note, however, that Vps26 shares structural similarity with β-arrestins (Shi et al., 2006; Collins et al., 2008; Aubry et al., 2009), and so a possible scenario that is at least consistent with the current data is that the PTHR1 docks to the retromer sorting proteins at a stage in the endosomal trafficking process at which it dissociates from arrestin, and that this retromer-arrestin exchange process results in the termination of signaling. The biochemical events that shift PTH-PTHR1-arrestin signaling complexes to inactive PTHR1-retromer complexes were recently elucidated. Thus, Gidon et al. (2014) showed that the termination of sustained cAMP signaling associated with internalized PTH-PTHR1 complexes involves a negative feedback mechanism by which the activation of PKA leads to the phosphorylation and hence stimulation of the v-ATPase proton pump, which resides on endosomal membranes and plays a major role in endosomal acidification. Thus, PKA activation of the v-ATPase results in acidification of the endosome, which, in turn, triggers the dissociation of the bound PTH ligand from the PTHR1 and a subsequent disassembly of the PTHR-arrestin signaling complex and concomitant assembly of the inactive PTHR1-retromer complex (Fig. 10) (Gidon et al., 2014).
4. Evidence for Endosomal Signaling by other G Protein–Coupled Receptors.
The prospect of endosomal signaling for the PTHR1 via Gs opens new questions about the mechanisms that maintain and regulate G protein signaling, both at the cell surface and within the endosomal compartment that likely could be relevant to the GPCR field more broadly. An important goal that most likely needs to be met before the hypothesis of endosomal G protein–dependent signaling can be confirmed, however, is to establish that the ligand-receptor–G protein complexes that reside in the endosomal membrane are in fact a direct source of the cAMP signal that is measured in the experiments. At present, the data show only a temporal correlation between the presence of an intracellular cAMP signal and the location of most, or perhaps nearly all, of the detectable ligand-receptor complexes within endosomal sites. It thus is conceivable that a small but undetectable population of activated receptors and G proteins remaining at the cell surface contribute also to the sustained cAMP signal observed.
A novel approach to addressing this problem was applied recently by Irannejad and colleagues (2013) in studies on the β2-adrenergic receptor (β2AR). This Gαs-linked family A GPCR does not mediate cAMP signaling responses that are as prolonged as those with the PTHR1, but the study nevertheless used it to examine whether internalized Gαs signaling could occur for the agonist activated receptor. The key to the approach was to express in the study cells, in this case HEK-293 cells, a single-chain camelid antibody, or nanobody, called Nb80 that was specific for the activate conformation of the β2AR (Steyaert and Kobilka, 2011). A second nanobody, NB37, specific for the active form of Gαs freed of nucleotides was also used, and each nanobody was tagged with green fluorescent protein to enable direct visualization and tracking of the subcellular location of the activated states of the receptor and G protein at times after addition of the agonist isoprenaline. The studies revealed that the activated β2AR and Gαs could both be detected at the plasma membrane as well as in early endosomes within a few minutes of ligand addition (Irannejad et al., 2013). These observations, coupled with the modest but significant decrease in the maximum level of cAMP induced by the agonist observed when β2AR internalization was blocked, support the view that internalized β2ARs can mediate Gαs activation and hence cAMP production, from the location of the endosome.
Interestingly, the activated β2AR and Gs proteins were not seen to be present in the early-stage clathrin-coated pits as they first appeared on the plasma membrane during the initial steps of endocytosis. This observation suggests that at least for the β2AR, there are two distinct phases of receptor/Gαs activation that occur at two distinct sites: the first at the plasma membrane immediately upon agonist binding and the second a few minutes later when the receptor has internalized to early stage endosomes (Irannejad et al., 2013). Such a process would seem compatible with the known capacity of β-arrestins to uncouple receptors from G proteins at the plasma membrane and thence mediate their movements into clatherin-coated pits (Krupnick et al., 1997). In the case of the PTHR1, for which β-arrestins can promote prolonged cAMP signaling and are associated with the receptor in endosomes, a second-phase of receptor/G protein activation may also involve a second round of β-arrestin recruitment to the endosomal complex. Clearly it would be of interest to use such a nanobody approach to track the subcellular locations of the activated states of the PTHR1 and Gαs after addition of various PTH and PTHrP ligands, but so far such studies have not been performed.
E. Regulation of the Type-1 Parathyroid Hormone Receptor by Sodium-Hydrogen-Regulating Factor Proteins
Although further work is needed to elucidate the complete mode of action by which the PTHR1 signals via the Gαs/cAMP pathway, including the relative roles of signaling at the cell surface versus signaling from within endosomes, it is clear that multiple cytoplasmic proteins can interact with the PTHR1 and thereby alter its subcellular location and/or signaling output. One class of such cytoplasmic interacting protein known to play an important role in modulating PTHR1 internalization and membrane dynamics are the sodium-hydrogen-regulating factor (NHERF) family of proteins, which play major role in trafficking proteins to and from the cell surface. The PTHR1 has thus been shown to interact with both NHERF-1 and -2 and have PDZ domains contained within the NHERF proteins and the last five residues of the receptor's C-terminal tail (Fig. 5) (Mahon et al., 2002, 2003; Sneddon et al., 2003). The interaction of the PTHR1 with NHERF proteins can regulate the docking of the receptor with the actin cytoskeleton via the EZRIN adaptor protein and to thereby dynamically modulate the movement of the ligand-activated receptor between the plasma-membrane and the intracellular domain (Wang et al., 2007, 2009; Wheeler et al., 2007, 2008; Ardura et al., 2011; Mamonova et al., 2012).
How different PTH and PTHrP ligands might influence the binding of the receptor to such intracellular regulatory and trafficking proteins and how such intracellular docking proteins regulate signaling of PTH•PTHR complexes, either at the cell surface or within the endosomal domains, are currently unclear. The elucidation of these determinants could, however, lead to the design of new PTH ligand analogs with altered signaling profiles that render them more effective agents for treating diseases such as hypoparathyroidism and even osteoporosis.
IX. Type-1 Parathyroid Hormone Receptor Mutations in Human Disease
Mutations in the PTHR1 have been associated with several human diseases, and the phenotypes involved are generally consistent with the receptor playing a particularly vital role in the growth and development of skeletal tissue. Thus, biallelic loss-of-function mutations in the PTHR1 have been identified in Blomstrand osteochondrodysplasia, a rare, neonatal lethal condition characterized by extremely advanced bone development (Jobert et al., 1998; Karaplis et al., 1998; Zhang et al., 1998; Karperien et al., 1999; Hoogendam et al., 2007). Four of the five PTHR1 gene mutations identified to date in Blomstrand osteochondrodysplasia result in premature translational termination due to either altered mRNA splicing (Jobert et al., 1998; Karperien et al., 1999) or coding frame errors (Karperien et al., 1999; Hoogendam et al., 2007), whereas one is a homozygous missense mutation of Pro132→Leu (Karaplis et al., 1998; Zhang et al., 1998). The affected proline-132 is highly conserved among the family B GPCRs and maps to the β3-strand in the core of the receptor's ECD (Fig. 5). Studies in transfected COS cells show that the P132L mutant PTHR1 is defective for PTH ligand binding (Karaplis et al., 1998; Zhang et al., 1998).
In the heterozygous state, the P132L mutation, as well as several other predicted loss-of-function mutations in the PTHR1 gene have been linked to familial cases of defective tooth eruption (Decker et al., 2008; Frazier-Bowers et al., 2010; Yamaguchi et al., 2011; Risom et al., 2013). Four other heterozygous PTHR1 point mutations: G121E, A122T, R150C, and R255H, the first three of which also map to the receptor's ECD region and impair receptor function, have been identified in Ollier disease, a development condition defined by the occurrence of multiple enchondromas (Hopyan et al., 2002; Couvineau et al., 2008). A homozygous stop codon mutation (R485-stop) in the PTHR1 that results in a receptor with a truncated C-terminal tail has been identified in a patient with Eiken syndrome, a very rare recessive skeletal dysplasia in which ossification is severely delayed (Duchatelet et al., 2005). In this case, the PTHR1-R485-stop mutant receptor has not been characterized in vitro, and so the extent to which the phenotype can be attributed to altered surface expression and/or altered function of the receptor remains unknown.
Gain-of-function activating mutations have been identified in patients with Jansen's chondrodysplasia, a disorder characterized by dwarfism and mineral ion imbalance (Calvi and Schipani, 2000). The identified point mutations occur at one of three, well conserved amino acid residue positions, each located at the cytoplasmic base of a transmembrane domain helix: His223 in TM2, Thr410 in TM6, and Arg458 in TM7 (Fig. 5). When transfected into COS-7 cell, the mutant receptors exhibit basal levels of cAMP signaling that are severalfold higher than those seen in cells transfected with the wild-type PTHR1, which presumably reflects a shift in the conformational status of the receptor toward an activated state that can couple to cytoplasimic G proteins (Schipani et al., 1996, 1997, 1999). In principal, at least, patients with Jansen's disease could be treated with a PTH or PTHrP ligand analog that functions as an inverse agonist (Carter et al., 2001), although this has never been tested.
X. Small-Molecule Ligands for the Type-1 Parathyroid Hormone Receptor
There has been considerable interest in developing a small-molecule ligand that can act as an orally active and potent agonist for the PTHR1, but as yet no such ligand has been reported. Several small-molecule ligands have been identified for the PTHR1 but most of these function as weak antagonists (Carter et al., 2007; McDonald et al., 2007), and only one compound, called AH-3960 (dibutyl-diaminomethylenepyrimidine-2,4,6-trione), exhibited agonist activity, albeit the potency of the compound for stimulating cAMP formation in cells was in the micromolar range compared with the low-nanomolar potency observed for PTH(1–34) (Rickard et al., 2006) (Table 2). The mechanisms of action of the compound ligands identified so far for the PTHR1 have not been determined, and so it is not clear whether they bind to the same or overlapping binding sites in the receptor as that used by PTH peptide ligands or to some other site and thereby function as allosteric modulators (Hoare, 2007). One antagonist compound, however, called SW106 [r-5-(2-E-cyclopropylvinyl)-t-3-ethyl-6,7-difluoro-5-(trifluoromethyl)benzo[e][1,4]-oxazepin-2(1H,3H,5H)-one], was found by screening for the capacity to inhibit binding of a modified PTH(1–14) radioligand analog to the PTHR1, and so this compound likely binds to the same TMD receptor site used by the N-terminal pharmacophore of the PTH agonist ligand (Carter et al., 2007). The findings with SW106 thus suggest that at least some portion of the true agonist ligand binding site in the TMD region of the receptor is at least accessible to small-molecule ligands. The reason for the apparent failure of screening approaches used so far to identify a potent small molecule agonist for the PTHR1 remains unclear, but as the methods for generating and screening compound libraries evolve and improve, they may well come closer to meeting this important goal.
Small-molecule ligands for the PTHR1
XI. Concluding Remarks
The PTH receptor type-1 displays the hallmark structural features of the class B GPCRs, but has the somewhat unique properties of mediating the functional actions of two distinct endogenous peptide ligands, PTH for the endocrine control of blood Ca and Pi levels and PTHrP for the paracrine control of tissue development. The receptor is expressed in target cells of bone and kidney, where it mediates effects on mineral ion transport, and in a number of developing tissues, including the skeleton, where it regulates morphogenetic programs of cell differentiation. The PTHR2 subtype, with only 51% identity to the PTHR1, mediates completely separate biologic functions, acting in the neuroendocrine system and during gonadogenesis, and it binds the distinct peptide ligand TIP39. Both PTHR1 and PTHR2 are evolutionary ancient, present at least as far back as the emergence of early vertebrates. Although the PTHR1 signals primarily through the Gαs/cAMP pathway, it can also activate a number of other second-messenger cascades, likely reflecting its capacity for conformational pleiotropy. One high-affinity conformation, called R0, mediates markedly prolonged cell-signaling responses, which translate into sustained actions in vivo. These sustained signaling responses appear to arise from, at least in part, a noncanonical signaling mechanism that involves receptor/G protein activation within the endosomal domain. Elucidation of the mechanisms underlying such altered ligand binding and signaling modes could potentially facilitate the discovery of new therapeutics targeted to the PTHR1. Such novel PTHR1 ligands, particularly orally active small molecules, are of considerable interest, as they could have utility toward several bone- and calcium-related diseases, including osteoporosis and hypoparathyroidism. Efforts to develop a potent small-molecule PTHR1 agonist, however, have so far been unsuccessful, likely in part because of the difficulty in mimicking the multisite binding mechanism used by the true peptide ligands.
Acknowledgments
The authors are grateful for the valuable advice and input provided by their many colleagues, including Drs. John T. Potts, Jr., Harald Jüppner, Henry M. Kronenberg, and Michael Mannstadt of the MGH Endocrine Unit, and Peter A. Friedman of the Department of Pharmacology at the University of Pittsburgh Medical Center.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Gardella, Vilardaga.
Footnotes
Grant support to conduct research related to this work was provided by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grants P01-DK11794 (project I; to T.J.G.) and R01-DK087688 and R01-DK102495 (to J.P.V.)]. NC-IUPHAR is supported in part by Wellcome Trust Grant 099156/Z/12/Z.
Abbreviations
- AH-3960
- dibutyl-diaminomethylenepyrimidine-2,4,6-trione
- β2AR
- β2-adrenergic receptor
- BPA
- para-benzoyl-l-phenylalanine
- CRF
- corticotrophin-releasing factor
- ECD
- extracellular domain
- ECL
- extracellular loop
- ERK1/2
- extracellular signal-regulated kinase
- FRET
- Forster Resonance Energy Transfer
- GCGR
- glucagon receptor
- GPCR
- G protein–coupled receptor
- ICL
- intracellular loop
- IP3
- inositol trisphosphate
- LA-PTH
- long-acting PTH
- NHERF
- sodium-hydrogen-regulating factor
- PDE4
- phosphodiesterase 4
- Pi
- inorganic phosphate
- PKA
- protein kinase A
- PKC
- protein kinase C
- PTH
- parathyroid hormone
- PTHR1
- type-1 parathyroid hormone receptor
- PTHR2
- type-2 parathyroid hormone receptor
- PTHR3
- type-3 parathyroid hormone receptor
- PTHrP
- PTH-related protein
- SW106
- r-5-(2-E-cyclopropylvinyl)-t-3-ethyl-6,7-difluoro-5-(trifluoromethyl)benzo[e][1,4]-oxazepin-2(1H,3H,5H)-one
- TIP39
- tuberoinfundibular peptide-39
- TM
- transmembrane helix
- TMD
- transmembrane domain
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




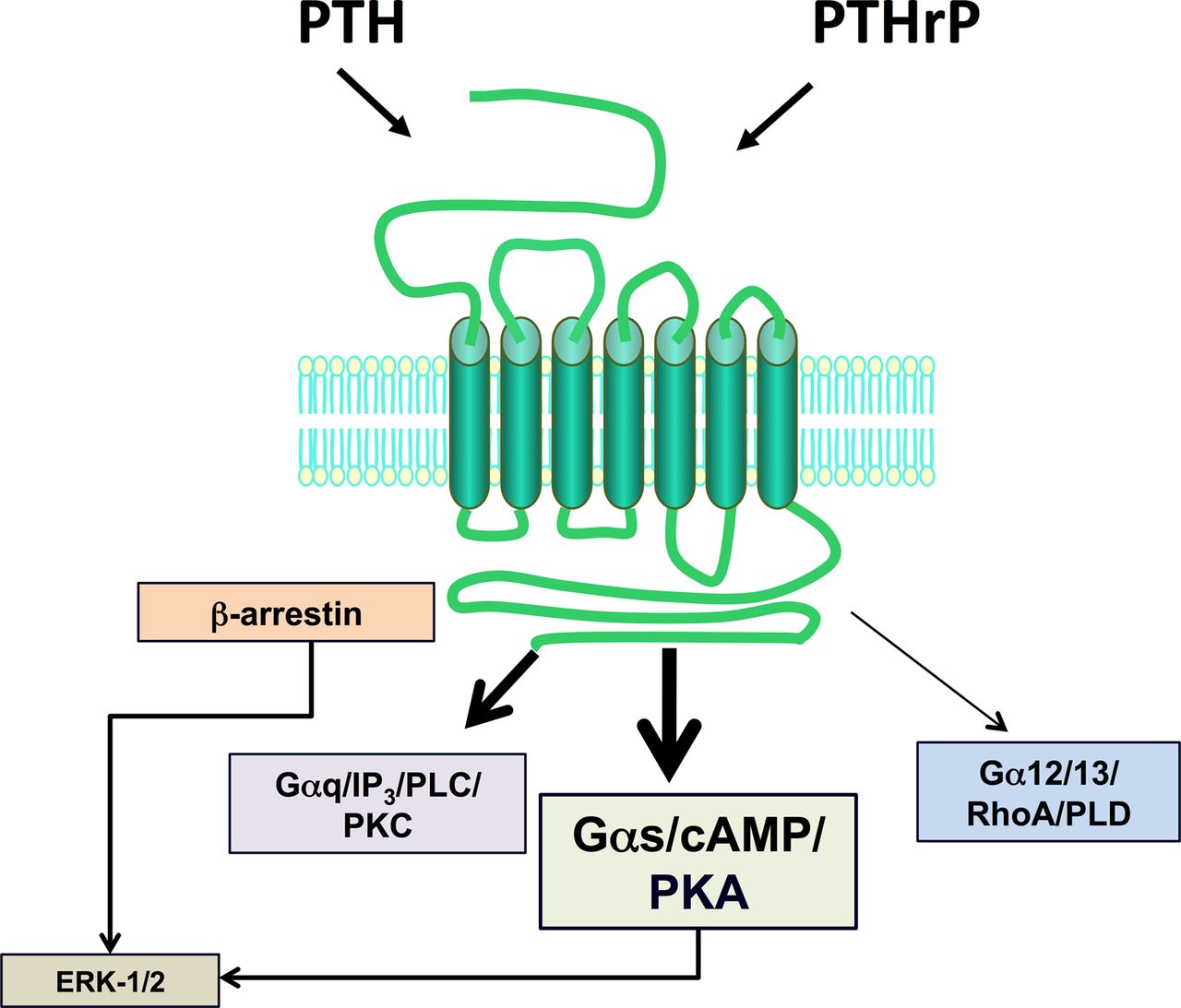{kind=link}
{kind=link}
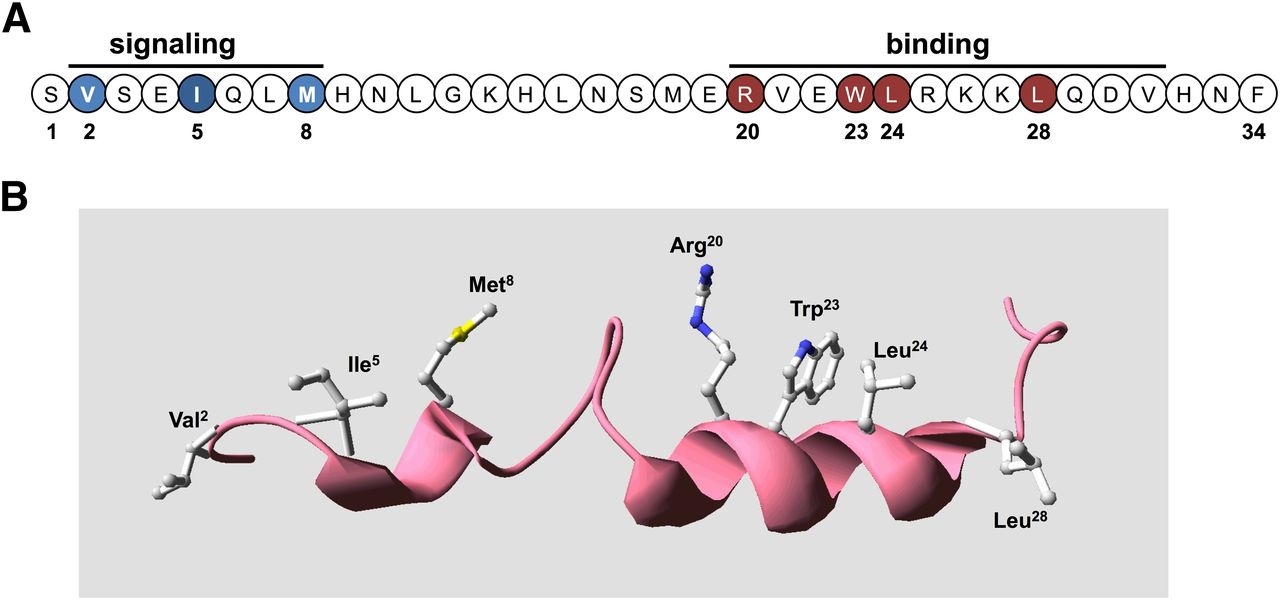{kind=link}
{kind=link}
{kind=link}
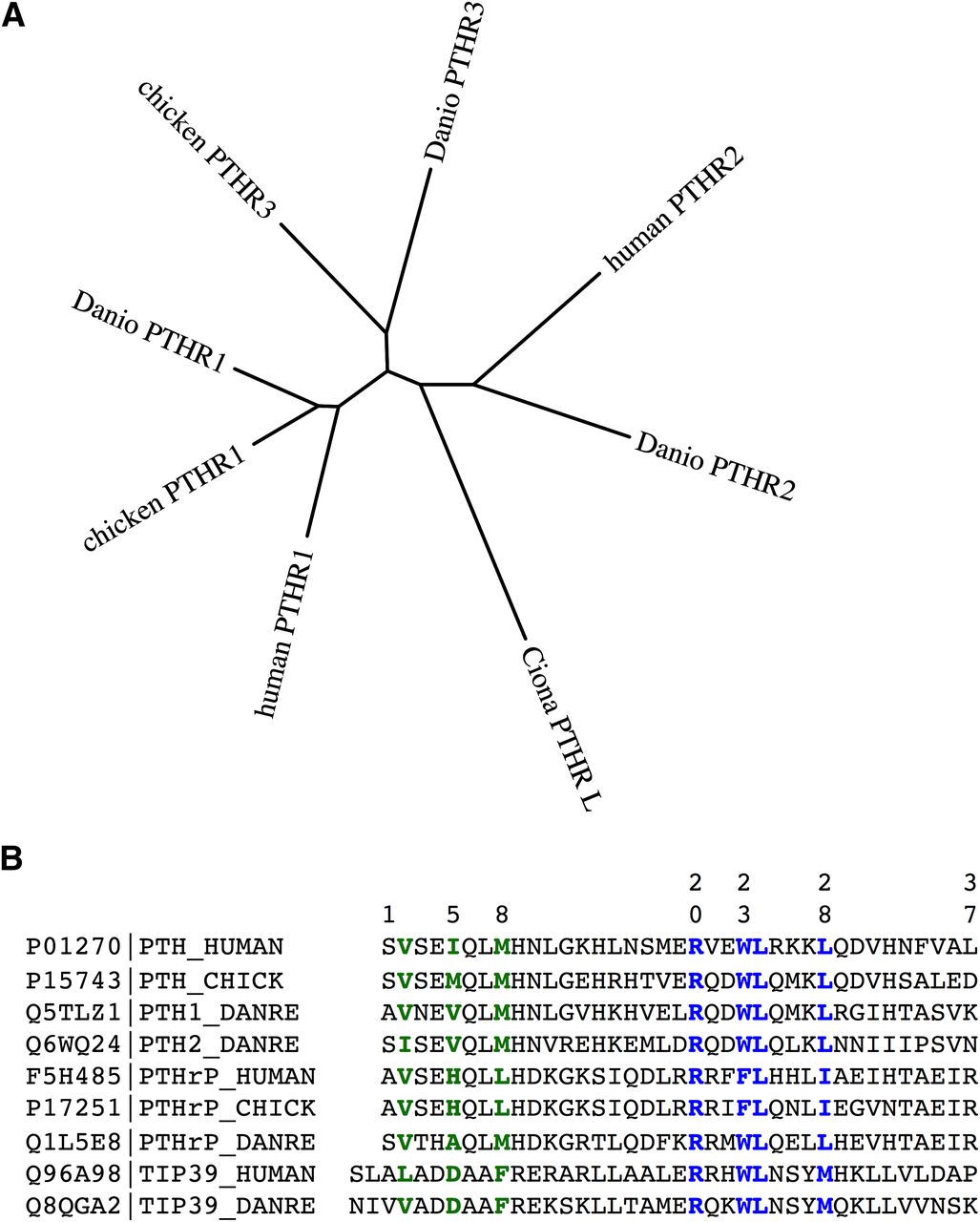{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Biologic Actions at the Type-1 Parathyroid Hormone Receptor In Vivo
- III. Structure-Activity Relationships in Parathyroid Hormone and Parathyroid Hormone–Related Protein Ligands
- IV. The Type-1 Hormone Receptor Gene Identification, Classification, and Structure
- V. The Parathyroid Hormone-2 Receptor and Tuberoinfundibular Peptide-39
- VI. Evolution of Parathyroid Hormone Receptors and Their Ligands
- VII. Structural Features of the Type-1 Parathyroid Hormone Receptor and Mode of Ligand Binding
- VIII. Mechanisms of Signal Transduction
- IX. Type-1 Parathyroid Hormone Receptor Mutations in Human Disease
- X. Small-Molecule Ligands for the Type-1 Parathyroid Hormone Receptor
- XI. Concluding Remarks
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters