


Abstract
The study of botulinum neurotoxins (BoNT) is rapidly progressing in many aspects. Novel BoNTs are being discovered owing to next generation sequencing, but their biologic and pharmacological properties remain largely unknown. The molecular structure of the large protein complexes that the toxin forms with accessory proteins, which are included in some BoNT type A1 and B1 pharmacological preparations, have been determined. By far the largest effort has been dedicated to the testing and validation of BoNTs as therapeutic agents in an ever increasing number of applications, including pain therapy. BoNT type A1 has been also exploited in a variety of cosmetic treatments, alone or in combination with other agents, and this specific market has reached the size of the one dedicated to the treatment of medical syndromes. The pharmacological properties and mode of action of BoNTs have shed light on general principles of neuronal transport and protein-protein interactions and are stimulating basic science studies. Moreover, the wide array of BoNTs discovered and to be discovered and the production of recombinant BoNTs endowed with specific properties suggest novel uses in therapeutics with increasing disease/symptom specifity. These recent developments are reviewed here to provide an updated picture of the biologic mechanism of action of BoNTs, of their increasing use in pharmacology and in cosmetics, and of their toxicology.
I. Introduction
Botulinum neurotoxins (BoNTs) are protein neurotoxins produced by neurotoxigenic strains of anaerobic and spore forming bacteria of the genus Clostridium (Clostridium botulinum, Clostridium butyrricum, Clostridium barati, and Clostridium argentinensis) (Smith et al., 2015). However, an open reading frame similar to the bont genes was identified within the genome of Weissella oryzae, a bacterium that shares some biologic niches with Clostridia (Mansfield et al., 2015). This BoNT-like is indeed a metalloprotease that cleaves vesicle-associated membrane protein (VAMP) like tetanus neurotoxin (TeNT) and several BoNTs do, but is serologically different (Zornetta et al., 2016). The BoNTs cause the flaccid paralysis of botulism by inhibiting neurotransmitter release mainly at peripheral cholinergic nerve terminals of the skeletal and autonomic nervous system (Burgen et al., 1949; Van der Kloot and Molgo, 1994; Poulain et al., 1995; Rossetto et al., 2014). Botulism is a disease of vertebrate animals, including humans, where presently, it is relatively rare owing to the improved techniques of food preparation that prevent the growth of anaerobes (Peck, 2006; Peck et al., 2011). BoNTs bind with high affinity to peripheral cholinergic nerve terminals and enter into their cytosol where they cleave SNARE proteins thus blocking the release of neurotransmitters (Rossetto et al., 2014; Rummel, 2015).
There are different forms of botulism related to the route of entry of the toxin into the body (intestine, anaerobic wounds, respiratory tract, intramuscular injection of excessive doses) but, in any case, the key pathologic symptom is a generalized peripheral neuroparalysis of variable extent that include both the skeletal and autonomic nervous systems. Such paralysis becomes evident first at the level of ocular muscles and then extends to the facial ones to reach respiratory muscles, causing respiratory failure. However, if the patient is mechanically ventilated and appropriately supported in an emergency room, usually recovery is complete, although it may take several months (Cherington, 1998; Johnson and Montecucco, 2008).
The BoNTs have been traditionally classified into seven serotypes distinguishable with animal antisera and designated with alphabetical letters from A to G (Smith et al., 2015). However, more recent molecular genetic analysis, including the use of next generation sequencing techniques, have led to the discovery of genes encoding for many novel BoNTs. They can be grouped within an existing serotype but are characterized by different amino acid sequences (Gene Bank and Uniprot databases). Although most, but not all, the known antigenic properties of these variants are conserved, they have been dubbed as subtypes and indicated with the letter of the serotype followed by a number (Rossetto et al., 2014; Montecucco and Rasotto, 2015; Smith et al., 2015). For example, for serotype A: BoNT/A1, BoNT/A2…BoNT/An; for serotype B: BoNT/B1, BoNT/B2…BoNT/Bn, etc. In addition, some chimeric BoNTs were identified and labeled accordingly: BoNT/DC, BoNT/CD, BoNT/FA. These chimeric neurotoxins are the result of past recombination events within the bont genes. The biologic significance of such a large and growing number of BoNTs has not been explained, but most likely, it is related to the different modalities of growth, transmission, and toxin production of neurotoxigenic Clostridia causing animal botulism (Eklund and Dowell, 1987; Smith and Sugiyama, 1988; Montecucco and Rasotto, 2015).
The BoNTs combine in their molecule several favorable pharmacological properties that have made them unique drugs. They are very potent and neurospecific, they have a limited diffusion when locally injected, and their action is reversible with time. These features have rendered BoNT/A1 the safest and most efficacious therapeutics for the treatment of a variety of human syndromes characterized by hyperfunction of selected nerve terminals. Their clinical use has been continuously expanding since their introduction in human therapy in the 1980s (Scott, 1980; Dressler, 2012; Hallett et al., 2013; Naumann et al., 2013b), and even more rapid is the growth of their use in a variety of cosmetic treatments (Wise and Greco, 2006; Carruthers et al., 2016; Gart and Gutowski, 2016).
The limited data available on the biologic properties of the novel BoNTs indicate that even minor differences in the amino acid sequence can significantly change their activity and toxicity (Wang et al., 2013; Whitemarsh et al., 2013; Kull et al., 2015). In addition, novel BoNT mutants endowed with ad hoc properties can be designed and produced via recombinant methods (Pirazzini et al., 2013b; Masuyer et al., 2014). Therefore, it is likely that new BoNTs with improved and/or different therapeutic targets/properties/indications will be discovered in the near future.
A. Genetics and Structure of Botulinum Neurotoxins and Their Progenitor Toxin Complexes
The bont genes are located within genetic mobile elements of phages or plasmids or in the chromosomal DNA. They code for 150-kDa proteins that fold in a three-domain structure (Fig. 1) (Lacy et al., 1998; Swaminathan and Eswaramoorthy, 2000; Kumaran et al., 2009). The bont gene is always positioned next to a gene termed ntnha (nontoxic nonhemagglutinin). The NTNHA protein is significantly similar to BoNT/A and /B sequences (∼20%), but lacks the HExxH zinc binding motif characteristic of Clostridial neurotoxin metalloproteases (Rawlings and Barrett, 1991; Schiavo et al., 1992b,c). However its structural similarity with BoNT/A is impressive, and the two proteins form a hand-in-hand–shaped heterodimer (Fig. 2A) (Gu et al., 2012; Eswaramoorthy et al., 2015). Such a structure suggests that NTNHA shields and protects the BoNT molecule from proteolytic and other chemical attacks (Miyata et al., 2009; Gu et al., 2012). This is particularly significant considering that this heterodimer is released within decaying biologic materials and it has to pass through the protease-rich gastrointestinal tract, which is the most common portal of entry of the BoNTs into the animal body.

Structure of BoNT/A1 and BoNT/B1 molecules. Crystal structures of BoNT/A1 (PDB ID: 3BTA) (Lacy et al., 1998) (A) and BoNT/B1 (PDB ID: 1EPW) (Swaminathan and Eswaramoorthy, 2000) (B) represented as space-filling models of the two opposite surfaces of each toxin molecule showing the organization of the three toxin domains: the neurospecific binding HC-C subdomain (green), the lectin-like HC-N subdomain (purple), the translocation HN domain (yellow), and the metalloprotease L domain (red). The pink cavity in the HC-C subdomains shown in the lower panels is the polysialoganglioside binding site. A peptide belt (shown in blue) surrounding the L domain and the interchain disulfide bond (white in the upper panels) linking the L and HN domain, which stabilize the structure, is also shown.

Structure of BoNTA1-NTNHA1 heterodimer and of the progenitor toxin complex (PTC). (A) Crystal structure of BoNT/A1 in complex with the NTNHA/A1 protein (PDB ID: 3V0B) (Gu et al., 2012) represented as space-filling models of the two opposite surfaces. For BoNT/A1, the L chain is in red, the HN domain is in yellow, and in green the HC domain. The BoNT/A1 protein binds “hand in hand” the NTNHA/A1 protein whose domain structure and organization are very similar to that of the toxin (see central inset for a simplified scheme). For NTNHA/A1 nL is in orange, nHN in pink, and nHC in light green. In blue and in light orange are the belts of toxin and NTNHA, respectively. Notice that NTNHA/A1 shields a large part of the BoNT surface. A similar structure has been determined for BoNT/E1 (PDB ID: 4ZKT) (Eswaramoorthy et al., 2015). (B) Space-filling representation of the large precursor toxin complex (PTC), which has a triskelion-like structure (Amatsu et al., 2013; Lee et al., 2013). BoNT/A1 (red) interacts with NTNHA/A1 (orange) but has no contacts with HA proteins. There are six HA33 proteins (blue), three HA17 proteins (light blue), and three HA70 proteins (pink) in each NTNHA/A1-BoNT/A1 complex. Because the HA proteins do not contact the BoNT/A1 molecule, they are unlikely to play any protective role on BoNT/A1, as previously proposed. Rather, the structure suggests a role as adhesin molecule (see text). Similar structures have been determined for BoNT/D and BoNT/B1 using single particle electron microscopy (Benefield et al., 2013; Hasegawa et al., 2007). The structure of (B) was assembled by joining the structure of the BoNT/A1-NTNHA/A1 heterodimer (PDB ID: 3V0B) and the structure of the triskelion (PDB ID: 3WIN).
The bont and ntnha genes are in close proximity to genes that code for neurotoxin-associated proteins endowed with hemagglutination activity (ha) in some strains, whereas in other strains they are next to genes named orfX. Both the HA proteins and the OrfX proteins form large molecular weight complexes with the BoNT-NTNHA dimer that are termed progenitor toxin complexes (PTCs) and that display a range of sizes. In the case of BoNT/A1, B1, and D the PTCs are formed by the BoNT heterodimer and HA proteins. They have been structurally characterized (Hasegawa et al., 2007; Benefield et al., 2013; Lee et al., 2013), and their spatial organization resembles that of a tripod (Fig. 2B). The HA proteins have little protein–protein contacts with NTNHA and none with the neurotoxin; therefore, they are unlikely to have any protective role versus the BoNT molecule, as proposed before, although a role of limiting the access of proteases to BoNT-NTNHA cannot be excluded. A key feature of the HA proteins of PTCs is the presence of multiple carbohydrate-binding sites (Nakamura et al., 2011; Amatsu et al., 2013; Lee et al., 2015) that are likely to be involved in the binding to the intestinal mucus layer and to the apical membrane of polarized intestinal epithelial cells or other cells of the intestinal wall through which BoNTs enter the lymphatic circulation and then the blood circulation (Fujinaga et al., 1997, 2000, 2004; Nakamura et al., 2008; Simpson, 2013). Detailed discussions of this process essential for the pathogenesis of alimentary and infant botulism can be found in Sugawara et al. (2010), Couesnon et al. (2012), Fujinaga et al. (2013), Lee et al. (2013), Simpson (2013), Lam and Jin (2015), and Rummel (2015).
The PTC complex is stable in acidic solutions, but the BoNT molecule is freed from the complexing proteins at the slightly basic pH values found below the intestinal polarized epithelial monolayer and in the intramuscular fluids (Eisele et al., 2011; Gu et al., 2012; Gu and Jin, 2013; Eswaramoorthy et al., 2015; Lam and Jin, 2015). This may become a problem in the case of hypertonic muscles where an acid pH can be generated.
B. Molecular Architecture of Botulinum Neurotoxins
Despite their amino acid sequence variability and immunologic differences, all BoNT serotypes display a similar molecular architecture. They are produced as inactive, single-chain polypeptides of 150 kDa, which are nicked by proteases within a loop subtended by a strictly conserved disulfide bond. This cleavage originates the mature (and pharmacologically active) toxin consisting of a light chain (L, 50 kDa) and a heavy chain (H, 100 kDa) connected by a disulfide bond, noncovalent interactions, and a unique segment composed by the N terminus of H, termed “belt”, which encircles the globular L domain, as shown in blue in Fig. 1 (Lacy et al., 1998; Swaminathan and Eswaramoorthy, 2000; Montal, 2010). Reduction of the single interchain S-S bond releases the L chain metalloprotease activity (Schiavo et al., 1992b, 1993c; Simpson et al., 2004).
The H chain consists of two 50-kDa domains (the amino-terminal part, HN, and the carboxy-terminal part, HC) and the molecules of BoNT/A1 and /B1, viewed in a plane, resemble the shape of a butterfly (Fig. 1). The structure of BoNT/E1 displays a different arrangement of the three domains with HC bended over the L chain (Fischer et al., 2008; Kumaran et al., 2009). The crystal structures of the remaining serotypes are not yet available for the entire molecule, but only for the separated L and the HC domains, whereas the structure of L-HN has been recently determined for BoNT/D (Masuyer et al., 2015). The similar tridimensional arrangement of isolated domains, together with the homologous amino acid sequences, suggests that the overall trimodular architecture is shared by all BoNTs, with BoNT/C, BoNT/D, and BoNT/G predicted to be similar to type A and B toxins, and type F similar to type E (Rossetto et al., 2014). In the holotoxin, the active site is partially occluded by the belt, thus preserving and preventing the catalytic activity until the conserved S-S bridge is reduced and the L chain is released in the nerve terminal cytosol (Lacy et al., 1998; Swaminathan and Eswaramoorthy, 2000; Brunger et al., 2007; Kumaran et al., 2009).
The C-terminal part of the HC domain (subdomain HC-C, 25 kDa, green in Fig. 1) mediates the interaction of BoNTs with unmyelinated areas of motoneurons, ensuring a rapid and strong interaction of the toxin with peripheral cholinergic nerve endings (Dolly et al., 1984; Binz and Rummel, 2009; Rossetto et al., 2014). HC-C is responsible for the neurospecific binding to a polysialoganglioside and to the luminal domain of a synaptic vesicle protein (Binz and Rummel, 2009; Rummel, 2013). Such binding leads to the ensuing internalization and trafficking of the toxin within endocytic compartments, which is initiated by the retrieval of synaptic vesicles after the release of their neurotransmitter content (Binz and Rummel, 2009; Rummel, 2013). The N-terminal part of HC (sub-domain HC-N, 25 kDa, purple in Fig. 1) folds similarly to sugar binding proteins of the lectin family, but its specific function is not known, although there is evidence indicating that it may improve BoNTs adhesion to the presynaptic membrane by interacting with anionic lipids (Muraro et al., 2009; Montal, 2010; Zhang and Varnum, 2012; Zhang et al., 2013). The HN domain (yellow in Fig. 1) plays a major role in the translocation of the L chain across the membrane of the endocytic vesicle into the cytosol. It consists of two long and four shorter, parallel, α-helices and of the belt segment (blue in Fig. 1).
C. Metalloproteolytic Activity
The L chain (red in Fig. 1) is a metalloprotease with its active site buried within the core of the structure. The discovery that tetanus neurotoxin (TeNT) and the BoNTs are metalloproteases with an atom of Zn2+ bound to the motif HExxH (Schiavo et al., 1992b,c), rapidly led to the subsequent identification of their unique substrates, which are the three SNARE proteins: VAMP/synaptobrevin, SNAP-25, and syntaxin. These proteins are cleaved at single peptide bonds within their cytosolic domains (Schiavo et al., 1992a, 1993a,b,c; Blasi et al., 1993a,b). These findings provided the first strong experimental evidence that the three SNARE proteins are major players in the process of exocytosis in general. Moreover, the observation that the cleavage of VAMP, a synaptic vesicle protein, was sufficient to cause neuroparalysis provided a final demonstration of the quantal hypothesis of neurotransmitter release (Katz, 1996). Very recently, a BoNT-like metalloprotease encoded by Weissella oryzae was found to cleave VAMP at a unique site within its juxtamembrane segment, which is essential for its function (Zornetta et al., 2016).
A very remarkable property of the metalloprotease activity of the BoNT L chains is their selectivity for the three SNARE proteins, which are cleaved at different peptide bonds (Fig. 3; Tables 1, 2, and 3). The molecular basis of such specificity has been clarified only for the BoNT/A1-SNAP-25 and BoNT/F1-VAMP complexes by solving the structure of their cocrystals (Breidenbach and Brunger, 2004; Agarwal et al., 2009). However, the overall picture is clear: specificity is due to multiple interactions of the L chains with their substrates, which include the cleavage site and exosites located along the sequence, both before and after the hydrolyzed peptide bond (Rossetto et al., 1994; Ahmed et al., 2001; Brunger et al., 2007). This explains why long peptide substrates are needed to test the proteolytic activity of the L chain in vitro and the current lack of specific and strong small molecule inhibitors of the BoNT metalloprotease activity (Rossetto et al., 2014).
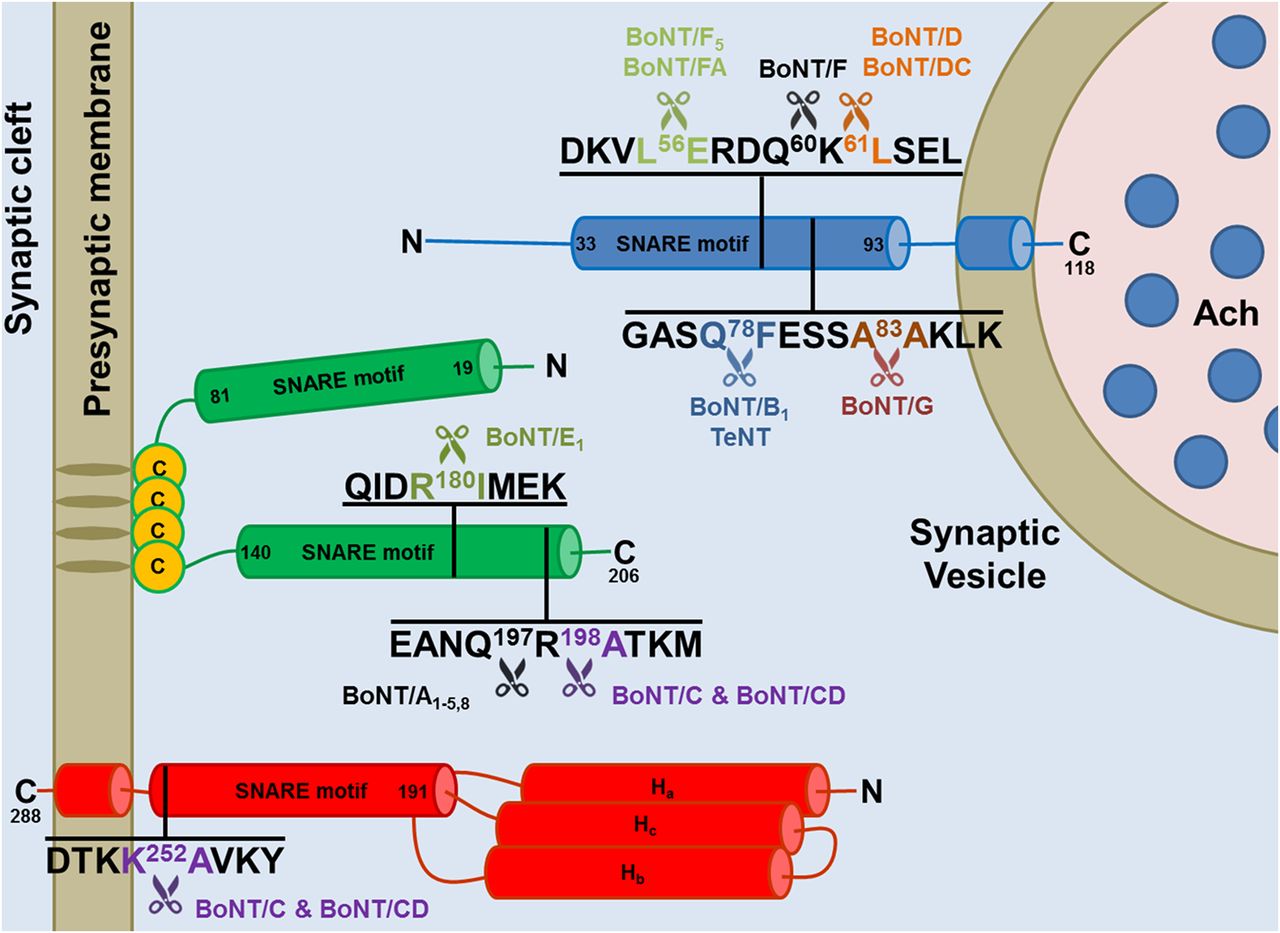
Cleavage sites of the neuronal SNARE proteins by the different BoNT types and subtypes. The BoNT proteolytic activity is highly specific and directed toward unique peptide bonds within the sequence of their respective SNARE protein targets. VAMP of the synaptic vesicle (in blue, isoform 1 is shown here) or SNAP-25 (in green) or syntaxin (in red, isoform 1B is shown here) mainly localized on the cytosolic side of the presynaptic membrane. Available evidence indicate that all the toxin subtypes and chimeric toxins cleave the same SNARE substrate, although different subtypes may cleave different peptide bonds. For example BoNT/F5 and BoNT/FA, a chimeric toxin derived from a genetic recombination between BoNT/F2, /F5, and A1 neurotoxin genes, cleave VAMP at a peptide bond different from the one cleaved by BoNT/F1. Notice that tetanus neurotoxin and botulinum B1 neurotoxin cleave the same target at the same site proving that the different symptoms of tetanus and botulism are not due to a different target molecule, but to different neuronal targets: the Renshaw cells of the spinal cord for tetanus neurotoxin and peripheral nerve terminals for BoNT/B1.
Sequence alignment of mouse, rat, and human VAMP isoforms and cleavage sites of the different BoNTs
The SNARE motifs of mouse, rat, and human VAMP isoforms have been aligned using http://www.uniprot.org/align/. The conserved cleavage sites of VAMP isoforms targeted by specific BoNT types and subtypes are in the same color of the respective toxins. The cleavage site of the newly identified BoNT-like metalloprotease of Weissella oryzae (WO) is also shown (purple). Conserved proteolytic sites whose susceptibility to cleavage is predicted, but not experimentally proven, are underlined with the color of the respective BoNT. Nonconserved cleavage sites are underlined in black as well as conserved cleavage sites, which were experimentally found to be noncleavable. When known, the recognition motif, used by the toxin to bind the substrate in addition to the cleavage sites (Rossetto et al., 1994), is underlined in red.
Sequence alignment of mouse, rat, and human SNAP25 isoforms and cleavage sites of the different BoNTs
The C-terminal SNARE motif of mouse, rat, and human SNAP isoforms have been aligned using http://www.uniprot.org/align/. The conserved cleavage sites targeted by specific BoNT types and subtypes are colored like the respective toxin. Conserved cleavage sites whose susceptibility to cleavage is predicted, but not experimentally proven, are underlined with the color of the respective BoNT. Nonconserved cleavage sites are underlined in black as well as conserved cleavage sites that were experimentally found to be noncleavable. Notice that mouse SNAP-23 has a cleavage site for BoNT/E different from the one present in SNAP-25, which is anyhow cleaved, whereas human SNAP-23, which has the same proteolytic site as SNAP-25, is not cleaved (Vaidyanathan et al., 1999). Similarly, mouse SNAP-23 was reported to be cleaved by BoNT/A, although the peptide bond is not conserved. At the same time, BoNT/C, whose cleavage site is conserved (also in human SNAP-23) does not cleave mouse SNAP-23. The cleavability of rat SNAP-23 was predicted on these premises. When known, the recognition motif used by the toxin to bind the substrate in addition to the cleavage sites (Rossetto et al., 1994) is underlined in red.
Sequence alignment of mouse, rat, and human Syntaxin isoforms and cleavage site of BoNTs
The C-terminal SNARE motifs of mouse, rat, and human syntaxin isoforms are aligned using http://www.uniprot.org/align/. BoNT/C conserved cleavage sites are shown in purple. Conserved cleavage sites whose susceptibility to cleavage is predicted, but not experimentally proven, are underlined in purple. Nonconserved cleavage sites are underlined in black.
II. Biology
A. Molecular Mechanism of Nerve Terminal Paralysis
BoNTs are typical examples of bacterial exotoxins targeting intracellular substrates. These toxins have evolved a structural organization designed to deliver the metalloprotease domain into the host cell cytosol and this has been achieved by exploiting several physiologic functions of nerve terminals. The mechanism of nerve terminal intoxication by the BoNTs is conveniently divided into five major steps (Fig. 4): 1) binding to nerve terminals, 2) internalization within an endocytic compartment, 3) low pH driven translocation of the L chain across the vesicle membrane, 4) release of the L chain in the cytosol by reduction of the interchain disulfide bond, and 5) cleavage of SNAREs with ensuing blockade of neurotransmitter release and neuroparalysis.

The nerve terminal intoxication by botulinum neurotoxins is a multi-step process. The first step (1) is the binding of the HC domain (green) to a polysialoganglioside (PSG) receptor of the presynaptic membrane (gray and black), followed by binding to a protein receptor. The currently known protein receptors are i) synaptotagmin (Syt, gray) for BoNT/B1, /DC, and /G; ii) glycosylated SV2 (black with its attached N-glycan in pink) for BoNT/A1 and /E1. Syt may be located either within the exocytosed synaptic vesicle or on the presynaptic membrane. The BoNT is then internalized inside SVs, which are directly recycled (2a) or inside SVs that fuse with the synaptic endosome and re-enter SV cycle by budding from this intermediate compartment (2b). The acidification (orange) of the vesicle, operated by the v-ATPase (orange), drives the accumulation of neurotransmitter (blue dots) via the vescicular neurotransmitter transporter (light blue). The protonation of BoNT leads to the membrane translocation of the L chain into the cytosol (3), which is assisted by the HN domain (yellow). The L chain (red) is released from the HN domain by the action of the thioredoxin reductase-thioredoxin system (TrxR-Trx, blue and dark blue) and Hsp90 (mud color), which reduce the interchain disulfide bond (orange) and avoid the aggregation of the protease (4). In the cytosol, the L chain displays its metalloprotease activity: BoNT/B, /D, /F, /G cleave VAMP (blue); BoNT/A and BoNT/E cleave SNAP-25 (green); and BoNT/C cleaves both SNAP-25 and syntaxin (Stx, dark red) (5). Each of these proteolytic events is sufficient to cause a prolonged inhibition of neurotransmitter release with consequent neuroparalysis.
1. Binding and Specificity.
After entering the lymphatic and blood circulations, following intestinal absorption or inspiration or injection, the BoNTs rapidly gain access to the perineuronal fluid compartment but do not cross the blood-brain barrier (Simpson, 2013). The known BoNTs bind with high affinity to the presynaptic plasma membrane of skeletal and autonomic cholinergic nerve terminals in numbers estimated to be, for BoNT/A1 or /B1, in the order of hundreds of molecules per square micrometers at the rat neuromuscular junction (NMJ) (Dolly et al., 1984). This restricted tropism is extraordinary, particularly considering that the presynaptic plasma membrane of cholinergic peripheral nerve terminals represents an infinitesimal part of the total surface area of cells and tissues exposed to body fluids. Such neurospecificity and affinity of binding, together with their catalytic activity, is at the basis of the BoNTs toxicity and, at the same time, of their pharmacological and therapeutic use.
This unique nerve terminal binding property of the BoNTs is due to their capacity to interact with two independent receptors of the presynaptic membrane: a polysialoganglioside (PSG) and the glycosylated lumenal domain of a synaptic vesicle protein that mediates the following step of internalization (Montecucco, 1986). Additional receptor(s) might be involved and contribute to the preference for cholinergic terminals (Montecucco et al., 2004; Kammerer and Benoit, 2014). It is also possible that cholinergic nerve terminals express unique N-glycans attached to synaptic vesicle (SV) glycoproteins that contribute to this preferential binding. Fibroblast growth factor and vanilloid receptors were recently reported to bind BoNT/A1 (Jacky et al., 2013; Li and Coffield, 2016), but their functional involvement in toxin binding has to be further validated.
The involvement of PSG in BoNT binding has been extensively investigated and is supported by a large wealth of experimental data (Rummel, 2013), including the demonstration that mice and cell lines devoid of complex PSG are largely resistant to BoNT (Kitamura et al., 1999; Bullens et al., 2002). This fact also contributes to explain why insects that are devoid of PSG are BoNT insensitive, a property that makes them reliable vectors of BoNT spread during outbreaks of animal botulism among birds and fishes (Montecucco and Rasotto, 2015).
The BoNTs-PSG interaction is rather well characterized and reviewed (Rummel, 2013). Briefly, BoNT/A, /B, /CD, /E, /F, and /G (and TeNT) possess within the HC-C subdomain a large cavity defined by the conserved motif sequence E(Q in BoNT/G)…H(K in BoNT/E and G in BoNT/G)…SXWY…G (Fig. 1, pink). The affinity of this PSG-BoNT binding has been estimated and the Kd values are in the 0.1–1 μM range (Rummel, 2013). At variance, BoNT/C, BoNT/D, and the mosaic neurotoxin BoNT/DC bind PSG at a binding site located in a similar position but defined by a different set of amino acids lateral chains and with lower affinity (Strotmeier et al., 2010; Rummel, 2013). Interestingly, BoNT/C, BoNT/D, and BoNT/DC (and TeNT) harbor a second PSG binding site in their HC-C domain, defined by conserved W and Y(F) residues (Strotmeier et al., 2010, 2011), which might bind the carbohydrate portion of a plasma membrane glycoprotein that is endocytosed from the presynaptic membrane. BoNT/DC was recently shown to bind a sialyl residue in a pocket of the HC domain and a cell binding mechanism involving a cooperative contribution of two ganglioside binding sites was proposed (Nuemket et al., 2011).
Together with sphingomyelin, cholesterol, and some proteins, PSG form anionic microdomains in the plasma membrane (Simons and Toomre, 2000; Sonnino et al., 2007; Prinetti et al., 2009). Accordingly, BoNTs bind complex PSGs whose large oligosaccharide head group projects at a distance from the membrane surface and it is flexible and negatively charged. There is evidence that anionic microdomains of the presynaptic membrane including PSG may orient the electric dipole associated to the BoNT molecules whose positive end is, significantly, located around the PSG binding site. This reorienting effect of the membrane on the approaching BoNT molecule would strongly increase its probability of PSG binding, making it a reaction controlled only by diffusion (Fogolari et al., 2009).
Following membrane binding, the BoNTs are internalized. The key observations that neuronal stimulation enhances the toxicity of BoNTs (Hughes and Whaler, 1962; Simpson, 1980, 1985; Black and Dolly, 1986; Keller et al., 2004) and that BoNT/B binds synaptotagmin in cultured cells (Nishiki et al., 1994) led to the suggestion that BoNTs endocytosis is facilitated by a protein receptor consisting of the luminal domain of a synaptic vesicle (SV) protein (Montecucco and Schiavo, 1995). This second binding provides the high affinity necessary to bind the very low amounts of BoNT estimated to be present in the circulation during botulism (10−13–10−14 M). During stimulation, the increased exoendocytosis rate causes a frequent exposure of the SV lumen, making the vesicle interior transiently available for binding. This also contributes to account for the efficacy of BoNT in treating human syndromes characterized by hyperactive nerve terminals, because the NMJs of these muscles have a higher rate of SVs exoendocytosis, which favors toxin uptake.
Within a few years, the protein receptors of other BoNTs were identified. BoNT/B1, BoNT/G, and BoNT/DC bind segments 39–50 of synaptotagmin I and 47–58 of synaptotagmin II (Nishiki et al., 1994; Dong et al., 2003; Rummel et al., 2007; Peng et al., 2012; Willjes et al., 2013). BoNT/A1 and BoNT/E1 bind different segments of the fourth lumenal loop of SV2, a multispanning integral protein of synaptic vesicles of unknown function (Dong et al., 2006; Mahrhold et al., 2006; Binz and Rummel, 2009; Benoit et al., 2014). Three isoforms of SV2 (A, B, C) are expressed at motor nerve terminals, but SV2C appears to be the one binding BoNT/A1 more efficiently than SV2A or B via its glycosylated fourth luminal domain (Mahrhold et al., 2006; Benoit et al., 2014; Mahrhold et al., 2016), whereas BoNT/E1 binds isoforms A and B, but not C (Dong et al., 2008).
The binding of BoNT/A1 to the glycosylated SV2C receptor is mediated by protein-protein and protein-glycan interactions that do not appear to influence each other. Of the five putative N-glycosylation sites of SV2C, only the Asn-559 is involved; this N-glycan is highly complex with a tetra antennary structure, which is likely to interact with an extensive area of HC/A1 (Mahrhold et al., 2016). The protein-protein HC/A-SV2C contacts involve mostly the backbones of the two proteins, through the pairing of two solvent-exposed β-strands, one from each partner (Benoit et al., 2014). Asn559 of SV2C is at the center of the BoNT/A1-SV2 area of interaction and its N-linked glycan inserts in a crevice made by one β-strand of the HC-N subdomain (segment 950–954) and two segments of the subdomain HC-C: the β-strand segment 1064–1066 and the C-terminal loop 1289–1292 (Fig. 5A) (Yao et al., 2016). As glycosylation patterns vary during development and among adult individuals (Knezević et al., 2009; Pucic et al., 2010; Lauc et al., 2016), such a feature might be responsible for the different onset and duration of neuroparalysis elicited in human patients by the same dose of injected BoNT/A1 as different amounts of bound toxin are likely to correspond to different numbers of L chains entering the nerve terminal cytosol. In addition, as invertebrate and vertebrate N-glycans are different (Moremen et al., 2012), this may contribute to the lack of sensitivity of invertebrates to BoNTs.

Close-up views of the binding interfaces between BoNT/A1 and BoNT/B1 to their synaptic vesicle protein receptors. (A) Two areas of interaction of BoNT/A1 with the synaptic vesicle protein glycosylated-SVC2 (PDB ID 5JLV). One main interaction is mediated by protein-protein contacts through the pairing of the backbones of two solvent-exposed β-strands (black dotted ellipsoid), one from each partner. Essential interactions are mediated by R1156 of BoNT/A1 making a cation-π stacking interaction with P563 of SV2C and also by R1294 of the toxin. The second main interaction is mediated by N559 whose side chain carries a N-glycan modification (shown as sticks), which fits within a crevice formed at the interface between HC-N (purple) and HC-C (green). Amino acids forming the groove are colored in cyan and labeled according to their location (P953, N954, S957, S1062, H1064, and R1065 from HC-N, in purple and T1145, Y1155, D1288, D1289, and G1292 from HC-C, in green). The cartoon also shows essential water molecules (black pellets) and the H bonds (yellow dotted lines), which mediate the interaction of BoNT/A1 with the N-glycan, suggesting the possibility that they serve to adapt the variety of N-glycans that are produced by different kind of neurons and/or by neurons of different individuals and animal species. (B) Interaction among BoNT/B1 and its synaptic vesicle protein receptors Synaptotagmin II (Syt-II) (PDB ID 4KBB). The interface of interaction is at the extreme bottom of BoNT/B and, at variance from BoNT/A1, involves exclusively HC-C (green). Syt-II is unstructured in solution but assumes an helical conformation upon binding to the toxin, forced by the interactions occurring at the level of two hydrophobic pockets. One pocket is formed by HC-C residues P1117, W1178, Y1181, P1194, A1196, P1197, F1204 with Syt-II residues M46, F47, and L50. The second pocket of HC-C is lined by residues K1113, S1116, P1117, V1118, Y1183, E1191, K1192, F1194, and F1204 with Syt-II residues F54, F55, E57, and I58. Only the most significant aminoacids involved in the interaction are shown and labeled. Note the H bond (black dotted line) formed by K1113 and E57, which may also interact electrostatically. The binding sites for the oligosaccharide portion of polysialoganglioside receptor are not shown, but in both BoNT/A1 and /B1 are located within the HC-C subdomain at a distance from the protein receptor binding sites in such a position that they do not interact with them (see text).
The structural basis of BoNT/B1 and of BoNT/DC binding to their synaptotagmin receptor have been determined and are shown in Fig. 5B (Chai et al., 2006; Jin et al., 2006; Berntsson et al., 2013a,b). Such binding is mediated by a pocket present in the HC-C subdomain formed by residues of the segment Pro1114-Phe1202 (BoNT/B1 numbering), which fits the α-helical segment 47–58 of synaptotagmin II located next to the lumenal surface of the SV membrane. Asn24 of synaptotagmin II is predicted to be glycosylated but the potential role of N-glycan linked to this residue on the binding of BoNT/B1 has not yet been tested, although this may be relevant with respect to the therapeutic use of BoNT/B1 in humans, because the pattern of glycosylation in the nervous system varies among individuals. The presence of Leu51 in human synaptotagmin II reduces significantly the binding affinity for BoNT/B1, /DC, and /G with respect to the corresponding Phe54 of mouse synaptotagmin II. This also explains the much higher dosage of BoNT/B1 necessary to achieve the same therapeutic effect of BoNT/A1 (Peng et al., 2012; Strotmeier et al., 2012).
The protein receptors of other BoNTs are not known in such details. BoNT/C might not have a protein receptor, because protease pretreatment does not affect its binding and internalization into cultured cells (Tsukamoto et al., 2005). Conflicting results have been reported for BoNT/D (Kroken et al., 2011; Peng et al., 2011; Rummel, 2013), but based on its similarity with BoNT/E, BoNT/F is expected to bind SV2, and some data support this possibility (Rummel et al., 2009).
2. Internalization into Nerve Terminals.
After intramuscular injection, BoNT/A1 is rapidly taken up and found within the lumen of SV at the neuromuscular junction (Colasante et al., 2013). In cultured primary neurons part of such internalization is clathrin mediated and most of the toxin is detected inside SV (Neale et al., 1999; Couesnon et al., 2009; Harper et al., 2011). Importantly, the average number of BoNT/A molecules per SV were estimated to be one to two (Harper et al., 2011; Colasante et al., 2013), a figure that matches the estimated copy number of SV2 molecules per vesicle (Takamori et al., 2006). The internalization of the other BoNTs has not been visualized by electron microscopy, but it is expected to occur as well via SV because these toxins bind a SV membrane protein.
SV exocytosis is strictly coupled to endocytosis, which can take place in different ways (Saheki and De Camilli, 2012; Cousin, 2015; Jähne et al., 2015; Kononenko and Haucke, 2015; Soykan et al., 2016). Before fusion with the presynaptic membrane at specialized release sites of nerve terminals (active zones), SVs are morphologically similar and, to be functional, they must have a defined proteic and lipidic composition (Takamori et al., 2006; Boyken et al., 2013). However, after fusion with the presynaptic membrane, endocytosed SVs may acquire molecular differences (Wienisch and Klingauf, 2006; Soykan et al., 2016). Linked to this and to the frequency of nerve terminal stimulation, SVs may undergo different forms of retrieval (Saheki and De Camilli, 2012; Jähne et al., 2015; Kononenko and Haucke, 2015; Soykan et al., 2016). In particular a functional SV, right after endocytosis, may be directly recycled and reacidified by the v-ATPase proton pump, which generates an electrochemical gradient driving the accumulation of neurotransmitter via a specific transporter (Parsons, 2000; Omote et al., 2011). Alternatively, some vesicles may fuse with endosomal compartments, where quality-controlled SVs of appropriate composition reform and re-enter the synaptic cycle. Under some conditions, early endosomal compartments can also be generated from the infolding of a large portion of the presynaptic membrane, and then SVs can reform by endosomal budding. Additionaly, it was reported that SV proteins (including synaptotagmins) of exocytosed SV can freely diffuse within the presynaptic boutons, where they are slowly confined in a periactive zone by endocytic adaptors and presynaptic diffusion barriers to be reinternalized by clathrin-dependent or bulk endocytosis mechanisms (Gimber et al., 2015).
Collectively, these different possibilities of SV endocytosis indicate that the trafficking of BoNTs may occur via different pathways, depending at least partially, on their receptors. This is supported by experiments performed with primary cultures of central nervous system neurons, showing a significant variability in the intoxication kinetics of different BoNTs (Keller et al., 2004; Sun et al., 2012). Moreover, the use of the endocytosis inhibitor 4-bromobenzaldehyde N-(2,6-dimethylphenyl)semicarbazone (EGA) (Gillespie et al., 2013) provided indirect evidence that different BoNTs may follow different trafficking routes after the initial endocytosis also in vivo (Azarnia Tehran et al., 2015). This would result in different entry times of the L chain into the cytosol and would explain the “rapid” entry of BoNT/A and /E and the slower one of BoNT/B determined in primary cultures of neurons (Keller et al., 2004; Sun et al., 2012; Colasante et al., 2013).
Interestingly, a BoNT may be taken up even after the inhibition of SV exocytosis by another BoNT in vitro (Keller et al., 1999, 2004; Pellett et al., 2015a) and in vivo (Eleopra et al., 1998; Adler et al., 2001; Meunier et al., 2003; Keller, 2006; Antonucci et al., 2008). Clearly these results are highly dependent on the SNARE isoform cleaved by the BoNT used to inhibit exocytosis and on the extent of cleavage, data that were not reported. Moreover, it is well established that PSG undergoes endocytosis by the ligand-receptor pathway (Sonnino et al., 1992), and therefore it cannot be ruled out that the sole BoNT-PSG interaction is sufficient to sustain neurotoxicity, particularly for those BoNTs that have two ganglioside binding sites. Taken together these results and considerations indicate that more than one way of uptake may be used at nerve terminals by the different BoNTs or by the same BoNT under different conditions.
a. Long distance effects of botulinum neurotoxins.
Peripheral neuroparalysis is the most evident symptom of botulism and is the one at the bases of the therapeutic use of BoNTs. However, indirect evidence that these neurotoxins could act at a distance from the injection site, i.e., within spinal cord and brain neuronal circuits, were reported long ago, and in some cases it was shown to be due to retroaxonal transport of BoNTs similar to that occurring with TeNT (Tyler, 1963; Polley et al., 1965; Garner et al., 1993; Priori et al., 1995; Santini et al., 1999; Gilio et al., 2000; Wohlfarth et al., 2001; Marchand-Pauvert et al., 2013; Matak and Lackovic, 2014; Mazzocchio and Caleo, 2015; Matak et al., 2016). Direct evidence of BoNTs retroaxonal transport after intramuscular injection was provided by the detection of cloramine T radioiodinated BoNT/A1 within the ventral roots ganglia and other spinal cord segments (Habermann, 1974; Wiegand et al., 1976). However, it was not clear whether retrotransported toxins remained active, because the method of radioiodination used inactivates the BoNTs. More recently, compelling evidence of BoNT/A1 retrotransport to the central nervous system (CNS) was provided by tracing the cleavage of the SNARE within CNS neurons after peripheral injection of the toxin, using an antibody very specific for the novel epitope generated by the BoNT/A1 cleavage of SNAP-25 (Antonucci et al., 2008; Matak et al., 2011, 2012; Restani et al., 2011; Matak and Lackovic, 2014; Mazzocchio and Caleo, 2015). The injection of BoNT/A1 in the rat whisker pad led to the appearance of truncated SNAP-25 in the somatodendritic area of primary efferent facial motoneurons (Antonucci et al., 2008). Cleaved SNAP-25 was also detected in the ventral horns of the spinal cord upon injection of BoNT/A1 in the gastrocnemius (Matak et al., 2012). Interestingly, BoNT/A1 retrograde transport can occur also via sensory neurons, as the injection in the whisker pad induces the appearance of truncated SNAP-25 both in the trigeminal nucleus caudalis (Matak et al., 2011) and in the dorsal horn of the spinal cord after subcutaneous or intramuscular injection in the hind limb (Marinelli et al., 2012; Matak et al., 2012, 2014). Notably, this suggests that BoNT/A1 undergoes retrograde transport from periphery to ganglia but also that an anterograde movement from ganglia to afferent innervations in the brain stem or in the spinal cord may occur (Restani et al., 2011). These long-distance effects are mediated by an active retroaxonal transport of catalytically competent toxins inside motor axons and not by their passive spread or to that of the cleaved product (Antonucci et al., 2008; Matak et al., 2011). Intriguingly, it was also found that BoNT/A1 can enter and cleave SNAP-25 in neurons which are even two synapses away from the injection site (Restani et al., 2012b), entailing that upon retrotransport the toxin can undergo sequential cycles of transcytosis and transport, remaining catalytically active. At variance, using neurons isolated from superior cervical ganglia of newborn rats, it was found that BoNT/A1 migrates within nerve terminal in a nonvesicular mode without inhibiting distal neurotransmission (Lawrence et al., 2012).
The prototype of the neurotoxins binding to peripheral nerve terminals and reaching the CNS is tetanus neurotoxin, which has become the marker of retroaxonal transport inside motor axons (Schwab et al., 1979; Rossetto et al., 2013). TeNT uses the neuromuscular junction as a portal of entry following its binding to nidogen, a protein of the basal lamina (Bercsenyi et al., 2014). Thereafter, it travels inside a compartment that links the clathrin-dependent endocytosis to the sequential activity of the two small GTPases Rab5 and Rab7, allowing the trafficking of the toxin from “signalling endosomes” (Zweifel et al., 2005; Schmieg et al., 2014) into the retrograde transport pathway of neurotrophins (Deinhardt et al., 2006). It is possible that BoNT/A1 is also recruited to the same nonacidic carriers exploited by TeNT and neurotrophins (Restani et al., 2012a). A recent study has implicated autophagosomal structures in the capture and transport along the axon of BoNT/A1 (Wang et al., 2015), a pathway that appears to be exploited by polio virus to be delivered in the CNS (Bird et al., 2014). However, these vesicle carriers may also be connected to degradative pathways as suggested by the partial costaining of BoNT/A1 with the autophagosome marker LC3 (Wang et al., 2015).
It should be noticed that no evident symptoms have been associated so far with BoNTs acting within the CNS upon peripheral injection in humans or in the animals tested, whereas there is evidence suggesting that they could contribute to the antinociceptive activity of BoNT/A (Matak and Lackovic, 2014; Mazzocchio and Caleo, 2015; Pellett et al., 2015d).
3. Membrane Translocation.
BoNTs translocate their L chain into the cytosol from an acidic intracellular compartment, and the process can be blocked by different amines and by bafilomycin A1, a specific inhibitor of the v-ATPase (Simpson, 1983; Williamson and Neale, 1994; Keller et al., 2004). However the lumen of the SV inside the nerve terminal is not experimentally accessible, making the study of this process difficult. Therefore, models that bypass the intracellular delivery of the toxin and induce the entry of the L chain of BoNTs from the cell surface across the plasma membrane were devised (Pirazzini et al., 2011; Sun et al., 2011). By using these systems, it was found that the translocation of L takes place between pH 4.5 and 6 (Pirazzini et al., 2011) and that the entire translocation process is rapid (few minutes at 37°C) and strongly temperature dependent (Pirazzini et al., 2013c). Little if any translocation occurs above pH 6, and this makes it unlikely that the L domain translocates from early endosomes, whose internal pH is only slightly acidic. On the other hand, BoNT/A1 has been localized within the SV lumen by immuno electron microscopy in cultured hippocampal neurons (Harper et al., 2011) and within the NMJ (Colasante et al., 2013). An essential component of SV is the electrogenic v-ATPase, which injects protons into the lumen, generating a transmembrane pH gradient ΔpH of 1.4 pH units and an electrical gradient ΔΨ of + 39 mV (Parsons, 2000). By using fluorescent synaptophluorins, the luminal pH of SV was estimated to be ∼5.8 pH units (Miesenbock et al., 1998). Accordingly, the translocation of L into the cytosol can take place from recycling SV or from SV budded from early endosomes. However, it cannot be excluded that other similarly acidic compartments such as late endosomes and lysosomes that could be reached by a BoNT upon endosome maturation, could provide the appropriate environment leading to L chain translocation, although it should be considered that these organelles contain proteases that may degrade the toxin.
Planar lipid bilayer studies have shown that at low pH several BoNTs and tetanus toxin form transmembrane ion channels (reviewed in Montecucco and Schiavo, 1995). Despite the intense effort of several laboratories, the molecular mechanism describing the transformation of the water-soluble BoNT molecule into a transmembrane ion channel that assists the translocation of the ∼440 amino acids of the L metalloprotease domain across the membrane is still missing. However, a remarkable set of patch-clamping experiments (Montal, 2010; Fischer, 2013; Fischer and Montal, 2013) and recent results obtained with the plasma membrane entry model (Pirazzini et al., 2011; Sun et al., 2011) have shed light on the initial molecular events of the process for BoNT/A1 and /B1. We have reviewed and modeled BoNT membrane translocation recently (Pirazzini et al., 2016), and, therefore, only the most relevant aspects are highlighted here.
The starting point is the BoNT molecule bound to the luminal side of the SV membrane via two interactions: with the polysialoganglioside and with the protein receptor. As the pH lowers, some conserved high pKa carboxylates of BoNT become protonated on the face of the molecule containing the interchain disulfide bond (Fig. 1, top), which acquires a net positive charge, as indicated by bioinformatics and mutagenesis analysis; remarkably, the opposite face is devoid of conserved high pK protonatable residues (Pirazzini et al., 2011, 2013b). Consequently, the BoNT molecule, with its positively charged surface, falls onto the anionic surface of the membrane generating a lipid-protein complex. The pH value close to the membrane is estimated to be at least one pH unit lower than in the lumen owing to the Guy-Chapman effect (Nordera et al., 1997), leading to the protonation of carboxylate residues of lower pKa values. This triggers a process of structural change involving the L and H chain together with membrane lipids, leading to the formation of an ion channel. During this process, both the H and L chain enter in contact with the hydrocarbon chain of lipids as determined by photoactive hydrophobic labeling (Montecucco et al., 1989). There is a general consensus that the H chain acts as a sort of transmembrane chaperone for the translocation of the L chain across the membrane (Koriazova and Montal, 2003; Montal, 2010; Fischer, 2013). Different molecular roles of the H chain can be envisaged, and two possibilities with a range of intermediate cases are mentioned here: 1) that of a protein conducting channel that translocates the unfolded L chain, as it occurs in the case of the protective and lethal factors of anthrax (Collier, 2009), 2) the toxin forms a “molten globule”, a protein state that is known to be capable of interacting with the hydrophobic membrane interior (Ptitsyn et al., 1990; van der Goot et al., 1991). Such molten globule would insert into the membrane together with anionic lipids and would deliver the L chain to the other side of the membrane, whereas the H chain would assemble an ion conducting channel. In any case, the belt has to be unfastened to permit the passage of the L chain on the cytosolic face of the membrane (Fischer and Montal, 2007). The understanding of the mechanism of membrane translocation is very important, because it is likely to be common to all BoNTs and may thus present a key target step for the development of pan-inhibitors of the entire family of BoNTs (Fischer et al., 2009; Pirazzini and Rossetto, 2017).
The number of BoNT molecules involved in membrane translocation is another undefined point. The finding that one SV contains one or two molecules of BoNT/A1 leaves open the possibility that one toxin molecule is sufficient to carry out the process and suggests that these toxins may have an in-built membrane translocating system, similarly to the bacterial system SecY (Park and Rapoport, 2012). Diphtheria toxin, a bacterial toxin with a structural architecture similar to that of BoNTs, also has a translocating domain that is mainly α-helical, and the available evidence supports a single molecule process, with few transmembrane helices forming an ion channel (Finkelstein et al., 2000; Gordon and Finkelstein, 2001; Leka et al., 2014). At variance, it was recently reported that a trimer might form the protein conducting transmembrane channel of BoNT/B1 in PC12 cells and BoNT/E1 at physiologic pH (Sun et al., 2012). Clearly, additional experiments are needed to clarify this essential step of the mechanism of action of the BoNTs.
4. Interchain Disulfide Reduction.
The importance of the interchain disulfide for the toxicity of clostridial neurotoxins is demonstrated by the fact that reduced toxins are inactive (Schiavo et al., 1990; de Paiva et al., 1993; Fischer and Montal, 2007). At the cellular level, Fischer and Montal (2007) demonstrated that the premature reduction of this S-S bond, at any stage before its exposure to the cytosol, aborts the L chain translocation. Also in the plasma membrane translocation model, the L chain has to be disulfide linked to the H chain to cross the membrane (Pirazzini et al., 2011). The detachment of the L chain from the cytosolic face of the membrane by reduction releases its metalloprotease activity in the cytosol (Fischer and Montal, 2007).
The lumen of most intracellular organelles is oxidizing and low pH prevents the reduction of disulfide bonds. At variance, the cytosol has a higher redox potential owing to the presence of several reducing molecules (NADH, NADPH, reduced glutathione, cysteine, etc.). The maintenance of an appropriate redox balance is particularly important for the activity of key proteins (Arner and Holmgren, 2000; Meyer et al., 2009; Hanschmann et al., 2013). The NADPH-thioredoxin reductase (TrxR)-thioredoxin (Trx) system is a major redox system of the cell that reduces protein disulfides. Its involvement in the BoNTs and TeNT entry into neurons was first identified using a pharmacologic approach (Pirazzini et al., 2013a), and then TrxR and Trx were shown to be extrinsic proteins of the cytosolic side of the SV membrane (Pirazzini et al., 2014), where translocation of the L chain is expected to occur. Several inhibitors of the TrxR-Trx redox couple prevent the display of the SNARE specific metalloprotease activity of the L chain of all serotypes of BoNTs, from A to G in cultured neurons. More importantly, these inhibitors largely prevent the BoNT-induced paralysis in mice in vivo, regardless of the serotype involved (Zanetti et al., 2015). The reduction of the single interchain disulfide bond is therefore a general and fundamental step of the BoNT [and TeNT (Pirazzini et al., 2013a; Zuverink et al., 2015)] mechanism of nerve terminal intoxication. As such it has to be considered a step per se in the sequence of passages leading from BoNT nerve terminal binding to neuroparalysis. More recently it was found that the chaperone Hsp90 is also present on the cytosolic face of SV and that it cooperates with TrxR and Trx in the entry of a folded and active L chain in the cytosol (Azarnia Tehran et al., 2017).
5. SNARE Protein Cleavage.
SNARE proteins form a large superfamily comprising many isoforms of VAMP (Rossi et al., 2004) and syntaxins but relatively fewer isoforms of SNAP. Specific isoforms of SNARE proteins selectively interact and form heterotrimeric coiled-coil complexes (SNARE complexes), which mediate most intracellular events of vesicle–target membrane fusion (Jahn and Scheller, 2006). The discovery that TeNT and BoNTs are metalloproteases (Schiavo et al., 1992b,c) and that TeNT and BoNT/B1 cleave the SV protein VAMP/synaptobrevin (Schiavo et al., 1992a) opened the way to the the rapid identification of the other SNARE proteins targets of the other BoNTs (Blasi et al., 1993a,b; Schiavo et al., 1993a,b,c). The cleavage by TeNT and BoNTs of proteins that were previously identified by molecular biology and biochemical methods (Elferink et al., 1989; Oyler et al., 1989; Bennett et al., 1992) explained the molecular basis of tetanus and botulism. At the same time, these findings provided the strongest evidence of the SNARE protein involvement in neurotransmitter release and, more generally, that the three SNAREs are the core proteins of the nanomachine that mediates vesicle fusion to target membrane.
The BoNT proteolytic activity is highly specific and directed toward unique peptide bonds within the sequence of the respective SNARE substrates (summarized in Tables 1, 2, and 3) (Binz, 2013; Blasi et al., 1993a; Pantano and Montecucco, 2014; Rossetto et al., 2014). All BoNTs, except the BoNT/As, cleave large portions of the cytosolic domains of their respective substrates (Fig. 3), preventing the formation of a stable SNARE complex and consequently of exocytosis (Hayashi et al., 1994; Sutton et al., 1998). At variance, BoNT/As generate a truncated SNAP-25, which is still capable of forming a stable SNARE complex (Hayashi et al., 1994; Otto et al., 1995) and has a life time within the nerve comparable to that of intact SNAP-25 (Foran et al., 2003). Remarkable work showed that BoNT/A truncated SNAP-25 (SNAP-251–197) by itself inhibits exocytosis (Huang et al., 1998), and results of several experiments indicate that the BoNT/A cleavage of a 10–15% fraction of total SNAP-25 both within the NMJ (Raciborska et al., 1998) and spinal cord neurons (Keller and Neale, 2001; Keller et al., 2004; Montecucco et al., 2005) is sufficient to cause paralysis. These results led to the suggestion that SNAP-251–197 acts as a dominant negative factor in the function of a multimeric radial super-SNARE complex because the C-terminal segment is necessary for protein-protein interactions underpinning the formation of a radial SNARE super-complex (Montecucco et al., 2005; Pantano and Montecucco, 2014). Electron microscopy data indicate that multimeric SNARE supercomplexes exist in the CNS (Rickman et al., 2005), and indirect evidence for the existence of an octameric neuroexocytosis radial nanomachine have been obtained (Megighian et al., 2013). A variety of experimental approaches have been used to estimate the number of SNARE complexes forming the nanomachine that mediates vesicle-target membrane fusion and a range of figures have been produced (reviewed in Pantano and Montecucco, 2014). Recent improvement in single particle cryo electron microscopy (Cheng, 2015) may soon reveal the structure of the nanomachine that mediates neuroexocytosis, allowing one to understand the molecular consequences of the cleavage of SNAP-25 by BoNT/A and BoNT/C. However, at the present stage it cannot be excluded that SNAP-25 exists in different pools within the nerve terminal and that a subpool consisting of 10–15% of total SNAP-25 is the one involved in neuroexocytosis. It is also possible that the C-terminal segment of SNAP-25 inserts in the lipid bilayer playing an essential role in membrane fusion.
The specificity of L chains for the SNARE proteins relies on extended and multiple protein-protein interactions with their specific substrate, which include the cleavage site and exosites (Rossetto et al., 1994; Pellizzari et al., 1996; Breidenbach and Brunger, 2004; Jin et al., 2007; Agarwal et al., 2009; Brunger and Rummel, 2009). Such extended enzyme-substrate interaction results in highly specific recognition and explains why no additional protein substrates of the BoNTs have yet been found. The selectivity of BoNTs is well shown by two examples that are relevant to the evolutionary biology of animal botulism. The replacement of a Gln with a Val at the P1′ position of the VAMP cleavage site by TeNT makes rats and chicken resistant to tetanus (Patarnello et al., 1993). BoNT/D is the most potent toxin on mice (lethal dose < 0.1 ng/kg) but poorly toxic for humans (Eleopra et al., 2013) and rats because one of their VAMP1 exosites is not conserved (Peng et al., 2014).
Different events of vesicle trafficking are mediated by different VAMP isoforms, and this fact has been highlighted by the recent identification of the VAMP isoforms involved in evoked and asynchronous release (Kavalali, 2015). To favor the use of the BoNTs as a simple knockout of specific SNARE isoforms and study their involvement in neurophysiological events, Tables 1, 2, and 3 list the known mouse, rat, and human isoforms of the three SNARE proteins and predict their susceptibility to the 10 neurotoxins whose cleavage sites are known.
B. Duration of the Neuroparalysis Induced by Botulinum Neurotoxins
1. Reversibility of the Neuroparalysis Induced by Botulinum Neurotoxins.
Compared with other bacterial and plant toxins, which kill cells, a very remarkable aspect of the neuroparalysis caused by the BoNTs is its complete reversibility, i.e., a botulism patient survives even a major intoxication provided that respiration is mechanically supported and supportive care is provided (Johnson and Montecucco, 2008). There are several reports of botulism patients that have survived botulism after many months in an emergency room. This is due to the fact that BoNTs are neither cytotoxic nor they cause any axonal degeneration, although toxicity in vitro has been reported for BoNT/C and by the combined use of BoNTs targeting different SNAREs (Williamson and Neale, 1998; Berliocchi et al., 2005; Zhao et al., 2010; Peng et al., 2013; Arsenault et al., 2014). However, in evaluating the latter, it should be considered that: 1) BoNTs were used at concentrations much higher than those causing botulism, 2) isolated neurons in primary cultures are more sensitive to any form of stress than neurons in situ where they are involved in multiple structural and physiologic interactions with neighbor cells and extracellular matrix, 3) no neurotoxicity of any kind was detected in human volunteers injected with a “therapeutic dose” of BoNTs (Eleopra et al., 1997, 2002, 2004). More importantly, the very extensive and long-term experience with BoNT/A1 and BoNT/B1 as therapeutics has provided no indications of neuronal damage after repeated treatments extended over many years (Naumann and Jankovic, 2004; Naumann et al., 2006; Ramirez-Castaneda and Jankovic, 2014). Therefore, the presently available data provide no evidence for any neurodegeneratve action of the BoNT tested so far when used at concentrations sufficient to paralyze the NMJ. This does not exclude the possibility that cytotoxic toxins might be found among the large number of novel, yet untested, BoNTs.
The duration of the paralysis varies extensively depending on: 1) type of BoNT, 2) dose, 3) animal species, 4) mode of administration, and 5) type of nerve terminal. Since BoNT duration of action is of paramount importance because it determines the duration of hospitalization of botulism patients and the duration of the therapeutic effects, this aspect of BoNTs biology and pharmacology will be discussed in some detail below.
The order of duration of action in mice and humans is: BoNT/A ∼ BoNT/C > BoNT/B ∼ BoNT/D, BoNT/F, and /G > BoNT/E (Foran et al., 2003; Eleopra et al., 2004; Keller, 2006; Morbiato et al., 2007) with the exception of BoNT/D that is poorly active in humans but very potent in mice (Eleopra et al., 2013). The duration of action of BoNTs is about three times longer in humans than in mice (i.e., BoNT/A 3–4 months versus 1 month, respectively), and skeletal muscles recover about three times faster than autonomic cholinergic nerve terminals (in humans 3–4 months versus ∼ 1 year for BoNT/A1).
It has been estimated that as few as 1,000 molecules of BoNT are sufficient to inactivate nerve transmission in a muscle (Hanig and Lamanna, 1979), rendering the L chain half-life difficult to study in vivo with the currently available biochemical methods. As a consequence, this investigation has been tackled mainly in vitro, by transfecting L chains tagged with protein reporters (Fernandez-Salas et al., 2004a,b; Tsai et al., 2010) and mainly on BoNT/A1 and BoNT/E1 L chains, because they display respectively the longest and the shortest persistence in vivo. Within the limits of the methods used, it appears that BoNT/A1 L chain has a longer lifetime than that of BoNT/E1 because it escapes the action of the cell degradation systems (Tsai et al., 2010). In fact, BoNT/E L chain is ubiquitinated and targeted to the ubiquitin-proteasome system, whereas BoNT/A1 L chain appeared refractory to this degradation pathway. This resistance may come from the ability of the L chain of BoNT/A1 to recruit deubiquitinases, specialized enzymes that remove polyubiquitin chains, thus sparing proteins from ubiquitin-proteasome system degradation (Shoemaker and Oyler, 2013). Ubiquitination of BoNT/B1 was also reported and associated with a decreased proteolysis of VAMP (Shi et al., 2009). Other factors that may come into play are the localization of the BoNT/A L chain on the cytosolic face of the membrane, the presence of a di-leucine motif at its C terminus, and the recruitment of septins (Fernandez-Salas et al., 2004a,b; Wang et al., 2011; Vagin et al., 2014).
Recent evaluation of BoNT/As L chain persistence in cultures of neuronal cells indicate that it can remain active for over a year as detected by cleavage of SNAP-25 (Whitemarsh et al., 2014); the functionality of the ubiquitin proteasome system in these neurons was not tested. Such a long lifetime should not surprise, because it has been reported that purified BoNT/A1 remains unaltered for 4 years at room temperature (Frevert, 2009). These findings support the generally accepted view that duration of the paralysis induced by the BoNTs reflects the lifetime of the L chain of a BoNT inside the neuronal cytosol (Shoemaker and Oyler, 2013; Pantano and Montecucco, 2014). However, the situation is different in the cases of the BoNT/As, the serotype predominantly used in human therapy (see next section), and of BoNT/C. Using animal ex vivo preparations or primary cultures of spinal cord neurons no correlation was found among the amount of SNAP-25 cleaved by BoNT/A and the extent of neuroparalysis, i.e., the cleavage of 10–15% of SNAP-25 was found to be sufficient to cause a complete blockade of neurotransmitter release (Raciborska et al., 1998; Keller et al., 2004), and 2–3% cleavage was sufficient to silence miniature postsynaptic currents (Beske et al., 2015). On the contrary, a good correlation was found in the case of BoNT/E, which removes a large part of the C-terminal half of SNAP-25, whereas BoNT/A and BoNT/C removes only nine and eight residues from the C terminus, respectively (Fig. 3). As mentioned above, the BoNT/A truncated SNAP-25 (SNAP-251–197), but not the BoNT/E truncated SNAP-25 (fragment1–180), is still capable of forming the SNARE complex. Other seminal findings were that SNAP-251-197 inhibits insulin secretion from insulinoma cells as BoNT/A does (Huang et al., 1998) and that a single mutation within the C terminus of SNAP-25 was able to impair exocytosis in chromaffin cells (Criado et al., 1999). The simplest explanation of these results is that there are different pools of SNAP-25 within nerve terminals and that only a 10–15% of total SNAP-25 is actively involved in neurotransmitter release and is accessible to the proteolytic action of BoNT/A and BoNT/C. However, this rationale does not explain the inhibitory activity of SNAP-251–197, and the fact that all SNAP-25 molecules are accessible to BoNT/E. SNAP-251–197 may persist for long periods of times after generation by BoNT/A (Keller et al., 1999; Foran et al., 2003; Meunier et al., 2003). Taken together, these data are consistent with the suggestion that SNAP-251–197 may contribute significantly to the long duration of action of BoNT/A by acting as a dominant negative component of a neuroexocytosis nanomachine, consisting of several SNARE complexes arranged radially whose assembly/function is critically dependent on an intact SNAP-25 C-terminus (Megighian et al., 2013; Pantano and Montecucco, 2014). Also the long duration of action of BoNT/C in human and mice is likely to be due to an inhibitory action of a long-living SNAP-251–198 fragment.
2. Reversibility of Botulinum Neurotoxin Action in Vitro and in Vivo.
The comparison of the very long duration of neurotransmitter release inhibition induced by local BoNT/A1 and BoNT/B1 injection at the human autonomic cholinergic nerve terminals (even more than a year) (Naumann et al., 2013b), and of the similar one found for BoNT/A in stem cell-derived neurons in culture (Pellett et al., 2015c), with the shorter duration of action of BoNT/A1 in skeletal nerve terminals (3/4 months in humans), indicates that other factors come into play in vivo to determine duration and that at least part of these factors are external to the nerve terminal itself. Indeed it is long known that the BoNT poisoned NMJ undergoes a profound remodelling (Duchen, 1971). Novel nerve terminals sprout from their unmyelinated motor axon terminal and, to a lesser extent, from the first node of Ranvier (Juzans et al., 1996; Meunier et al., 2002). These nerve sprouts follow projections that emerge from perisynaptic Schwann cells, which multiply and migrate from the original NMJ to other sites of the sarcolemma soon after inactivation of the motor axon terminal. New contacts with muscle fibers are formed. Also the postsynaptic component undergoes critical changes with spreading of ACh receptors over the sarcolemma, without formation of the organized clusters characteristic of the mature NMJ (Sanes and Lichtman, 1999). The new synapses, although immature, can sustain vesicle recycling (de Paiva et al., 1999; Meunier et al., 2002), but are poorly efficient in ACh release (Rogozhin et al., 2008), providing a limited contribution to the recovery of the neurotransmission from nerve to the muscle fiber. Once a certain level of functionality is reestablished at the original site, terminal and nodal sprouts are pruned, and the newly formed synaptic specializations are eliminated (Johnson and Montecucco, 2008). Apparently, the neuroparalysis due to injection of BoNT/A followed by recovery of function can take place many times, one after the other, without loss of NMJ function, and this is at the basis of the repeated injections of this neurotoxins in human therapy (Borodic, 2007).
The duration of BoNTs activity has a paramount significance with respect to their therapeutic use and to the length of recovery of botulism patients in intensive care units and ensuing rehabilitation. On the therapeutic side, long lasting BoNTs require fewer injections and lower doses, limiting the possibility of immunization. On the other hand, a potent but short lasting BoNT could be desirable in some pathologies such as disjointed bone fractures, where a short duration might be more useful in ameliorating the course and outcome of the illness. Accordingly, there is a growing area of research that aims at changing binding specificity, affinity, and duration of BoNT action to obtain tailor-made therapeutic agents and more sophisticated tools to be used in cell biology studies (Chen and Barbieri, 2009; Ferrari et al., 2011; Masuyer et al., 2015; Sikorra et al., 2016).
III. Pharmacology
A. Introduction
The exquisite selectivity of BoNTs for nerve endings, their extreme potency in inhibiting neurotransmitter release, their limited diffusion from the site of injection, and their reversibility have been exploited for therapeutic purposes with great success (Dressler, 2012). The therapeutic potential of BoNT/A1 was unveiled in the 1970s, by pioneering studies performed by Scott and colleagues who treated strabismus in monkeys and then in humans by injecting the toxin into the orbicularis muscles (Scott et al., 1973; Scott, 1980). This innovative therapeutic indication paved the way to the therapeutic use of BoNT/A1 for a wide range of human disorders characterized by hyperactivity of nerve terminals of whatever origin. As soon as the use of this new drug was approved by the Food and Drug Administration (FDA) for the treatment of blepharospam in 1989, the range of therapeutic applications expanded and the process is still ongoing. Meanwhile, novel BoNT/A1 preparations were approved, and the use of BoNT/B1 for cervical dystonia was licensed by the FDA in 2000. The use of BoNT/A1 in cosmetics was approved for the first time by the FDA in 2002. There is also an increasing use of BoNT for the treatment of a variety of animal pathologies, including the rather common horse laminitis and dog prostate disorders (Chuang et al., 2006; Lin et al., 2007; Carter and Ben Renfroe, 2009; Hardeman et al., 2013). In addition, one can foresee that novel therapeutic BoNTs will be identified by analyzing the biologic properties of the large number of known BoNTs and of those that are being discovered.
BoNTs are very particular therapeutics endowed with unique properties among pharmaceutical drugs. They are “natural products”, purified from living bacteria of the genus Clostridium, but they are also “biopharmaceuticals”, i.e., exogenous proteins with a well-defined biologic activity. Actually, BoNTs are very special biopharmaceuticals, because they are the only ones exerting their pharmacological activity by modifying a single target protein within the cytosol of neurons. Intriguingly, this also makes them “lead compounds” whose engineering may result in derivatives or biologic platforms capable of expanding their current range of use (Band et al., 2010; Masuyer et al., 2014, 2015; Sikorra et al., 2016).
Another important aspect of BoNT/A1 and BoNT/B1 as therapeutics is that, regrettably, no information is available on their pharmacokinetics at the doses used for therapy. In fact, even the relatively high doses used in the treatment of hypercontracting large muscles correspond to very low amounts of protein (few nanograms), making pharmacokinetics studies very difficult. Moreover, BoNT/A1 and /B1 are injected locally, rapidly bind, and affect the injected muscle, but have a limited effect to nearby muscle around the site of injection. The unbound toxin is expected to be washed away and diluted in the lymphatic circulation, becoming incapable of binding elsewhere because of a decreased concentration and consequent lack of binding. These conclusions derive from studies in mice and rats using rather higher dosages (Carli et al., 2009; Simpson, 2013). Some further considerations are made in section IV.
Another unique feature of BoNTs is the impossibility of reversing their effects, i.e., once the toxin has reached the interior of neurons its activity cannot be reversed until it has naturally ceased. No drugs permeable to the neuronal plasma membrane and very effective in blocking their metalloproteolytic activity are as yet available (Rossetto et al., 2014; Pirazzini and Rossetto, 2017).
B. Present Botulinum Neurotoxin Formulations
Several BoNT preparations are licensed for clinical use, all of them are based on serotype A1, whereas only one is based on serotype B1 (Albanese, 2011). Three major branded products are commercially available worldwide and contain BoNT/A1 deriving from Clostridium botulinum strain Hall: onabotulinumtoxinA marketed as Botox/Vistabel by Allergan Inc. (Irvine, CA), abobotulinumtoxinA (ABO) marketed as Dysport/Azzalure by Ipsen (Paris, France), incobotulinumtoxinA (INCO) marketed as Xeomin/Bocouture by Merz Pharmaceutical Gmbh (Frankfurt, Germany); their characteristics are reported in Table 4. Three manufacturers are based in Korea and in China and produce three other BoNT/A brands marketed mainly in Asia. Medy-Tox (Korea) has the Meditoxin/Neuronox/Siax brands and Hugel Inc. (Korea) has a further type A BoNT dubbed Botulax (also Zentox or Regenox in other countries). Lanzhou Institute for Biologic Products (Lanzhou, China) produces a licensed BoNT/A1 available since 1997 termed BTXA, which is distributed by Hugh Source, in Hong Kong, and by other local/regional companies under different trade names. One BoNT/B1 (rimabotulinumtoxinB) product has been made available for more than a decade with the trade name Myobloc/Neurobloc. The product is presently marketed by US WorldMeds in USA and by Eisai Europe Limited in Europe (Pickett, 2014).
Comparison of botulinum neurotoxin products marketed in Europe and North America
Units are manufacturer specific and are not interchangeable.
Botox and Dysport are purified Precursor Toxin Complexes (PTCs), i.e., neurotoxin complexes including the BoNT/A1 molecule (referred to as active pharmaceutical ingredient by regulatory agencies), NTNHA, and the HA proteins (Fig. 2B); at variance, Xeomin contains only the purified BoNT/A1 (Fig. 1A) (Frevert, 2009, 2015; Pickett, 2014). All these commercial products contain human serum albumin (HSA) as an excipient to diminish toxin loss during lyophylization, to prevent protein aggregation and surface adsorption, to improve toxin stability and extend shelf life (Pickett, 2014). The toxin preparations are freeze dried (Dysport and Xeomin) or vacuum dried (Botox) and sold as lyophilized products to be reconstituted with sterile saline buffer, yielding a solution of slightly acidic, but variable, pH (Frevert, 2010; Dressler et al., 2016). Despite the different formulations, these three toxin products display the same limited diffusion from the site of injection (Carli et al., 2009), which is anyway dependent on the injected volume and influences the degree of paralysis in mice (Kutschenko et al., 2016). This indicates that the best pharmacological result is achieved when BoNTs reach all the nerve terminals innervating the target muscle(s) and that the amount of toxin needed for a certain application should be diluted according to the size of the muscle/area to be injected.
Table 4 also reports the characteristics of Myobloc/Neurobloc, which contains PTCs of BoNT/B1 (Callaway, 2004) produced by the C. botulinum strain Bean. It is marketed as a ready to inject solution of pH 5.6, containing 0.05% HSA. Myobloc/Neurobloc was approved by the FDA in 2000 for the treatment of patients who had become immune to BoNT/A1 (Brin et al., 1999), because the two serotypes are immunologically distinct (Atassi, 2004). However, larger doses of BoNT/B1 are required to achieve a comparable therapeutic effect (Botox/Neurobloc dose ratio 1:40–50), and its duration is shorter with respect to that of BoNT/A1 at skeletal muscles, but not at the autonomic nervous system where efficacy of the two neurotoxins is comparable with a dose ratio of 1:25–30/A1:B1 (Bentivoglio et al., 2015). As mentioned above (section II.A), recent data on receptor binding have shown that human synaptotagmin II does not have high affinity for BoNT/B1 owing to a single amino acid substitution in the toxin binding site (Strotmeier et al., 2012). This reduced affinity contributes to explain why high doses of BoNT/B1 are required to achieve therapeutic effects similar to those elicited by low doses of BoNT/A1.
Dose equivalence is a very important issue, especially considering the different BoNT/A1 formulations. Like for all biologic medicines, the potency of BoNT preparations is expressed as Units (U), where 1 U corresponds to 1 LD50 in the mouse bioassay (Schantz and Johnson, 1990; Sesardic, 2012). Although dosing is described in terms of units, there is evidence that the clinical effect of 1 unit is not interchangeable between formulations because of differences in the bioassay methodologies used by different producers (Rosales et al., 2006). These differences may be ascribed to the mouse strain used, the different housing conditions of the animals, different times of injection during the day (mice are nocturnal animals with an opposed light/dark cycle with respect to humans), the amounts of albumin and type and amounts of other excipients present in the various formulations. More importantly, diluent buffer was found to have a very significant effect on the biologic activity of therapeutic formulations of botulinum toxin, which are about twofold more potent in buffers containing bulking agent such as gelatin (Hambleton and Pickett, 1994; McLellan et al., 1996). Dysport and Botox are the most divergent, having nonparallel dose-response curves, and thus differing in relative potencies. The comparison of data in the clinical literature provides a dose conversion ratio between Botox and Dysport of 1:2.5–3 (Dashtipour and Pedouim, 2016; Scaglione, 2016), whereas Botox and Xeomin are reported as equivalent in potency in most cases (Frevert, 2015). A very recent study employing the wheel-running mouse model, calculated a dose conversion ratio between Botox and Dysport of 1:2 and between Xeomin and Botox of 1:0.75–1:0.5, (Kutschenko et al., 2016). However, it is not clear to what an extent these data are comparable to the mouse bioassay. Even less comparable are the toxin effects on the physiologic performance of the entire motor sytem with the clinical efficacy in mitigating the variety of humans hypercholinergic disorders, measured as patient satisfaction/relief. Accordingly, the results obtained with animal models should be considered with caution and not translated directly into clinical practice.
Currently, a large effort is ongoing to develop neurons in culture to test the potency of BoNTs and one manufacturer has obtained FDA approval for a cell-based potency assay as a substitute of the animal-based assay in the manufacturing of BOTOX (http://www.allergan.com/responsibility/animal-testing). Clearly, cultured neurons do not replicate the complexity of the intoxication pathway in vivo, but one may assume that their use provides information about the neuronal toxicity and allows one to determine the reproducibility of the BoNT production in terms of lot-to-lot manufacturing consistency. This reduces the number of animals to be used provided that one is able to define exactly the reproducibility over time of its cultured neuron test system. Clearly, using data generated by different laboratories using different products and different cell-based assay conditions (reagents, cells, media, etc.) will render it difficult to make meaningful comparison across products.
Although only BoNT serotypes A1 and B1 have reached clinical use, other serotypes have been tested in humans. BoNT/C was demonstrated to have a neuroparalytic profile very similar to that of BoNT/A1, with a similar duration of action on human skeletal muscles. BoNT/C represents a valid therapeutic alternative in patients treated with commercially available products who have developed an immune response with consequent secondary treatment failure (Eleopra et al., 1997, 2002, 2004, 2006).
A Japanese pilot study has compared the effect of a BoNT/A2 preparation with that of BoNT/A1 (onabotulinumtoxinA) on human volunteers by measuring the compound muscle action potentials of injected extensor digitorum brevis muscles. A comparable onset and duration of action among them was found without significant adverse effects (Mukai et al., 2014). Currently, a Phase 2 clinical trial performed in Japan is evaluating efficacy and safety of the use of BoNT/A2 for the treatment of poststroke lower limb spasticity (https://clinicaltrials.gov/show/NCT01910363) (Kaji et al., 2015).
C. Immunogenicity of Botulinum Neurotoxin Formulations
Botulinum neurotoxins and associated proteins present in commercial preparations may elicit antibody formation when injected into patients. Antibodies formed against the accessory proteins do not interfere with the biologic activity of the toxin, and they are therefore nonneutralizing antibodies, whereas antibodies formed against the neurotoxin (primarily against the heavy chain) may prevent or not prevent its biologic activity. Those antibodies that prevent it are neutralizing antibodies and are expected to interfere with the clinical efficacy of the product (Dolimbek et al., 2007; Atassi et al., 2012). However, all BoNT serotypes can be considered poor antigens, particularly with respect to the cousin molecule tetanus neurotoxin. Indeed, very few patients develop neutralizing antibodies, although BoNT therapy is often continued over many years. The immunologic human response to BoNT/A1 is very low, whereas it is higher for BoNT/B1 most likely because it is injected in much larger amounts. The reported frequency of anti-BoNT antibodies detection in injected patients ranged from 0 to 3% for BoNT/A1, depending on the formulation used, and from 10 to 44% for BoNT/B1 (Dressler and Rothwell, 2000; Dressler and Bigalke, 2005; Naumann et al., 2013a,2013b). Various factors can influence the immunogenicity of the botulinum neurotoxin therapeutics, including product-related factors like the manufacturing process, the toxin source, the antigenic protein load, and, perhaps more important, the presence of inactive or denatured toxin acting as a toxoid. Moreover, treatment-related factors such as the toxin dose, frequency of injections, and prior exposure via other routes can impact on the immunogenic response. Another important factor is the site of injection as anatomic regions rich in lymph nodes, such as the neck, are more likely to produce an immune response. In view of the potential risk for secondary treatment failure it is advisable, in clinical practice, to follow the general rules for minimizing the risk of developing an adaptive immune response, including the use of the lowest effective doses and employing the longest clinically acceptable interinjection interval (Naumann et al., 2013b). Although the recommended minimum intervals between BoNT/A1 treatment is 12 weeks, injection intervals as short as 6 weeks with Xeomin have been recently shown to be well tolerated and not associated with increased safety concern (Jost et al., 2015).
D. Clinical Applications of Botulinum Neurotoxins
After Scott’s seminal studies in ophtalmology, botulinum toxins entered the therapeutic arsenal of the neurologist, first with the purpose of relieving hypercontraction of small muscles of eyelid and of the face (blepharospasm and hemifacial spasm) and larger dystonic muscles of the head and neck (cervical and oromandibular dystonia) and later to treat limb movement disorders, including use-dependent cramps (occupational dystonia) and limb spasticity (Table 5). Because BoNTs not only block the skeletal cholinergic neuromuscular transmission but also the cholinergic autonomic innervation controlling sweat, tear, salivary glands, and smooth muscles and sphincters, hyperhidrotic disorders and urologic or gastrointestinal disorders also benefit from the use of botulinum toxins (Comella and Pullman, 2004; Dressler, 2012; Hallett et al., 2013; Naumann et al., 2013b; Ramirez-Castaneda and Jankovic, 2014; Simpson et al., 2016). The myorelaxant and atrophic effect associated with an ill-known analgesic effect of BoNT brings relief from pain and BoNT/A1 has been tentatively used in different pain syndromes (Table 5).
Therapeutic uses for botulinum neurotoxin
BoNT/A1-induced paralysis can be detected after 2–3 days from injection and reaches its maximum value within 1–2 weeks; it maintains this level for a variable period of time depending on the anatomic site and then gradually declines to return to the original value within 3–4 months for skeletal nerve terminals and about 1 year for autonomic ones.
The assessment of BoNT efficacy as therapeutics for a given pathologic condition is not an easy task and requires the comparison of many different clinical studies, often performed by different research teams in different clinical centers. Differences among individual patients and variables such as dosing, dilutions, number of injections per site, targeting [visual, electromyography (EMG), or ultrasound-guided] influence the outcome and decrease comparability of data among different centers. Consequently, many meta-analyses and quantitative study designs used to systematically assess previous research studies were recently performed on BoNT clinical applications to have a consolidated and quantitative review of a large and often complex, sometimes apparently conflicting, body of literature. Accordingly, we have reviewed rigorous meta-analyses on different human pathologic conditions treated with commercial BoNT preparations, and the results are reported here for the current therapeutic applications of BoNT/A1 and BoNT/B1.
1. Dystonias.
Dystonias are a heterogeneous group of disorders characterized by sustained involuntary muscle contractions, frequently causing repetitive twisting movements, abnormal postures, and pain (Albanese and Lalli, 2009, 2012). Localized injections of BoNT provide a transient symptomatic relief in primary and nonprimary focal dystonia syndromes, as demonstrated by several randomized controlled studies and by a large number of uncontrolled studies (Hallett et al., 2013; Guidubaldi et al., 2014).
In general, the therapeutic benefits of BoNT in dystonias derive from a decreased acetylcholine release from alpha motoneurons at the NMJ and the ensuing relaxation of the affected skeletal muscle. However, substantial evidence suggests that BoNT injected peripherally may also influence indirectly CNS function (Gracies, 2004). In fact, the BoNT inhibition of intrafusal muscle fibers by blocking also the gamma motoneurons, reduces muscle spindle afferent input to the CNS and may contribute to the therapeutic effects of BoNT in focal dystonias (Giladi, 1997; Hallett, 2000; Rosales and Dressler, 2010). Moreover there is considerable evidence that BoNTs, particularly at high doses, can reach higher structures in the brain. This is an area that deserves further investigation as a detailed understanding of direct central effects of BoNTs will provide valuable information for present and future therapeutic uses of BoNTs, particularly in the case of lower limb spasticity, which requires the use of high doses (Caleo and Schiavo, 2009; Mazzocchio and Caleo, 2015).
BoNT injection is considered the treatment of choice for most focal and segmental dystonias and in hemifacial spasm. The efficacy of BoNT for the treatment of these movement disorders, which include blepharospasm, cervical dystonia, hemifacial spasm, oromandibular dystonia, focal limb dystonias, and laryngeal dystonia has been recently reassessed by a thorough meta-analysis study, which included a total of 42 clinical trials (Hallett et al., 2013; Del Sorbo and Albanese, 2015).
Blepharospasm is a form of dystonia of the periocular muscles that produces forced eyelid closure, sometimes leading to functional blindness. FDA approved BoNT/A1 for the treatment of blepharospasm in 1989. Typically, 30–60 U of Botox or Xeomin, 60–180 U of Dysport or 1200–3600 U of Myobloc/Neurobloc are applied to the orbicularis oculi, procerus and corugator supercilii muscles bilaterally. Several studies demonstrated the efficacy and safety of BoNT therapy for this indication (Hallett et al., 2013). Response rates of around 90% make blepharospasm one of BoNT’s most successful indications. Adverse effects are usually mild and always transient and include local hematoma, ptosis, and diplopia (Dressler, 2012).
In cervical dystonia, also referred to as “spasmodic torticollis”, where dystonia of the neck and shoulder girdle muscles produces impairment of head control and pain, there is adequate evidence of BoNT efficacy, based on data from eight randomized, controlled, Class I clinical trials (Hallett et al., 2013). In clinical practice, the average total dose injected in patients with cervical dystonia is 100–300 U Botox or Xeomin, 400–800 U Dysport, or 10,000–20,000 U Myobloc/Neurobloc. These doses can vary considerably as the recommended range has to be adjusted depending on the individual patient’s features and sensitivity to the toxin. BoNT/A1 is the preferred treatment, whereas BoNT/B1 is recommended for A-resistant patients, because injections are generally more painful, possibly owing to the acidic pH of its solution, efficacy is shorter and immunogenicity higher (Bentivoglio et al., 2015).
Focal limb dystonia refers to dystonia affecting one arm or leg. Primary dystonia of the upper extremity commonly begins during selective, usually highly skilled, and repetitive motor tasks and are also referred as occupational or attitudinal dystonia. Typical upper limb dystonias include musician’s cramps and writer’s cramps where BoNT/A1 has been reported to be effective (Karp, 2004). However, due to the functional complexity, the usually high performance levels required and the narrow therapeutic window in the forearm muscles, functional outcome of treatments is sometime disappointing compared with those of blepharospasm or cervical dystonia. This also derives from the difficulty in obtaining the requested quality of voluntary movement without some weakness. BoNT application under electromyographic or ultrasound guidance is recommended as it improves the outcome.
The lower extremity is commonly affected in subjects with genetic dystonia (e.g., DYT-1 Dystonia), whereas upper limb and foot dystonia can be either idiopathic, in the context of a generalized dystonia, or symptomatic, as in Parkinson’s disease or in juvenile cerebral palsy. Successful treatments with BoNT have been reported but no controlled trials are available (Schneider et al., 2006). Higher doses of BoNT may need to be injected in the limb than in hand dystonia because motor control is less refined.
Hemifacial spasm describes synchronous unilateral contractions presumably caused by vascular compression of the facial nerve and is generally treated with lower doses than blepharospasm (Bentivoglio et al., 2009). Although experience with BoNT in hemifacial spasm treatment mainly originates from open-label trials, there is no doubt on its efficacy and safety (Guidubaldi et al., 2014). Notwithstanding the limited number of studies available, it appears that other nondystonic disorders including tics and tremors also benefit from BoNT therapy (Lotia and Jankovic, 2016).
2. Spasticity.
Spasticity describes the combination of a central paresis together with various forms of muscle hyperactivity, including dystonia, rigidity, and spasms often associated with pain. Most frequent etiologies include cerebral stroke, multiple sclerosis, traumatic brain injury, spinal cord injury, and infantile cerebral palsy. The goal of spasticity treatment is to reduce motor overactivity to improve movement without worsening weakness. BoNT treatment of spasticity can reduce muscle tone, improve function, facilitate nursing, and prevent contractures and decubitus. However, successful spasticity management requires a multiprofessional task force where all medical and surgical treatments need to be combined with physical interventions. Rehabilitation treatment of poststroke patients (i.e., muscle stretching exercises, eventually combined with neuromuscular electrical stimulation), when begun immediately after injection enhances the efficacy of BoNT therapy (Ward, 2002). However, recent systematic reviews have concluded that BoNT/A1 injections have to be considered as the pharmacological treatment of choice in focal spasticity to improve limb position, functional ability and to reduce pain (Esquenazi et al., 2013; Simpson et al., 2016), whereas limited data are available on the efficacy of BoNT/B1 in spasticity. BoNT therapeutic effects are better established for spasticity in the upper rather than lower limb (Esquenazi et al., 2013). It usually involves large BoNT doses: typically 300–500 U Botox or Xeomin or 600–1000 U Dysport for arm or leg spasticity. Recent studies reported treatment of poststroke patients with up to 800 U of Botox, 1800 Dysport, and up to 1200 U of Xeomin without signs of systemic toxicity (Dressler et al., 2015; Santamato et al., 2015). A higher dilution results in larger injection volumes and higher degree of paralysis (Kutschenko et al., 2016), probably because of larger spreading to neuromuscular junctions remote from injection site (Gracies et al., 2009).
3. Autonomic Disorders.
Botulinum neurotoxins inhibit neuroexocytosis from cholinergic nerve terminals of the sympathetic and parasympathetic autonomic nervous systems. The use of BoNT within the broad category of autonomic indications includes hypersecretory disorders, such as hyperhidrosis, sialorrhea, and chronic rhinorrhea, and smooth muscle hyperactivities in urologic and gastrointestinal disorders. Owing to an apparently higher affinity for cholinergic autonomic nerve endings, BoNT/B1 is as efficacious as BoNT/A1 in the therapy of secretory disorders both in terms of extent of inhibition and duration of the beneficial effects and at lower relative doses than those required in skeletal muscles (Bentivoglio et al., 2015).
Hyperhidrosis is medically benign but may be a socially devastating condition characterized as it is by excessive sweating, which may be occurring focally within the axillary region or it may extend to palms and soles. A formalized assessment of the American Academy of Neurology confirmed that BoNT therapy is safe and effective for axillary hyperhidrosis (Naumann et al., 2013b); total doses per axilla of 50–100 U Botox or Xeomin, 100–200 U Dysport or 2500 U NeuroBloc/Myobloc may be used (Bentivoglio et al., 2015).
Hypersalivation (or sialorrhea) refers to the presence of excessive saliva in the mouth, which may cause drooling with consequent severe embarrassment for the affected people. In several cases hypersalivation is secondary to other pathologic conditions including Parkinsonian syndromes, motoneuron diseases (amyotrophic lateral sclerosis) and cerebral palsy and is caused by impaired swallowing of saliva. The total therapeutic dose injected into the parotid glands for sialorrhea is tipically 2500 U for NeuroBloc/Myobloc, whereas BoNT/A1 ranges from 50 U for Botox to 75–450 U for Dysport (Naumann et al., 2013b).
Allergic rhinitis is a common disorder, one that is not disabling by itself, but it imposes a substantial burden in medical costs and indirect costs due to loss of productivity (Simoens and Laekeman, 2009). BoNT/A1 may be a good alternative for the treatment of rhinitis patients who are unresponsive to other treatment methods. Some restrictions such as painful injections and needing of specialized experience for adminitration prevent its wide use (Ozcan and Ismi 2016). BoNT for the treatment of allergic rhinitis is supported by a level B recommendation (probably effective) (Naumann et al., 2013b).
4. Urologic Pathologic Conditions.
The therapeutic applications of BoNT in urology include detrusor sphincter dyssynergia, lower urinary tract symptoms due to benign prostatic hyperplasia, and detrusor overactivity (both neurogenic and idiopathic). These conditions are common among patients with spinal cord lesions of different types (multiple sclerosis, spinal cord tumors, other spinal cord diseases, traumatic spinal cord injuries). The mechanism(s) underlying the therapeutic actions of BoNT in urologic disorders is not fully understood but a complex inhibitory action on receptors, neuropeptides, and neurotransmitters involved in the pathophysiology of the overactive bladder has been proposed (Chancellor et al., 2008). In the bladder, BoNT is thought to act primarily by inhibiting acetylcholine release from parasympathetic nerve endings inducing detrusor muscle relaxation (Schurch et al., 2000; Fowler et al., 2008). In addition, basic and clinical evidence suggests that BoNT may have sensory inhibitory effects unrelated to its actions on acetylcholine release (Apostolidis et al., 2005). Indeed, BoNT/A1 reduces the release of glutamate, of substance P, and of calcitonin gene-related peptide from the peripheral terminals of afferent bladder neurons (Duggan et al., 2002; Rapp et al., 2006) and of neurotrophins from the urothelium (Chancellor et al., 2008). The BoNT inhibition of secretion of these neuropeptides and ATP as well as cyclooxygenase-2 products, which are inflammatory response mediators released from nociceptive sensory endings in response to noxious stimuli (Khera et al., 2004; Chuang et al., 2008), would explain the therapeutic benefit of BoNT injection in painful bladder conditions (Smith et al., 2004; Wang et al., 2016).
BoNT/A1 injection for overactive bladder treatment is also associated with a significant decrease of purinergic receptors P2X3 and capsaicin receptors TRPV1, two receptors involved in nociception in the suburothelial nerve fibers (Apostolidis et al., 2005; Liu et al., 2014). These changes may reflect a direct activity of BoNT on the afferent innervation of the bladder and/or a secondary effect on the efferent innervation of the detrusor. Moreover, evidence of BoNT/A retrograde transport to the CNS after bladder injection in rats has been recently provided (Papagiannopoulou et al., 2016). Further studies are needed to elucidate this transport and its significance with respect to possible changes induced by BoNTA1 in the CNS after bladder injection.
Most studies have reported injection doses of 100–300 U of Botox, with 100 U being a typical starting dose for idiopathic detrusor overactivity and higher doses for those patients with insufficient response to an initial injection or those with neurogenic detrusor overactivity (Gomelsky and Dmochowski, 2014; Cheng et al., 2016). The injected volume is a key factor in the spread and action of BoNT/A in the bladder, with a wider diffusion of the toxin when higher volumes are injected (Coelho et al., 2012). In an experimental animal model of severe bladder pain, intrathecal injection of Botox has a strong analgesic effect, and this route of administration should be further explored for “intractable” forms of pain (Coelho et al., 2014).
5. Pain.
Despite current availability of a large number of analgesic drugs, management of chronic pain is still a challenge for clinicians. Over the past decade, BoNT treatment has shown efficacy in a large spectrum of human pain disorders, and the potentially relevant use of BoNT/A1 has been reported as efficacious in some form of chronic pain refractory to other treatments. The established ability to produce neurotoxins by recombinant protein expression (Bade et al., 2004; Band et al., 2010; Masuyer et al., 2014, 2015) and to modify them to create engineered neurotoxins with enhanced specificity for nociceptive nerve terminals will expand the therapeutic utility of BoNT for pain treatment. In this context, an innovative class of biopharmaceuticals obtained by replacing the native binding domain with another protein to redirect the light chain to a different nerve or cell has been recently proposed (Chaddock et al., 2004; Pickett, 2010; Masuyer et al., 2014).
The analgesic effects of BoNT/A1 were first reported in 1985 in a pilot study of BoNT/A treatment of cervical dystonia, characterized by abnormal, involuntary neck and shoulder muscle contractions and often resulting in significant, disabling musculoskeletal pain (Tsui et al., 1985). Early in its use as a therapeutic agent, BoNT/A1 was observed to provide pain relief in spastic and nonspastic muscle conditions in humans such as myofacial pain syndromes, temporomandibular disorder and bruxism, low back pain, prostatic pain, tension type and migraine headache, different types of neurophatic pain syndromes and many others (Wheeler and Smith, 2013). However, no effect was reported in a model of pain perception in human skin (Schulte-Mattler et al., 2007).
The association between BoNT/A1 and pain relief was originally thought to correlate only to its effect on muscle overcontraction or contractures. However, it is particularly interesting that the analgesia provided by BoNT injection occurs before muscle paralysis and outlasts any muscle weakness. It is now well documented that the analgesic effects of BoNT/A1 are related not only to its paralytic effect, but also to an effect on the nociceptor system (Wheeler and Smith, 2013). The widely reported antinociceptive effect of BoNT/A1 would be primarily mediated by the blockade of neuropeptides and inflammatory mediators release, and by the inhibition of plasma membrane exposure of pain sensors at peripheral level (as described previously for bladder pain). Indeed, in cultured sensory neurons, the TNF-α induced surface trafficking of TRPV1 and TRPA1 channels is mediated by SNAP25, VAMP-1 and syntaxin-1 and is inhibited by the serotypes of BoNTs that selectively cleave their respective SNAREs (Meng et al., 2014, 2016). This BoNT/A1 inhibitory effect would reduce peripheral sensitization and afferent input to the spinal cord, thereby dampening indirectly central sensitization. However, a growing number of experimental and clinical data on BoNT/A1 antinociceptive action cannot be explained adequately only by the inhibition of the release of peripheral neurotransmitter/inflammatory mediators. It is envisaged that the central effect may be directly involved, via retro and anterograde axonal transport along the branches of nociceptive neurons (Matak and Lackovic, 2014; Luvisetto et al., 2015; Mazzocchio and Caleo, 2015; Pellett et al., 2015d). Several studies reported toxin bilateral actions in distant regions after unilateral injection of BoNT/A1, suggesting that peripherally administered BoNT/A1 reaches the CNS region via axonal transport to target neurotransmission of pain sensory circuits. Enzymatic activity of BoNT/A1 has been immunohistochemically detected in spinal cord or brain stem areas, to which sensory and motoneurons in BoNT/A1-injected peripheral regions are projected (Matak and Lackovic, 2014; Luvisetto et al., 2015). Based on these and other evidence it was suggested that the BoNT/A action on pain is dominantly a central effect (Matak and Lackovic, 2014). Moreover, recently, in a rat model of neuropathic pain, BoNT/A was shown to influence microglial activation and to restore neuroimmune balance by reducing the levels of pronociceptive factors (IL-1 β and IL-18). In addition, it appears to increase the level of antinociceptive factors (IL-10 and IL-1RA) in the spinal cord and in the dorsal root ganglia (Zychowska et al., 2016). These findings suggest that neuroimmunological changes are also involved in BoNT/A-mediated analgesia.
Within the pain medicine field, only chronic migraine is an approved FDA indication for BoNT/A1. All other areas are currently considered off label, although for several of them, the available literature already strongly suggests efficacy. Concerning the evidence-based efficacy, the lack of standardized guidelines for BoNT application and dosage for pain management, and the lack of appropriate definition of study primary outcomes, in addition to the small sample size and limited number of randomized controlled clinical trials, have led to contradictory or negative findings in some forms of pain (Jabbari and Machado, 2011). Only some examples of chronic pain conditions for which prospective and controlled data are available on BoNT efficacy are presented in this paragraph.
a. Neuropathic pain.
This is one of the most common forms of human pain, which derives from damage or dysfunction of the peripheral or central nervous systems; in some cases it may result from nociceptive injury (Argoff, 2002). Failure of response to current analgesic medications is not uncommon. The data on type A1 toxin (mostly Botox has been used in these studies) are encouraging and indicate efficacy or probable efficacy in three major and common forms of neuropathic pain, namely, postherpetic neuralgia, posttraumatic neuralgia, and painful diabetic neuropathy. For example, in one study, to reduce the tension of the tendons passing through the tarsal tunnel that were causing pain, 120 U per muscle (dilution 200 U/ml) were injected under electromyographic guidance into all the muscles involved. The treatment was repeated after 3 months and resulted in a defined reduction of the number of dysethesias with a clear gait improvement associated to the BoNT/A injections (Buonocore et al., 2017). In addition, a recent systematic review of the therapeutic value of BoNT/A subcutaneous or intradermal injection for the therapy of trigeminal neuralgia concluded that this treatment is to be considered as an alternative option to the surgery (Morra et al., 2016). Much remains to be learned about the most effective technique of injection, most effective dose, optimal dilutions, and differences among different BoNTs in the treatment of neuropathic pain.
b. Primary headaches.
These diseases are categorized in migraine or tension headaches. Migraine is a disabling disease for the affected patients and it is differentiated in episodic (<15 headaches per month) or chronic (≥15 headaches per month). Chronic migraine is the only pain disorder with approved BoNT/A1 use (Chen, 2012), notwithstanding the fact that the mechanism of action is still not completely elucidated (Luvisetto et al., 2015). Migraine is thought to be caused by brain stem neuronal hyperexcitability, cortical spreading depression, abnormal release of neurotransmitters/neuropeptides, and trigeminal system activation (Pietrobon and Moskowitz, 2013). Other data support the role of peripheral activation via the sensory nerves surrounding blood vessels of the head and neck (Olesen et al., 2009) and the role of vasodilatation of the extracranial arteries (Shevel, 2011).
An extensive meta-analysis involving 23 studies and more than 5000 patients has analyzed the BoNT/A1 efficacy in episodic migraine, chronic migraine, chronic daily headache and tension type headache (Jackson et al., 2012). It was concluded that BoNT/A1 may reduce the mean number of headache days per month in chronic migraine and chronic daily headache, in comparison with placebo. BoNT/A1 was not beneficial in the treatment of episodic migraine or tension-type headache (Simpson et al., 2016).
6. Other Applications.
The therapeutic possibilities of BoNTs are manifold and certainly not yet exhausted. Controlled studies for many indications are frequently lacking because of the limited incidence of some disorders and symptoms.
a. Gastroenterology and proctologic disorders.
BoNT in the treatment of spastic gastrointestinal disorders has gained widespread acceptance over the last 15 years, especially for achalasia and chronic anal fissures. Its administration is generally safe and relatively noninvasive compared with alternative therapies (Brisinda et al., 2015).
Achalasia is an idiopathic esophageal motility disorder characterized by incomplete relaxation of the smooth muscles of the lower esophageal sphincter (LES) with swallowing and aperistalsis in the esophageal body. Because the pathologic hallmark of achalasia is a loss of nitric oxide-releasing inhibitory neurons in the esophageal myenteric plexus resulting in unopposed excitatory activity, BoNT injection counterbalances the increased muscle contraction by decreasing LES pressure, and hence enables passive esophageal emptying and improve the symptoms (dysphagia, regurgitation, and chest pain) (Pasricha et al., 1995; Lake and Wong, 2006). BoNT injection into all four LES quadrant (80–100 U Botox or 240–400 U Dysport in total) is the most common initial endoscopic therapy for achalasia in the United States, owing to its excellent safety profile, high initial response rate, and ease of administration. However, BoNT injection does not compare favorably in terms of duration of action with invasive treatments such as pneumatic dilation or laparoscopic Hellers myotomy, which are successful in 80% of patients; moreover, repeated treatments were shown to make subsequent Heller myotomy more challenging (Zaninotto et al., 2004; Smith et al., 2006). Thus, BoNT/A1 injection has not been recommended as a first-line therapy for achalasia, although it has an important role to play in elderly patients and in patients with comorbidities who are not candidates for definitive therapy (Ramzan and Nassri, 2013; Pandolfino and Gawron, 2015).
Chronic anal fissure is an ischemic ulceration related to sphincter hypertonia. Lateral internal sphincterotomy, the most common treatment of chronic anal fissure, may cause permanent injury to anal sphincter, leading to fecal incontinence. Extensive studies begun long ago (Gui et al., 1994; Maria et al., 1998) have led to BoNT injection becoming the standard treatment. BoNT is more than a symptomatic treatment of anal fissure as it produces a reversible sphincterotomy reducing the sphincter pressure and, in addition, it influences the nitric oxide synthase and other agents, thus interfering with the pathogenic mechanisms of the chronic fissure. The exact site and dose of injection remains somewhat unsettled with most studies injecting the internal anal sphincter and others injecting the external anal sphincter laterally to the fissure with 2.5–10 U Botox or 10–40 U Dysport (Jost, 2006; Brisinda et al., 2015).
b. Depression.
Depression is a psychiatric condition affecting millions of people worldwide and it can be treated with many psychotropic substances, which cause side effects, are difficult to be standardized and whose therapeutic outcomes are often unsatisfactory (Gilmer et al., 2005; Rush et al., 2006; Helton and Lohoff, 2015). A single injection of BoNT/A1 in the glabellar frown lines was reported to improve symptoms in ten patients suffering of depressive disturbs (Finzi and Wasserman, 2006). Thereafter, three main randomized control trials were reported; the toxin was administered as monotherapy or as adjunctive treatment and patients were evaluated after 3–6 weeks (Wollmer et al., 2012; Finzi and Rosenthal, 2014; Magid et al., 2014). Results corroborated the initial evidence of a beneficial effect of BoNT/A1 for the treatment of depression after injection in the glabellar frown muscles of the forehead (Magid et al., 2015). Mood improvement in BoNT/A1-treated patients is explained in the facial feedback hypothesis as linked to the emotional perception to facial expression (e.g., smiling led to happiness and frowning led to sadness). In other words, the neuroparalytic effect of the toxin may improve the proprioceptive perception, neutralizing the sadness by neutralizing frowning (Magid et al., 2014, 2015). On the other hand, reduction of depressive symptoms was reported to continue after BoNT/A1 cosmetic effect was ceased, entailing that the antidepressive effect may not be totally dependent on toxin paralytic action (Magid et al., 2014). It is therefore tempting to speculate a possible direct effect of the toxin on central circuits. Intriguingly, functional magnetic resonance imaging showed that BoNT/A1 injection in frown muscles leads to a functional uncoupling of brain stem centers with the amygdala, resulting in a reduction of neuronal activity within the central circuitries of emotion (Hennenlotter et al., 2009; Kim et al., 2014). Although this application field is only at the beginning, and further studies are necessary to validate BoNTs efficacy in the treatment of depression, these initial results are encouraging, particularly considering that they are based on the readaptation of the well-established and safe procedure of BoNT injection for cosmetic purposes.
7. Cosmetic Uses.
The present major use of BoNT/A1 in humans is related to a variety of cosmetic indications (Carruthers et al., 2016). Recently published statistics from the International Society of Aesthetic Plastic Surgery show that BoNT/A1 injection is the most popular of all cosmetic procedures worldwide (Frevert, 2015). Current estimates suggest that approximately half of the medical production of BoNT/A1 is used in aesthetic medicine. The serendipitous observation that BoNT/A1 smoothed facial lines when used therapeutically (Carruthers and Carruthers, 1992) led to study the toxin effect on glabellar lines, which are perceived as a sign of aging and of negative emotions. The repetitive contraction and activity of the procerus and the corrugator supercilii muscles involved in facial expression is mainly responsible for glabellar lines and can be attenuated by BoNT/A1 injection into three to five sites of these muscles (Gendler and Nagler, 2015). A recent meta-analysis showed that a 20 U total dose of BoNT/A1 is remarkably effective and safe for the treatment of glabellar lines (Guo et al., 2015). BoNT/A1 formulations have been FDA approved for glabellar lines (Vistabel, Allergan; Azzalure, Dysport, and Bocouture, Merz), and very recently Bocouture in Europe has been approved also for the treatment of horizontal frown lines and lateral periorbital lines. However, BoNT/A1 can be used to reduce all wrinkles in all skin areas of the face and the neck as long as they are based on increased muscle tone. Best results are obtained when BoNT is combined with filler substances to correct nondynamic aspects of the skin lines (Dressler, 2012). Repeated injections have not been reported to cause permanent damages to the superficial muscles involved and this is in line with the findings obtained in the treatment of dystonic muscles (please see next paragraph).
E. Adverse Effects
A key feature of BoNT/A1 is a minimal effect even at short distances from the site of injection, a property that ensures optimal results in most of the clinical applications. However, the possibility of local diffusion or potential leakage into the systemic circulation exists and depends on a number of factors including volume and speed of injection, dose, and site of injection. The paralytic effect outside the target muscle is due to local diffusion and is generally limited to adjacent muscles, which, for example, could cause ptosis, heavy brow, or a frozen face in facial aesthetics (de Almeida and De Boulle, 2007). However, it should be considered that the diffusion of BoNT from the site of injection is accompained by its progressive dilution in larger and larger volumes of extracellular fluids. Accordingly, the injected toxin becomes so diluted as to be no longer capable of binding to the presynaptic membrane.
A systematic review and meta-analysis of randomized clinical trials did not find reports of serious side effects due to BoNT treatment and indicated that the overall rate of adverse events, from mild to moderate, was 25% in BoNT/A-treated subjects, compared with 15% in the placebo group (Naumann and Jankovic, 2004). Autonomic side effects are rare and occur far more often after BoNT/B1 than after BoNT/A1 treatment, probably owing to a higher affinity of BoNT/B1 for cholinergic autonomic nerve endings (Dressler and Benecke, 2004). Indeed the mutation on human synaptotagmin II that decreases its affinity for BoNT/B is not present on human synaptotagmin I (Strotmeier et al., 2012), which is the isoform mainly expressed in peripheral autonomic and sensory innervations but not in motor endplates (Li et al., 1994).
Importantly, the long and very extensive use of BoNT therapy indicates that it is remarkably safe when administered by authorized medical personell, using licensed toxin preparations. In contrast, the handling of BoNTs by unskilled/nonauthorized persons or the use of counterfeit or unapproved agents that have permeated the market worldwide, or their combination, may place patients at risk for potentially devastating consequences. In fact, four cases of iatrogenic botulism have been described following cosmetic injection with high dose of an unlicensed BoNT/A1 preparation (Chertow et al., 2006).
F. Contraindications and Drug Interactions
Contraindications to BoNT therapy are limited to a hypersensitivity to the toxin or an infection at the site of injection, but adequate hypersensitivity tests have not been yet developed. An increased monitoring is recommended in patients treated simultaneously with aminoglycosides, anticholinergics, or other neuromuscular-blocking agents. Due to higher affinity for autonomic nerve terminals, BoNT-B should be applied carefully in patients with pre-existent autonomic dysfunction, additional anticholinergic treatment, and in conditions where anticholinergics are contraindicated (Dressler and Benecke, 2004).
G. Botulinum Neurotoxin in Pregnancy
There is no definitive evidence that BoNT-A treatment during pregnancy carries a risk. There are growing numbers of reports of the outcome of pregnancy following botulism or after therapeutic injection that have not led to fetal harm (Pearce, 2014). However, in absence of a long-term follow up, BoNT is placed in pregnancy risk category C and the FDA advocates that it should be “administered during pregnancy only if the potential benefit justifies the potential risk to the fetus.”
IV. Toxicity of Botulinum Neurotoxins
The toxicity of the botulinum neurotoxins is the highest among presenty known toxic agents (Gill, 1982; Middlebrook and Dorland, 1984). This is due to 1) their neurospecificity, 2) the enzymatic nature of the N-terminal domain whose activity can inactivate one molecule of substrate after the other, and c) the essential role of their proteolytic substrate, i.e., the SNARE proteins that mediate the fundamental physiologic function of neurotransmitter release at peripheral nerve terminals. In this respect, it is very remarkable that BoNTs are very safe pharmacologic and cosmetic agents used in million and million of doses each year worlwide. Indeed, the therapeutic use of BoNTs is characterized by a very positive record of efficacy and safety when the BoNT is administered by trained personnel (Lew et al., 1997; Brin et al., 1999; Simpson et al., 2008; Dressler et al., 2013; Attal et al., 2016; Dashtipour and Pedouim, 2016).
The other face of the coin is the possible use of BoNTs in bioterrorism (Arnon et al., 2001; Villar et al., 2006), although the actual efficacy of BoNTs as bioterrorist weapons appears to be very limited, and, in any case, much lower than that of some viruses or of Bacillus anthracis spores.
Toxicology of BoNTs is presently a complicated issue to review, owing to several limiting factors. The number of BoNTs is continuously growing and only the amino acid sequence is known for most novel BoNTs through genomic databases. Toxicology data are available only for a limited number of these BoNTs and only for few vertebrate species. A further complication is the fact that available toxicological studies have been performed with BoNTs preparations of different purity and storage conditions; in addition, the BoNTs can be produced as recombinant proteins in Escherichia coli (Bade et al., 2004; Band et al., 2010; Pirazzini et al., 2013b; Sikorra et al., 2016). Moreover, different strains of the same animal species may be polymorphic with respect to the SNARE protein isoforms (Peng et al., 2014) and for the glycosylation of BoNT receptor proteins. Additionally, it is long known that the high dilutions that are generally necessary when handling toxins of such a high potency may influence the outcome of toxicity assays. This is overcome by adding small amounts of gelatin or purified albumin or colloids, which were found long ago to increase stability and reproducibility of pure botulinum and tetanus neurotoxins toxicity assays after dilution for reasons that are not fully clear (Wentzel et al., 1950; Wright, 1955; Hambleton and Pickett, 1994; McLellan et al., 1996). Possibly, these so called “carriers” prevent self-aggregation or attachement of the diluted proteins to plastics or glass. In fact all commercial preparations of BoNT/A1 and BoNT/B1 contain human albumin (Pickett, 2014).
Another major aspect that should be considered is that the large majority of botulism cases occur among animals in the wilderness, particularly among fish and birds (Eklund and Poysky, 1974; Smith and Sugiyama, 1988; Montecucco and Rasotto, 2015). Even a minor dose of BoNT that is not enough to kill the caged animal is sufficient to cause initial symptoms, such as diplopia, that will lead eventually to death because of an unsustainable loss of the physical performance required to survive in the wild.
With these limitations in mind, we have attempted to condensate the present knowledge in Table 6, which reports data of toxicity of different BoNTs in Mus musculus, taken as reference animal, although different mouse strains were used across studies. Moreover, it should be noted that lethality assessed in the laboratory may be influenced by the specific conditions used, including cage density, time of injection during the day, diet, that are not usually reported. Table 6 gives the mouse lethal dose 50 values (MLD50), i.e., the dose that causes 50% death in caged mice within 4 days, as determined by intraperitoneal injection. The toxicity of BoNT/A1 in humans is not known but an estimation can be made by extrapolating the toxicity data obtained in monkeys, whose intramuscular LD50 is reported to be 39 U/kg (Scott and Suzuki, 1988) and the intravenous LD50 is 40 U/kg (Herrero et al., 1967), with U corresponding to one mouse LD50. Considering 6 kg the median weight of a Rhesus monkey and 70 kg that of a human, the intramuscular LD50 would be 234 U and 2730 U in Rhesus monkeys and in humans, respectively. In the case of BoNT/F1, subcutaneous lethal dose in Rhesus monkey was estimated to be 25 U/kg and 4 × 106 U/kg for the oral route of administration (Dolman and Murakami, 1961).
Toxicity of BoNTs in mice upon i.p. injection expressed as LD50 (ng/kg)
The available data on the intravenous and intraperitoneal routes of BoNTs injection are comparable in terms of general toxicity, whereas the intramuscular route of injection in mice is less effective (Aoki, 2001). It was estimated that in less than 20 minutes, part of the injected toxin has already bound and internalized within local neurons in such a way that it cannot be neutralized any longer by antitoxin antibodies (Simpson, 2013). However, very few pharmacokinetic studies on distribution, metabolism, and excretion of BoNTs have been performed and all of them employed multilethal doses of toxin (Simpson, 2013). It is therefore not clear to what an extent they can be valid for the case of local injection of minimal doses of toxin as it occurs when BoNT is used as a drug. On the other hand, as mentioned above (section III.A), no information is available on the pharmacokinetic of BoNT products used clinically, for reasons mostly related to the very low therapeutic doses that are below detection in circulating fluids.
BoNTs toxicity is lower via the oral route of delivery. In fact, BoNTs have to pass through acidic and protease-rich anatomic regions before being absorbed in the first portion of the intestinal tract; this lowers considerably the number of intact BoNT molecules that reach the lymphatic and blood circulations. Consequently, the oral/intraperitoneal toxicity ratio varies considerably for the different toxin serotypes and for the animal species taken into account. As an example, humans seem to be refractory to BoNT/C envenomation through the oral route, because basically no records but one (Oguma et al., 1990) of BoNT/C-botulism are reported, whereas they are higly sensitive to the toxin when injected intramuscularly (Eleopra et al., 1997, 2002, 2006). Moreover, the type of toxin preparation, i.e., toxin alone, or in complex with NTNHA or as PTC, strongly influences the toxicity of the oral route. In fact, the structure of the BoNT/NTNHA dimer, shown in Fig. 2A, clearly indicates that NTNHA protects a large part of the BoNT surface from proteases and the attack of chemically reactive molecules (Gu et al., 2012; Eswaramoorthy et al., 2015; Lam and Jin, 2015). Accessory proteins are commonly believed to protect BoNTs, but their most important role in toxicity appears to be that of mediating the binding of PTC to the intestinal mucus and to the polarized epithelial monolayer (Ohishi et al., 1977; Ohishi and Sakaguchi, 1980; Couesnon et al., 2012; Amatsu et al., 2013; Fujinaga et al., 2013; Lee et al., 2013; Lam and Jin, 2015; Lee et al., 2015; Rossetto et al., 2014; Rummel, 2015). The accessory proteins do not influence BoNT toxicity via the intraperitoneal, intravenous, and intramuscular routes, because PTC rapidly dissociates at the neutral pH of circulating fluids (Eisele et al., 2011; Frevert, 2015); therefore, they do not contribute to the therapeutic/cosmetic effect of the different BoNT preparations commercially available.
The inhalational route has not caused botulism, except from one laboratory case (Holzer, 1962; Park and Simpson, 2003), and is considered only with respect to a possible use of BoNT in bioterrorist attack. In Rhesus macaques, the ratio of toxicity intraperitoneal/inhalation was estimated to be around 11 for BoNT/A1 (Sanford et al., 2010). The situation appears to be similar in humans, where the lethal dose was estimated to be between 10 and 13 ng/kg for inhaled BoNT/A1 (Arnon et al., 2001).
A frequently used toxicity assay is the one introduced by Boroff and Fleck (1966), which measures the time to death of mice injected in the tail vein with a very high dose of BoNT (103–105 MLD50). The animal dies within dozens of minutes from injection. It should be noted that no post mortem pathologic examinations were reported by Boroff and Fleck (1966). This assay is very pratical, because it diminishes the experimental time, but it provides figures that are only partially comparable with the mouse lethal dose values. In fact, at the very high doses of toxin used, BoNTs are likely to bind to noncholinergic nerve terminals and to other cells, i.e., the chromaffin cells of the surrenal glands or neurons of the dorsal ganglia. These cells are very sensitive in vitro to the low nanomolar concentrations of BoNTs commonly used. Apparently, these cells exhibit lower affinity for BoNT receptors with respect to cholinergic nerve terminals implicated in botulism and therefore do not contribute to death in botulism. However, additional bindings and interactions of BoNTs with chromaffin cells, ganglion cells, and others cells such as heart sympathetic and parasympathetic neurons (Lamanna et al., 1988) are likely to contribute to the very early death in the Boroff and Fleck assay because of the extremely high does used.
In recent years, assays in vitro using primary neuronal cultures or neurons induced from pluripotent stem cells or cell lines derived from tumors have been used to test the activity of BoNTs (Pellett et al., 2007; McNutt et al., 2011; Whitemarsh et al., 2012; Beske et al., 2015). Clearly the complexity of the in vivo situation is not recapitulated by in vitro systems. For example, the toxin is not diluted by the circulating fluids after addition to a neuronal culture. More importantly, in vitro, the architecture and some cell components of the peripheral synapses, such as muscle, Schwann, or gland cells are lacking. These “accessory cells” are main determinants of the duration of action of a BoNT at the neuromuscular junction (section III.B). A striking example is given by the comparison of the 3 month duration of the BoNT/A1-induced paralysis at the NMJ synapse, whereas cleavage of SNAP-25 was reported to last over 1 year in some neurons in culture (Whitemarsh et al., 2014). In addition cultured neurons may lack some receptor components present in vivo; for example, Neuro2A or PC12 cells have different ganglioside profiles. Apart from these limitations, neurons in culture are very useful to test many aspects of the biochemical and cellular action of BoNTs from binding to internalization and from membrane translocation to SNARE cleavage. They provide some comparative toxicity data, which, however, cannot be extended simply to dose values to be used in human therapy or in the evaluation of cases of botulism.
The pharmacological aspects of a product for human therapy such as botulinum toxin are very relevant and must be determined. Although greatly needed, classic pharmacokinetic studies on adsorption, distribution, metabolism, and excretion for BoNT products used clinically, have not been performed for reasons mostly related to the very low therapeutic doses and therefore to limitations of detection.
V. Conclusions
The botulinum neurotoxins represent a unique case of molecular Janus, the Greek mythological creature with two faces. The first face became apparent with the finding that these proteins are the most poisonous poisons for humans and other high vertebrates. As such the botulinum neurotoxins have been studied in laboratories involved in research on biologic warfare, with work primarily aimed at the neutralization of the potential bioterrorist use of these neurotoxins using novel immunotherapies, vaccines, and drug inhibitors.
Their very high toxicity is no longer surprising now that we know the major aspects of their molecular and cellular mode of action. The complementary efforts of basic scientists and clinicians turned light on the other Janus face, i.e., the therapeutic potential. The effort of many laboratories and pharmaceutical companies together with the wisdom of ruling agencies has made botulinum neurotoxins a therapy of choice for many human diseases, and the list of syndromes that are successfully treated with botulinum neurotoxins is still increasing. The ever-expanding therapeutic and cosmetic use of the botulinum neurotoxins is accompanied by a remarkably high record of safety as compared with the other pharmaceuticals, provided that they are used by trained medical personnel.
It is easy to anticipate that additional pathologic conditions might be treated with botulinum neurotoxins following the development of three lines of research: 1) definition of the molecular, cellular, tissue, and pharmacological properties of the many novel botulinum toxins that are being discovered; 2) molecular design and biotechnology production of botulinum neurotoxins endowed with novel biologic properties; 3) understanding the mechanisms that regulate intraneuronal localization and duration of action of botulinum neurotoxins in vivo.
Acknowledgments
We thank the reviewers and the editor for insightful comments, criticisms, and suggestions. We thank Dr. J. E. Keller for a very relevant comment.
Authorship Contributions:
Wrote or contributed to the writing of the manuscript: Pirazzini, Rossetto, Eleopra, and Montecucco.
Footnotes
The authors' research was supported by Fondazione Cariparo, the Axonomics project of Provincia di Trento, the Ministery of Defence, the University of Padova and the University of Udine.
Abbreviations
- BoNT
- botulinum neurotoxins
- CNS
- central nervous system
- FDA
- Food and Drug Administration
- H
- heavy chain
- HSA
- human serum albumin
- HC
- carboxy-terminal part of heavy chain
- HC-C
- C-terminal part of the HC domain
- HC-N
- N-terminal part of HC
- HN
- amino-terminal part of heavy chain
- L
- light chain
- LES
- lower esophageal sphincter
- NMJ
- neuromuscular junction
- NTNHA
- nontoxic nonhemagglutinin
- PSG
- polysialoganglioside
- PTC
- progenitor toxin complexes
- SV
- synaptic vesicle
- TeNT
- tetanus neurotoxin
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- VAMP
- vesicle-associated membrane protein
- Copyright © 2017 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵




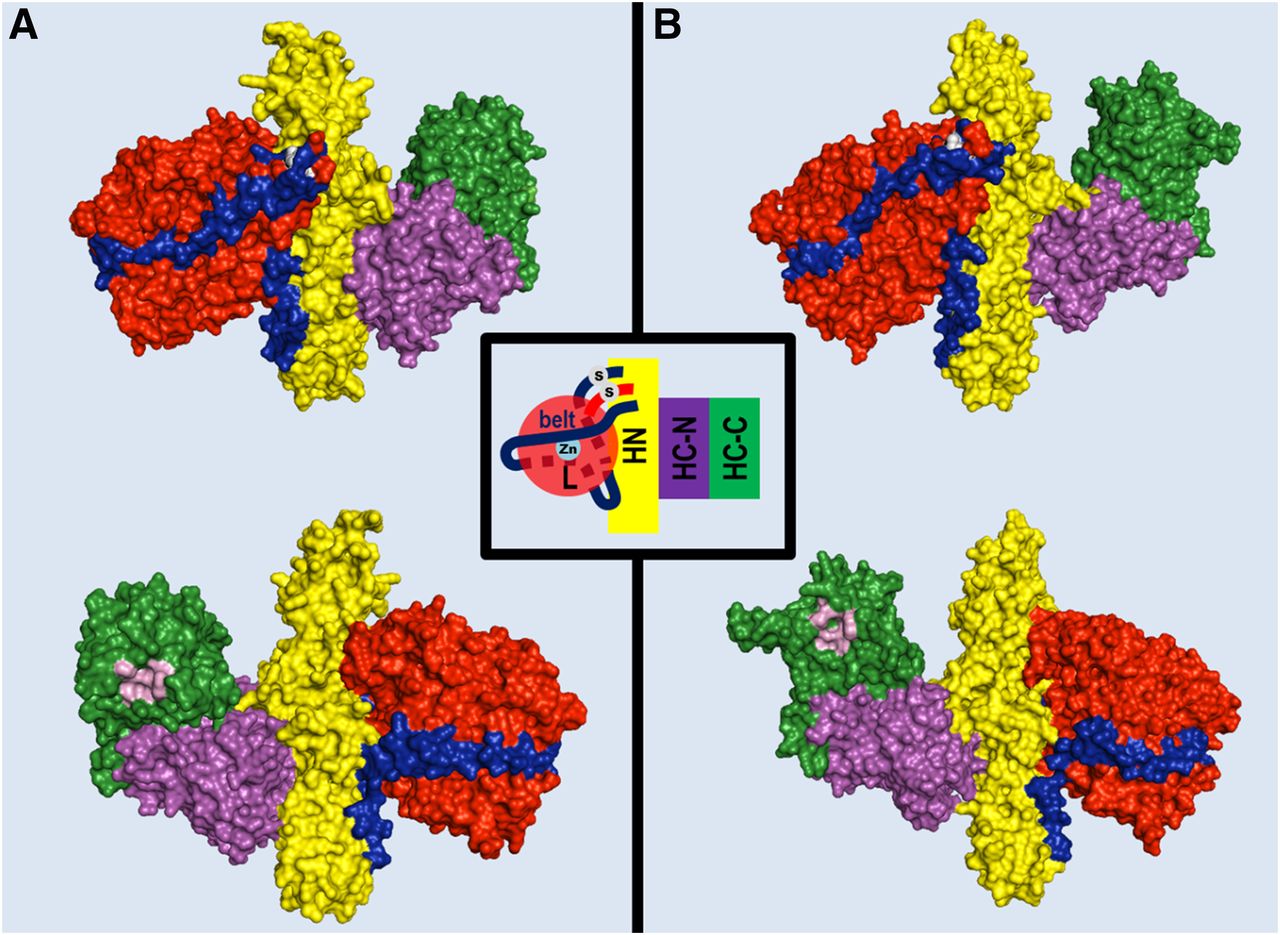{kind=link}
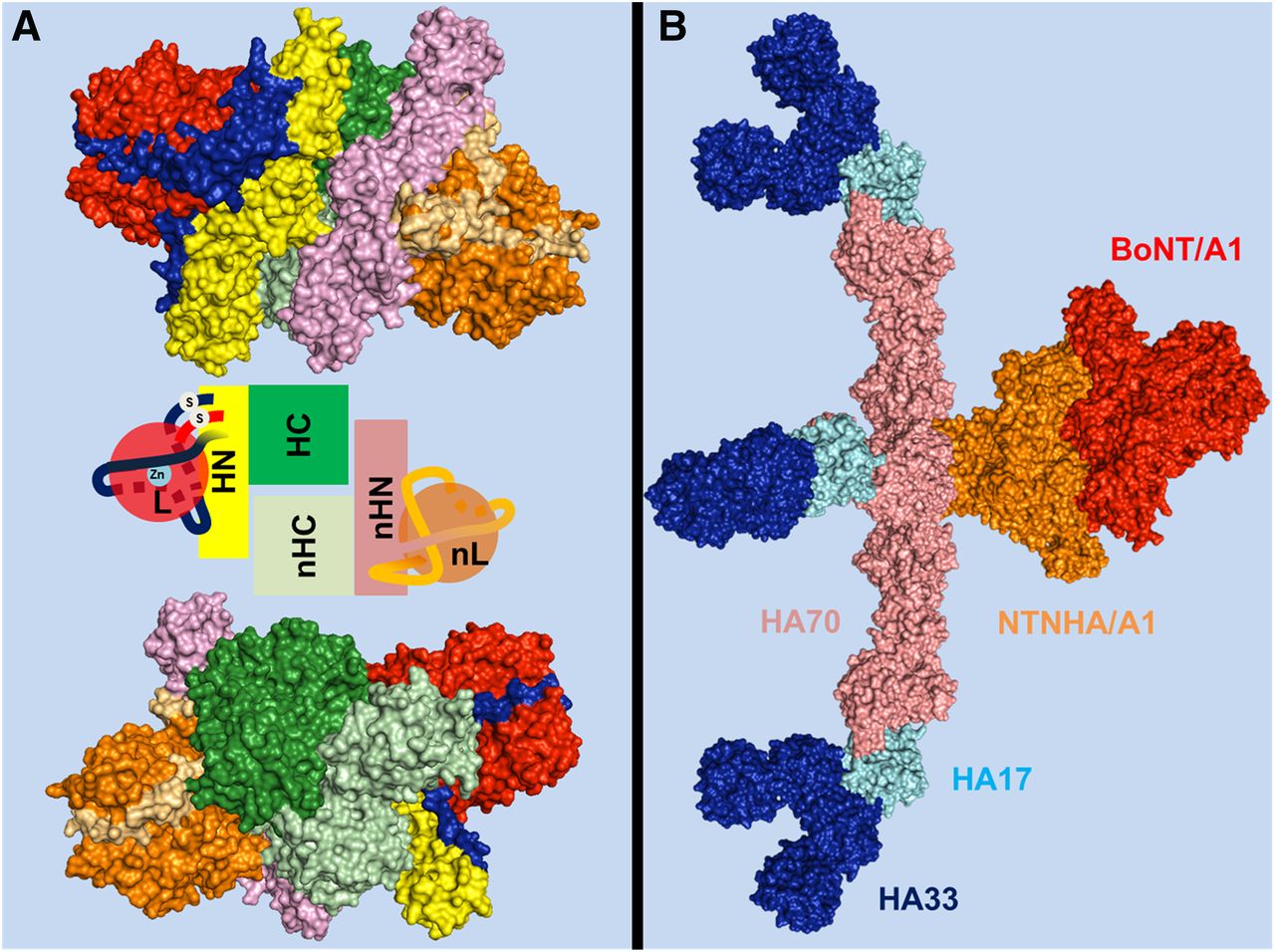{kind=link}
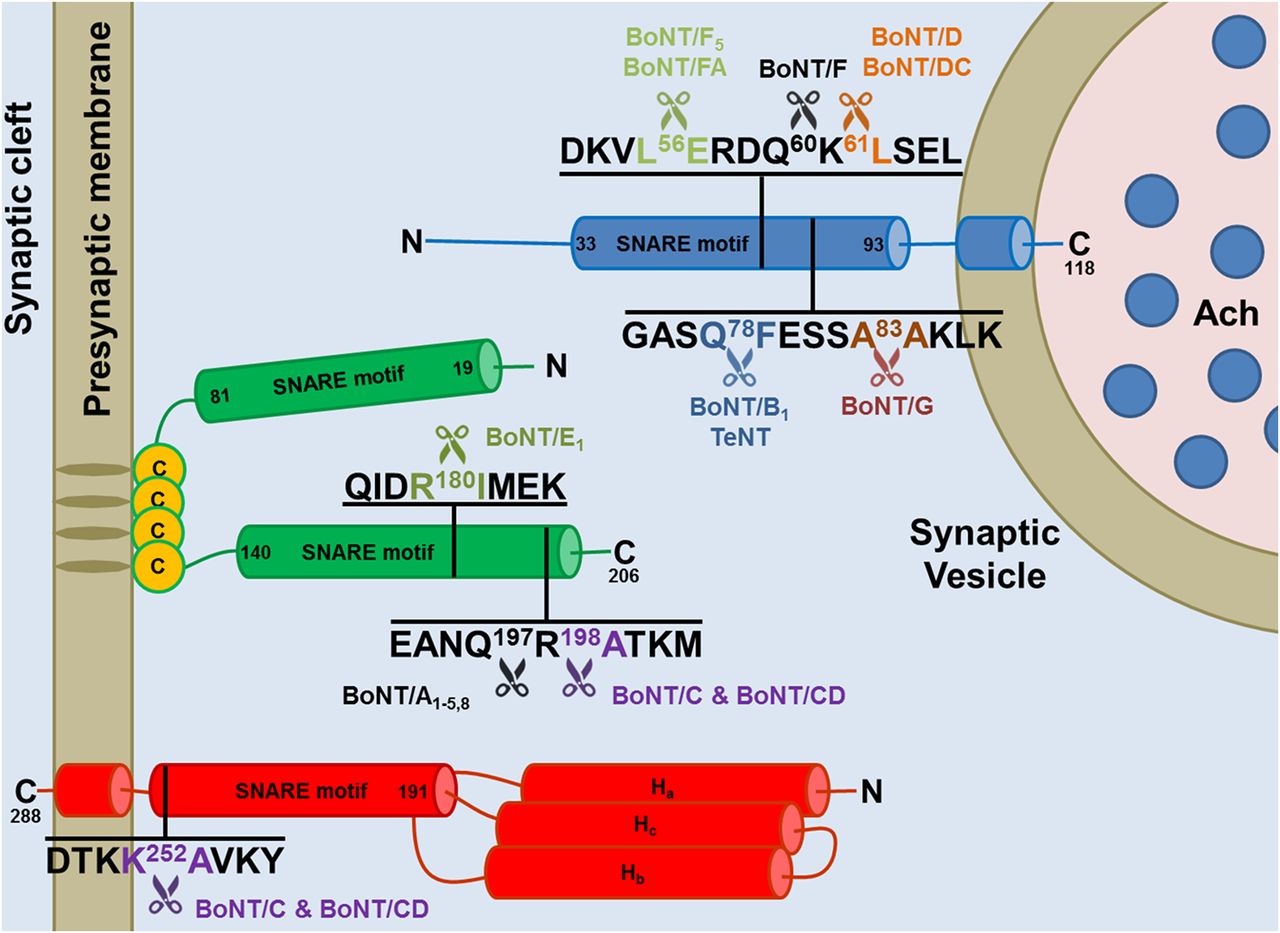{kind=link}
{kind=link}
{kind=link}