


Abstract
GABAA receptors are the major inhibitory transmitter receptors in the brain. They are ligand-gated chloride channels and the site of action of benzodiazepines, barbiturates, neuroactive steroids, anesthetics, and convulsants. GABAA receptors are composed of five subunits that can belong to different subunit classes. The existence of 19 homologous subunits and their distinct regional, cellular, and subcellular distribution gives rise to a large number of GABAA receptor subtypes with distinct pharmacology, which modulate different functions of the brain. A variety of compounds have been identified that were claimed to modulate selectively individual GABAA receptor subtypes. However, many of these compounds have only incompletely been investigated or, in addition to a preferential modulation of a receptor subtype, also modulate other subtypes at similar concentrations. Although their differential efficacy at distinct receptor subtypes reduced side effects in behavioral experiments in rodents, the exact receptor subtypes mediating their behavioral effects cannot be unequivocally delineated. In addition, the discrepant in vivo effects of some of these compounds in rodents and man raised doubts on the applicability of the concept of receptor subtype selectivity as a guide for the development of clinically useful drugs. Here, we provide an up-to-date review on the currently available GABAA receptor subtype-selective ligands. We present data on their actual activity at GABAA receptor subtypes, discuss the translational aspect of subtype-selective drugs, and make proposals for the future development of ligands with better anxioselectivity in humans. Finally, we discuss possible ways to strengthen the conclusions of behavioral studies with the currently available drugs.
I. Introduction
A. γ-Aminobutyric Acid Type A Receptors and Their Heterogeneity
γ-Aminobutyric acid (GABA) is the major inhibitory transmitter of the central nervous system and exerts its action via two types of receptors, GABAA and GABAB receptors. Whereas GABAB receptors are metabotropic receptors that activate inwardly rectifying K+ channels and/or inhibit high voltage-activated Ca2+ channels (Bettler et al., 2004), GABAA receptors belong to the Cys-loop family of pentameric ligand-gated ion channels that includes the nicotinic acetylcholine-, serotonin type 3- , and strychnine-sensitive glycine receptors (Galzi and Changeux, 1994; Olsen and Sieghart, 2008), a Zn2+-activated ion channel (Davies et al., 2003), and an invertebrate glutamate-gated chloride channel (Hibbs and Gouaux, 2011). GABAA receptors are anion-selective channels, and increased chloride permeability generally reduces neuronal excitability. But these receptors also can conduct other anions with variable permeability ratios relative to chloride. For instance, HCO3− flux could be physiologically relevant under certain conditions (Kaila et al., 1997; Olsen and Sieghart, 2008).
Twenty distinct GABAA receptor subunit genes (6α, 4β, 3γ, δ, ε, π, θ, 3ρ) have been identified in the vertebrate nervous system, including a putative β4 subunit gene that originally was identified in chicken (Bateson et al., 1991), but also has been demonstrated in humans (Levin et al., 1996; Berezhnoy et al., 2007). These genes as well as several alternatively spliced isoforms of the respective subunits, for instance γ2L and γ2S, give rise to a possible enormous heterogeneity of GABAA receptor subtypes (Barnard et al., 1998; Berezhnoy et al., 2007; Olsen and Sieghart, 2008). Recently, the extent of heterogeneity has been extensively discussed together with criteria for the unequivocal identification of native GABAA receptor subtypes. So far, no sufficient new information has been accumulated that would justify the addition of a new member to the list of 26 GABAA receptor subtypes “identified” or “existing with a certain probability” (Olsen and Sieghart, 2008). The majority of GABAA receptors is composed of two α, two β, and one γ subunits (αβγ receptors, for receptor nomenclature, see Alexander et al., 2015; Olsen and Sieghart, 2008). In these receptors, alternating α and β subunits are connected by a γ subunit (Tretter et al., 1997). Depending on the subunit composition and arrangement, these receptors exhibit different pharmacology, and due to their distinct regional and cellular distribution (Wisden et al., 1992; Fritschy and Mohler, 1995; Pirker et al., 2000; Hörtnagl et al., 2013) each receptor subtype also contributes to the modulation of distinct functions of the brain (Sieghart, 1995; Olsen and Sieghart, 2008). Whereas GABAA receptors composed of α1βγ2, α2βγ2, α3βγ2 are predominantly located synaptically and contribute to phasic inhibition, receptors composed of α4βγ2, α5βγ2, α6βγ2, or αβε subunits are partially located extrasynaptically and therefore, contribute to both phasic and tonic inhibition of neurons (Wagner et al., 2005; Glykys and Mody, 2007a; Belujon et al., 2009; Chen et al., 2017). In contrast, GABAA receptors composed of αβ or αβδ subunits seem to be exclusively located extrasynaptically and hence only exert a tonic inhibition of neurons (Brickley and Mody, 2012).
In addition, at least some GABAA receptor subtypes not only can be modulated by GABA but also exhibit some spontaneous gating in the absence of GABA and thus also contribute to tonic inhibition (Wlodarczyk et al., 2013). These include α6β2γ2 (Knoflach et al., 1996), α6β2δ (Hadley and Amin, 2007); α4β3γ2 and α4β3δ (Tang et al., 2010); α4β1δ, α4β2δ, α6β1δ, α6β3δ, α1β3δ (Karim et al., 2012b); or receptors containing an ε subunit (Neelands et al., 1999; Wagner et al., 2005). Furthermore, rat or murine homo-oligomeric β1 receptors exhibit spontaneous gating that could be inhibited by picrotoxin (Sigel et al., 1989; Krishek et al., 1996) and modulated by pentobarbital and propofol (Krishek et al., 1996). These receptors, however, were insensitive to GABA. In contrast, bovine (Krishek et al., 1996) or human β1 receptors (Sanna et al., 1995) that also exhibit spontaneous gating were GABA sensitive. GABA sensitivity of homo-oligomeric β1 receptors thus seems to be species dependent. Homo-oligomeric β3 receptors are also spontaneously open ion channels that are insensitive to GABA (Cestari et al., 1996; Wooltorton et al., 1997) and can be modulated by various allosteric modulators, such as pentobarbital, etomidate, alphaxalone, propofol, and chlormethiazole (Slany et al., 1995; Wooltorton et al., 1997). Interestingly, however, these receptors could be further activated by histamine (Saras et al., 2008; Seeger et al., 2012). Homo-oligomeric β3 receptors are thus not GABA-gated but histamine-gated channels (Saras et al., 2008; Hoerbelt et al., 2016). In contrast to the receptors mentioned above and composed of αβγ2 or αβδ subunits that were classified as “identified in the nervous system,” receptors containing ε subunits were classified as “tentatively occurring in the brain” (Olsen and Sieghart, 2008). Homo-oligomeric β1- or β3-containing receptors so far have not been demonstrated in the brain due to a lack of specific tools that could identify them among a variety of hetero-oligomeric αβ, αβγ, or αβδ receptors. However, especially homo-oligomeric β3 receptors are easily formed in recombinant expression systems (Slany et al., 1995; Cestari et al., 1996; Miller and Aricescu, 2014) and there is no reason to believe that they cannot also be formed in the nervous system (Hoerbelt et al., 2016). In any case, drugs that can modulate spontaneously open receptors will also modulate tonic inhibition.
It has to be kept in mind, however, that depending on the chloride concentration in the inside of neurons, that is enhanced by the Na+-K+-2Cl− cotransporter NKCC1 and reduced by the K+-Cl− cotransporter KCC2, opening of the GABAA receptor associated Cl− channel can elicit a depolarization and hyperpolarization, respectively. During early development of the nervous system NKCC1 predominates, resulting in a high Cl− concentration in the inside of neurons and thus in a Cl− efflux on activation of GABAA receptors (Ben-Ari et al., 2012; Kaila et al., 2014). At this time, GABA is an excitatory transmitter. During the establishment of the excitatory glutamate system in the brain the expression of KCC2 increases, the Cl− concentration in the inside of neurons decreases and activation of GABAA receptors causes Cl− influx and thus a hyperpolarization. In the adult brain, activation of GABAA receptors in most cases results in a hyperpolarization, and thus GABAA receptors become the major inhibitory transmitter receptors in the nervous system. But even then, depolarizing actions of GABA can also occur in some neurons or even in individual compartments of neurons, depending on the subcellular localization of NKCC1 and KCC2 as well as on the intracellular distribution of large impermeable anions that are predicted to cause a discrete balance of chloride ions (Astorga et al., 2015; Knoflach et al., 2016).
B. γ-Aminobutyric Acid Type A Receptor Structure and Pharmacology
GABAA receptors are the site of action of a variety of pharmacologically and clinically important drugs, such as benzodiazepines, barbiturates, anesthetics, neuroactive steroids, and convulsants (Sieghart, 1995; Berezhnoy et al., 2007). Whereas GABA exerts its channel opening effects via two binding sites located at the two extracellular interfaces of β and α subunits (β+α− interfaces), (Smith and Olsen, 1995), benzodiazepines bind to the homologous interface of an α and a γ subunit (α+γ− interface) (Sigel and Buhr, 1997; Ernst et al., 2003). Ligands acting via the benzodiazepine binding site cannot directly open the GABAA receptor-associated chloride channel, but only allosterically enhance (positive allosteric modulators) or reduce (negative allosteric modulators) GABA-induced currents. A third group of benzodiazepine site ligands does not significantly change the conformation of GABAA receptors. These ligands are antagonists (silent, neutral, or null modulators) at the benzodiazepine binding site. They exhibit no direct effects at GABA-induced currents but are able to block the action of positive or negative allosteric modulators acting at this site.
In addition to the GABA and the benzodiazepine binding sites there are multiple other binding sites at GABAA receptors. At least 16 solvent accessible spaces have been identified in the extracellular and transmembrane domain of a GABAA receptor structural model (Ernst et al., 2005). Five are located at the five extracellular interfaces between subunits, among those the benzodiazepine- and the two GABA-binding sites, five at the transmembrane interfaces, five are located within the four-helix-bundles forming the transmembrane domain of individual subunits, and at least one is located within the channel pore, mediating the action of some convulsants (Ernst et al., 2005). Furthermore, ligand-bound crystal structures of bacterial homologs of GABAA receptors (GLIC, isolated from Gloeobacter violaceus; ELIC, isolated from Erwinia chrysanthemi), the structures of a glutamate-gated channel (isolated from the nematode Caenorhabditis elegans) for an overview, see Sieghart, 2015; and the recently published crystal structure of the homo-oligomeric β3 GABAA receptor (Miller and Aricescu, 2014) indicated the existence of additional ligand-binding sites in the transmembrane and extracellular domains (Sieghart, 2015; Puthenkalam et al., 2016). All these solvent accessible spaces could function as drug binding pockets. They differ from each other in their size and their hydrophilic and hydrophobic properties, depending on the types of amino acid residues contributing to their formation (Ernst et al., 2005). In addition, neighboring subunits can influence the conformation of the pockets via subunit-subunit interactions, indicating that even pockets formed by the four-transmembrane helices of two identical α or β subunits within GABAA receptors could be different from each other. And some of the pockets might accommodate more than one drug at distinct positions (Sieghart, 2015; Puthenkalam et al., 2016). In addition, binding of a drug to its pocket(s) can influence the conformation of other pockets of the receptors, resulting in allosteric interactions with other drugs (Puthenkalam et al., 2016). There are multiple examples for such allosteric interactions of drugs in GABAA receptors (Sieghart, 1995).
Possible drug binding sites in the intracellular domain of GABAA receptors so far could not be investigated because the structure of the intracellular domain of these receptors currently is not known. In addition, drugs could bind to the surface of GABAA receptors and by that influence their flexibility (Baur et al., 2005, 2013). And binding sites of ions such as Zn2+, Cu2+, or La3+ in many cases are formed by specific amino acid residues that differ in distinct receptor subtypes (Sieghart, 2015). In contrast to the GABA and the benzodiazepine binding sites that are located in the extracellular domain of GABAA receptors, barbiturates, anesthetics, and neuroactive steroids seem to bind to sites in the transmembrane domain (Sieghart, 2015; Forman and Miller, 2016; Feng and Forman, 2018). These compounds not only allosterically modulate GABA-induced currents but at higher concentrations also can directly activate the ion channel intrinsic to GABAA receptors in the absence of GABA. In recent years, more than 100 compounds have been identified that are able to modulate GABA-induced currents or to activate directly GABAA receptors via binding sites different from the benzodiazepine or the GABA binding site, respectively. For most of these compounds, their binding sites at GABAA receptors so far have not been identified.
Drugs that exclusively modulate GABA-induced currents in an allosteric way are limited in their activity to those GABAA receptors that are active in a certain task and therefore have less adverse effects than GABA site agonists or drugs that are able directly to open the chloride channel of most if not all GABAA receptors. Benzodiazepines were the first compounds identified only to allosterically modulate GABAA receptors, and due to their anxiolytic, anticonvulsant, sedative, hypnotic, and muscle relaxant action and their clinical importance, much of the ensuing effort was directed toward the development of benzodiazepine site ligands with a more selective action.
The classic benzodiazepines possess comparable affinities and efficacies for GABAA receptor subtypes composed of α1βγ2, α2βγ2, α3βγ2, or α5βγ2 subunits and thus produce comparable behavioral effects. Over the years, however, a variety of benzodiazepine binding site ligands from different structural classes have been developed that are able to differentially interact with these diazepam-sensitive α1βγ2, α2βγ2, α3βγ2, or α5βγ2 receptor subtypes. Their reduced side effect profile in behavioral experiments in rodents seemed to confirm the molecular genetic evidence indicating that α1βγ2 receptors partially mediate the sedative, anterograde amnestic, and anticonvulsive actions, and α2βγ2 receptors partially mediate the anxiolytic-like action (Löw et al., 2000). α2βγ2 together with α3βγ2 receptors seem to mediate some antinociceptive and muscle relaxant actions (Crestani et al., 2001; Knabl et al., 2008; Ralvenius et al., 2015), and α5βγ2 receptors mediate the cognitive effects of benzodiazepine site ligands (Collinson et al., 2002; Crestani et al., 2002; Rudolph and Möhler, 2014). Moreover, from such experiments it was concluded that selective targeting of distinct GABAA receptor subtypes not only may provide an anxiolytic-like effect without sedation, but also unveil additional effects, which can be starting points for the development of an innovative treatment of pain, cognitive disorders, stroke, schizophrenia, depression, or Down syndrome (Rudolph and Knoflach, 2011; Rudolph and Möhler, 2014).
C. Aim of the Present Review
Unfortunately, however, many of the compounds claimed to be GABAA receptor subtype selective have only been incompletely investigated or, in addition to a preferential modulation of one receptor subtype, also significantly modulate other receptor subtypes at similar concentrations. Although their differential efficacy at distinct receptor subtypes reduced side effects and was beneficial for certain applications, the exact receptor subtype(s) mediating the behavioral effects of such compounds cannot be unequivocally delineated. Uncritical acceptance of a compound as being subtype selective, therefore, leads to poorly supported conclusions on GABAA receptor subtypes, eliciting compound-induced behavior, that add confusion to the literature. In addition, the discrepant in vivo effects of some of these ligands in rodents and humans recently raised doubts on the applicability of the concept of receptor subtype-selectivity as a guide for the development of clinically useful drugs (Skolnick, 2012).
Here, we provide an up-to-date review on the currently available GABAA receptor subtype-selective ligands. In sections II, III, and IV, we discuss methodological aspects important for the development of such drugs. In section V we critically discuss compounds claimed to be GABAA receptor subtype-selective and provide information on their actual interaction with various receptors, on their behavioral actions in vivo, as well as on their receptor occupancy during their behavioral actions, if available. Since presenting all the reasonably characterized compounds with some receptor subtype selectivity would have been a highly repetitive endeavor, compounds discussed were selected either because of their frequent use in behavioral and pharmacological studies, or because they are the currently most selective compounds within a compound class for which a reasonable data set is available. Most of these ligands exert their actions via the benzodiazepine binding site. Some of them, however, act via other allosteric binding sites at GABAA receptors. And a few ligands have been identified that can be used to selectively activate or inhibit certain GABAA receptor subtypes by acting via their GABA binding site. In section VI we discuss the translational aspect of subtype-selective drugs as exemplified by the discrepant in vivo effects of some of these ligands in rodents and man (Skolnick, 2012). In section VII we make proposals for the future development of ligands with improved anxioselectivity as well as discuss possible ways to strengthen the conclusions of behavioral studies with the currently available “receptor subtype-selective drugs.”
II. Receptor Subtype-selective Binding Versus Subtype-selective Efficacy
Since the discovery of the high affinity “central” benzodiazepine binding site in the brain (Braestrup and Squires, 1977; Möhler and Okada, 1977) that later on turned out to be an allosteric modulatory binding site at GABAA receptors (Karobath and Sperk, 1979; Sieghart and Karobath, 1980), more than 100 distinct compound classes have been identified that could inhibit high affinity binding of [3H]diazepam, [3H]flunitrazepam, or [3H]flumazenil to brain membranes. Most of these compounds have never been investigated for a possible GABAA receptor subtype selectivity because receptor subtypes and techniques to investigate them were not available at the time of their synthesis. Evidence for a molecular heterogeneity of GABAA receptors was available soon after the discovery of the benzodiazepine binding site (Sieghart and Karobath, 1980). However, recombinant receptor subtypes could only be expressed and investigated several years later after the individual subunits had been cloned and sequenced (Schofield et al., 1987; Levitan et al., 1988; Pritchett et al., 1989). In those early days, electrophysiological investigations of recombinant GABAA receptors were only rarely performed and putative receptor subtype-selectivity of drugs was deduced from their differential inhibition of [3H]flunitrazepam or [3H]flumazenil binding at the recombinant diazepam-sensitive α1,2,3,5βγ2 or diazepam-insensitive α4,6βγ2 GABAA receptor subtypes, respectively (McKay et al., 2004; Selleri et al., 2003, 2005; Yin et al., 2010), or from two or three of these receptor subtypes, only (Guerrini et al., 2007; Lager et al., 2008). However, there are multiple examples of both, drugs exhibiting a comparable affinity but a differential efficacy and drugs exhibiting a differential affinity but a comparable efficacy for modulating GABA-induced currents of all these receptors (Atack, 2010a,b; Vinkers et al., 2010). It has to be kept in mind that it is the potency and efficacy of a drug for modulating GABAA receptor subtypes that is decisively important for its action and not its affinity for a certain binding site (Korpi et al., 2002).
This conclusion is strengthened by increasing evidence that most if not all currently available drugs can interact with several binding sites at the same GABAA receptor subtype (Sieghart, 2015; Puthenkalam et al., 2016). Interaction of the drug with some of these binding sites can be silent, whereas interaction with other binding sites can induce or stabilize a conformational change eliciting some drug effects. The most impressive examples for such a pattern are the pyrazoloquinolinones, such as CGS 9895, which more than three decades ago were identified as high affinity ligands for the benzodiazepine site of GABAA receptors (Bennett, 1987). These compounds exhibited anxiolytic and anticonvulsant effects but produced less sedative and muscle relaxant effects than benzodiazepines. Only recently it was demonstrated that CGS 9895 and most of its structural analogs are high affinity silent modulators (null modulators, antagonists) at the benzodiazepine binding site but mediate their low potency action at various GABAA receptor subtypes via a novel binding site at the previously not investigated α+β− interface of GABAA receptors (Ramerstorfer et al., 2011; Varagic et al., 2013b). Thus it is the functional selectivity resulting from the interaction of a drug with all its binding sites at the respective receptor subtype and at the concentration used, which is important for the action of a drug. But of course, a compound highly selective for the benzodiazepine site of a certain GABAA receptor subtype might also exhibit a high functional selectivity for this subtype if its action primarily is mediated via the benzodiazepine site.
III. Receptor Subtype-selective Efficacy—Dependence on the Conditions of Measurements
A possible functional receptor subtype selectivity can only be identified by a comparison of drug effects on GABA-induced currents elicited at various GABAA receptor subtypes. Unfortunately, however, depending on the expression system used for generating recombinant GABAA receptor subtypes (Xenopus laevis oocytes or cell line type, permanent or transient transfection, ratio of cDNAs used for transfection, etc.), in addition to the desired receptor subtype, homo- or heterooligomeric combinations of subunits can be formed, some of which might not occur in vivo (Olsen and Sieghart, 2008; Boileau et al., 2010; You et al., 2010). Depending on the exact mixture of recombinant receptors formed in the expression system the measured pharmacological properties can be different. And differences in electrophysiological protocols between laboratories also can impact efficacy and potency estimates (de Lucas et al., 2015). Finally, the GABA concentration used for measuring allosteric modulation of GABA-induced currents in recombinantly expressed receptors, strongly influences the extent of modulation observed. In different studies, GABA concentrations eliciting 3%–5% (EC3, Baur and Sigel, 2007; Ramerstorfer et al., 2010), 10% (EC10; Popik et al., 2006), 20% (EC20; Carling et al., 2006), or even 50% (EC50; Harvey et al., 2002; Yin et al., 2010) of the maximal currents were used, making comparison of the data from individual studies even more difficult.
While synaptic GABAA receptors are only partially saturated by GABA released from single vesicles in the course of miniature inhibitory postsynaptic currents (Perrais and Ropert, 1999; Hájos et al., 2000; Rumpel and Behrends, 2000), they are completely saturated by GABA released by repetitive action potentials. GABA concentrations within the synaptic cleft were estimated to rapidly rise to 1.5–3 mM and decay within a few hundred microseconds (Mozrzymas et al., 2003). When receptors are saturated by GABA, allosteric modulators no longer can enhance the current amplitude but only prolong the action of GABA. To estimate the action of a drug under these conditions, one would have to increase the time of measurement. However, the extent of GABA-induced current modulation would then strongly be influenced also by the desensitization of the receptor, a phenomenon more dominant at higher GABA concentrations and longer measurement times (Jones and Westbrook, 1995). This effect will be even more dramatic with drugs that allosterically accelerate current decay (Dillon et al., 1993, 1995; Simeone et al., 2017).
GABAA receptors have a high density at synapses, but synapses constitute only a small part of the cell surface. Despite their lower density at the extrasynaptic membrane, the overall abundance of extrasynaptic receptors is higher than that of synaptic receptors (Nusser and Mody, 2002; Kasugai et al., 2010). Since GABAA receptors are inserted into the membrane at extrasynaptic sites (Bogdanov et al., 2006), all GABAA receptor subtypes, even the synaptic receptors, are also, at least temporarily, present extrasynaptically. The GABA concentrations acting at extrasynaptic GABAA receptors (0.2–2.5 μM; Glykys and Mody, 2007b) activate extrasynaptic as well as synaptic receptors, although to a different extent (Mortensen et al., 2012; Karim et al., 2013). For the majority of recombinant αβγ2 GABAA receptors, such concentrations are between GABA EC3 and EC40. Some, but not all αβ and αβδ receptors, however, are more sensitive to GABA. In these receptors, 0.2 μM GABA concentrations already elicit 10%–50% and 2.5 μM GABA concentrations 20%–80% of the maximal GABA-induced currents (Mortensen et al., 2012; Karim et al., 2013). Nevertheless, it has been demonstrated that even α1β2γ2 receptors, which exhibit a quite low GABA sensitivity, can be activated by 0.5 μM GABA, and the resulting GABA EC1 currents can be modulated by allosteric modulators (Li and Akk, 2015).
In addition, measurements at low GABA concentrations (GABA EC3) have technical advantages over measurements at higher GABA concentrations. For instance, allosteric modulation measured as a percentage of increase in the GABA-induced current in the presence of a drug is much stronger at GABA EC3 than at higher GABA concentrations, because it is referred to a smaller GABA-induced current. Differences in the efficacy of a drug for modulating GABA-induced currents at different receptor subtypes are thus also more evident at low than at high GABA concentrations. Measurements are also more reproducible, because GABA EC3 is not in the linear range of GABA stimulation. Slight differences in the GABA concentration from the actual GABA EC3 thus do not matter as much as similar variations at GABA EC10, EC20, or EC50. Given all these arguments, it is suggested to measure allosteric modulation of GABAA receptors at GABA EC3 whenever technically feasible to increase the sensitivity of measurements and to allow a better comparison of pharmacological data.
IV. Importance of Concentration-response Relationships of “Receptor Subtype-selective Ligands”
A. Referring Compound Actions to that of Standard Benzodiazepines Distorts Original Data
In an effort to compare the actions of compounds at various receptor subtypes with those of chlordiazepoxide (Blackaby et al., 2006; Carling et al., 2006; Jennings et al., 2006), diazepam (Griebel et al., 2001; Alhambra et al., 2011; de Lucas et al., 2015), or zolpidem (Griebel et al., 2001), their efficacy was often also given relative to the maximal efficacy of these reference drugs measured in the same oocyte or cell culture system. Although such presentation allows for an immediate estimation of whether the compound exhibits a stronger or weaker effect at a receptor subtype than the reference compound, it also distorts the actual efficacy of the compound. For instance, if a compound exhibits a comparable efficacy at two receptor subtypes, its relative efficacy at these receptor subtypes will be different if the efficacy of the reference compound is different at these receptors. This is the case, for instance, for diazepam, which exhibits a higher maximal efficacy for α3β3γ2 than for α2β3γ2 or for α1β3γ2 and α5β3γ2 receptors (Puia et al., 1991; Ramerstorfer et al., 2010). Furthermore, data presented relative to diazepam cannot be compared with those presented relative to chlordiazepoxide or zolpidem in the absence of information on the efficacy of the reference compounds used to calculate the results. Thus, unfortunately, important information is lost due to such data presentation, and the actions of the examined compounds cannot be compared with those of other ligands. To fully benefit from a comparison with standard benzodiazepines, the authors should also provide the original concentration-response curves of the compounds at the individual receptor subtypes, as for instance in Dias et al. (2005), Atack et al. (2006b, 2011b), Ren et al. (2010), and Christian et al. (2015).
B. Maximal Efficacy Hides a Possible Subtype Selectivity at Lower Concentrations
An apparent lack of receptor subtype selectivity of most of the published “receptor subtype-selective” compounds, however, can be deduced from three recent reviews that compared their affinity for the benzodiazepine binding site and their maximal relative efficacy at various GABAA receptor subtypes (Atack, 2010a,b; Vinkers et al., 2010). From these overviews it is clear that even compounds that preferentially modulate a certain receptor subtype exhibit substantial activity also at one or more other receptor subtypes when the maximal efficacy at each receptor subtype is attained. It has been established previously that, depending on the behavioral task involved, a 1%–25% in vivo GABAA receptor occupancy by diazepam (Gardner, 1992; Lippa et al., 2005) may be sufficient to elicit discernible anxiolytic or other behavioral effects. A 15%–30% receptor occupancy (Atack et al., 2010) or an efficacy greater than 0.1 relative to 1 μM diazepam (Alhambra et al., 2011) elicits sedation and sleep. Thus even small effects of drugs at receptor subtypes must not be ignored. A 10% or 20% increase in living costs significantly matters in daily life, and a 10% or 20% enhancement of GABA-induced currents probably also will be able to elicit some but not all effects that can be elicited by a stronger enhancement of GABA currents. The in vivo effects of a drug therefore cannot be exclusively assigned to that receptor which can be modulated by the drug with the highest efficacy.
C. Concentration of Compounds Eliciting In Vivo Effects
The maximal efficacy of a drug is usually measured at a very high drug concentration. In many cases, concentrations 1000-fold the Ki value estimated from benzodiazepine binding studies were used, resulting in drug concentrations of 1–10 µM (Street et al., 2004; Carling et al., 2006; Goodacre et al., 2006). Such concentrations, at least for benzodiazepine site ligands, only rarely can be achieved in the brain after pharmacological (rather than toxicological) doses. It has been demonstrated that the oral dose of diazepam required to occupy 50% of all diazepam-sensitive GABAA receptors in rodents was as low as 1 to 2 mg/kg, resulting in total brain concentrations of 1 μmol/l and above (Greenblatt and Sethy, 1990; Müller Herde et al., 2017). But only the unbound (free) fraction of the total drug concentration in the brain may bind to receptor sites, and it has been demonstrated that the free concentrations of drugs are best correlated with their pharmacological effects in the brain (Hammarlund-Udenaes, 2010). The unbound fraction of benzodiazepine site ligands can differ substantially and amounts to 3.6% for diazepam or 47.1% for zaleplon (Summerfield et al., 2007). Under therapeutic conditions, benzodiazepines thus usually achieve free brain concentrations far below 1 µM.
The free drug concentration at the receptor easily can be estimated from in vivo receptor occupancy data of compounds and their concentration-response curves at individual recombinant receptor subtypes. Assuming that a 50% receptor occupancy under therapeutic conditions corresponds with the concentration of the compound that generates 50% of its maximal effect at the most abundant GABAA receptor subtype (α1βγ2) in the brain, the active concentration of the compound at this receptor and in the brain, as well as the extent of modulation of the individual receptor subtypes under these conditions, can be estimated. The data discussed below indicate that most of the compounds so far investigated are eliciting their in vivo effects at low nanomolar concentrations. It is then clear that most if not all receptor subtypes are fully modulated by the drug at a close to 100% receptor occupancy. Therefore, it is reasonable to investigate the receptor subtype selectivity in a concentration range that also can be achieved in the brain in vivo and that elicits the behavioral actions of the drug. The best and most reliable way to measure receptor subtype selectivity, thus, is a comparison of the functional concentration-response curves for all possible receptor subtypes, because there might be concentration ranges of a compound in which it exhibits high receptor subtype selectivity. Only by knowing these concentrations and the doses required for achieving these free concentrations in the brain, it is possible selectively to modulate the respective receptor subtype(s) in in vitro and in vivo experiments, respectively.
V. Currently Available Compounds Claimed to Be γ-Aminobutyric Acid Type A Receptor Subtype-Selective
A. Subtype-Selectivity Claimed for Incompletely Investigated Compounds
Even when the effects of compounds at recombinant GABAA receptor subtypes were investigated by electrophysiological studies, sometimes not all diazepam-sensitive receptor subtypes were investigated and in many cases only the maximal ligand effects, but no concentration-response curves, were shown at the individual receptor subtypes. Receptor subtype selectivity was then claimed when a compound exhibited a higher positive or negative maximal efficacy at the given receptor as compared with other receptors, although other receptors also were significantly modulated by the compound (Street et al., 2004; Goodacre et al., 2006). In addition, a receptor subtype selectivity was even claimed based on a relatively selective binding to the benzodiazepine site of a receptor subtype and a single maximal efficacy at this one receptor (Achermann et al., 2009; Buettelmann et al., 2009).
B. Compounds Claimed to Selectively Modulate α1βγ2 Receptors
Combined molecular genetic and pharmacological approaches demonstrated that α1βγ2 GABAA receptors partially mediate the sedative, anticonvulsant, and anterograde amnestic properties of diazepam (Rudolph et al., 1999; McKernan et al., 2000; Ralvenius et al., 2015). Drugs selectively modulating α1βγ2 GABAA receptors should thus exhibit sedative and anticonvulsant properties.
1. Zolpidem
The imidazopyridine zolpidem (Fig. 1) exhibits a 10-fold higher affinity for the benzodiazepine binding site of α1βγ2 than for that of α2βγ2 or α3βγ2 receptors and an exceptionally low affinity for α5βγ2 receptors (Sieghart, 1995). From that it was concluded that zolpidem is an α1βγ2-selective compound. However, from the concentration-response curves of zolpidem (Fig. 1) it can be deduced that there is only a small concentration range (between 1 and 30 nM) in which zolpidem only modulates recombinant GABAA receptors composed of α1βγ2 receptors (Ramerstorfer et al., 2010).
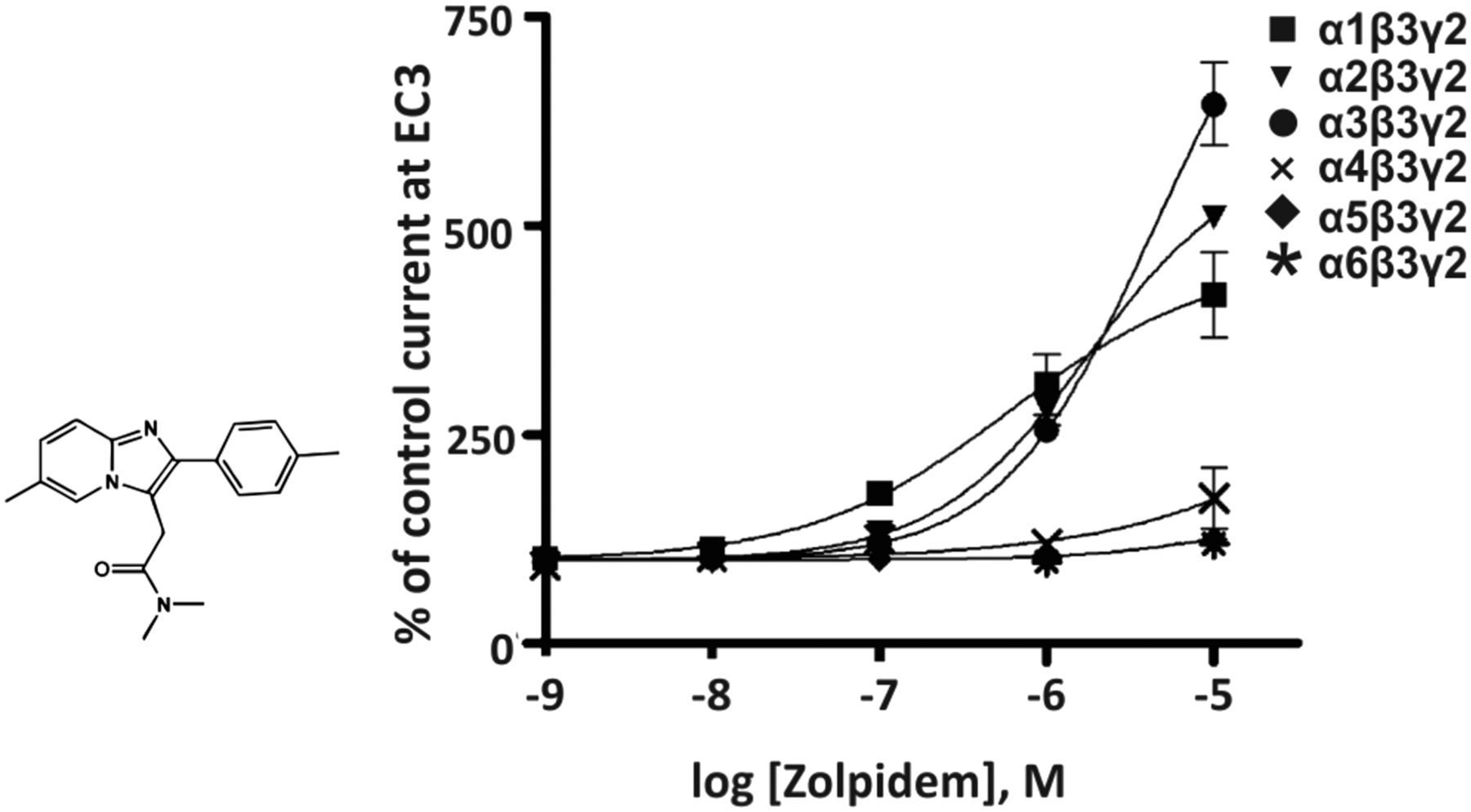
Chemical structure and concentration-response curves of zolpidem at various recombinant GABAA receptor subtypes from the rat expressed in Xenopus laevis oocytes and measured at GABA EC3. The concentration-response curve of α5β3γ2 receptors is overlapping with that of α6β3γ2 receptors. Modified from Ramerstorfer et al., Eur J Pharmacol, 636,18-27, Elsevier, 2010.
At 100 nM concentrations, zolpidem already significantly enhanced GABA-induced currents at α2β3γ2 or α3β3γ2 receptors from 100% to 132% or 121%, respectively, and at concentrations above 100 nM, which presumably are achievable in vivo at a moderate to high zolpidem dose, zolpidem only preferentially, but not selectively, modulates α1β3γ2 receptors. So, by carefully controlling the concentration of zolpidem, for instance in electrophysiological experiments using cell cultures or brain slices, or by local application of zolpidem in in vivo experiments, it is possible to use this drug for a selective modulation of α1β3γ2 receptors. As expected, due to its preferential action at α1-containing receptors, zolpidem exhibits sedative, hypnotic, and anticonvulsive actions in rodents and humans. The muscle relaxant action of zolpidem might be mediated by α2- and α3-containing GABAA receptors (Ralvenius et al., 2015) while any anxiolytic-like action of zolpidem is behaviorally nonspecific and confounded by sedation (Savić et al., 2004).
Interestingly, zolpidem at clinically relevant concentrations was recently demonstrated to enhance GABA-induced currents in α1β3 receptors composed of 3α1 and 2β3 subunits in a flumazenil-sensitive manner (Che Has et al., 2016). These receptors contain an α1-α1 interface and thus differ from α1β3 receptors composed of 2α1 and 3β3 subunits that contain a β3-β3 interface. Diazepam also was able to modulate the 3α1:2β3-containing α1β3 receptors, but the efficacy of diazepam was significantly lower for this receptor than that of zolpidem, and no modulation by either zolpidem or diazepam was detected at the 2α1:3β3 receptor. To the best of our knowledge this is the first example of a stoichiometry-dependent action of a drug. Results indicate that zolpidem is acting via a binding site at the α1+α1− interface, which obviously mimics the classic α1+γ2− benzodiazepine site. Receptors composed of α1β3 subunits are expressed in the rat brain (Mortensen and Smart, 2006; Olsen and Sieghart, 2008). Studies on recombinant α1β3 receptors have indicated that they exhibit a stoichiometry of 2α1:3β3 subunits (Tretter et al., 1997; Baumann et al., 2001). However, other studies have indicated that recombinant α1β2 (Boileau et al., 2005) or α6β2 (Im et al., 1995) receptors might be composed of 3α and 2β subunits. As discussed in Che Has et al. (2016), zolpidem is not a typical GABAA receptor hypnotic. Unlike benzodiazepines, zolpidem modulates tonic GABA currents in the rat dorsal motor nucleus of the vagus (Gao and Smith, 2010), exhibits residual effects in mice carrying the point mutation γ2F77I that drastically reduces interaction of zolpidem with the benzodiazepine binding site of GABAA receptors (Cope et al., 2005; Ramerstorfer et al., 2010), and improves speech as well as cognitive and motor functions in human patients with severe brain injury (Che Has et al., 2016). The receptors by which zolpidem mediates these effects are not known. It is thus quite possible that α1β3 receptors composed of 3α1:2β2 subunits might mediate at least some of these effects. In any case, these surprising observations on the action of zolpidem at α1β3 receptors add to previous observations on the existence of additional benzodiazepine binding sites at GABAA receptors (Sieghart, 2015). In addition, they indicate that not all actions of benzodiazepines or benzodiazepine site ligands can be explained by their allosteric modulation of the classic benzodiazepine binding site at the α+γ− interface of αβγ2 GABAA receptors. It has to be kept in mind that these compounds in addition to their interaction with the benzodiazepine site might be able to elicit some of their effects via other GABAA receptor subtypes that do not carry that site, or even via other effector systems.
2. Indiplon
The pyrazolopyrimidine indiplon (Fig. 2) has a nanomolar affinity for the benzodiazepine binding site of GABAA receptors in various rat brain regions (Sullivan et al., 2004) and was claimed to exhibit a 10-fold selectivity for α1β2γ2 over α2-, α3-, or α5-containing receptors in electrophysiological experiments at recombinant GABAA receptors from the rat (Petroski et al., 2006). But the concentration-response curves in Fig. 2 indicate that this at best is true up to 1 nM concentration. At 10 nM concentrations, all receptor subtypes investigated become positively modulated by that compound. At high concentrations, indiplon is a high efficacy positive allosteric modulator (full benzodiazepine site agonist) at all GABAA receptor subtypes investigated. Indiplon exhibits sedative-hypnotic properties in rodents and humans.
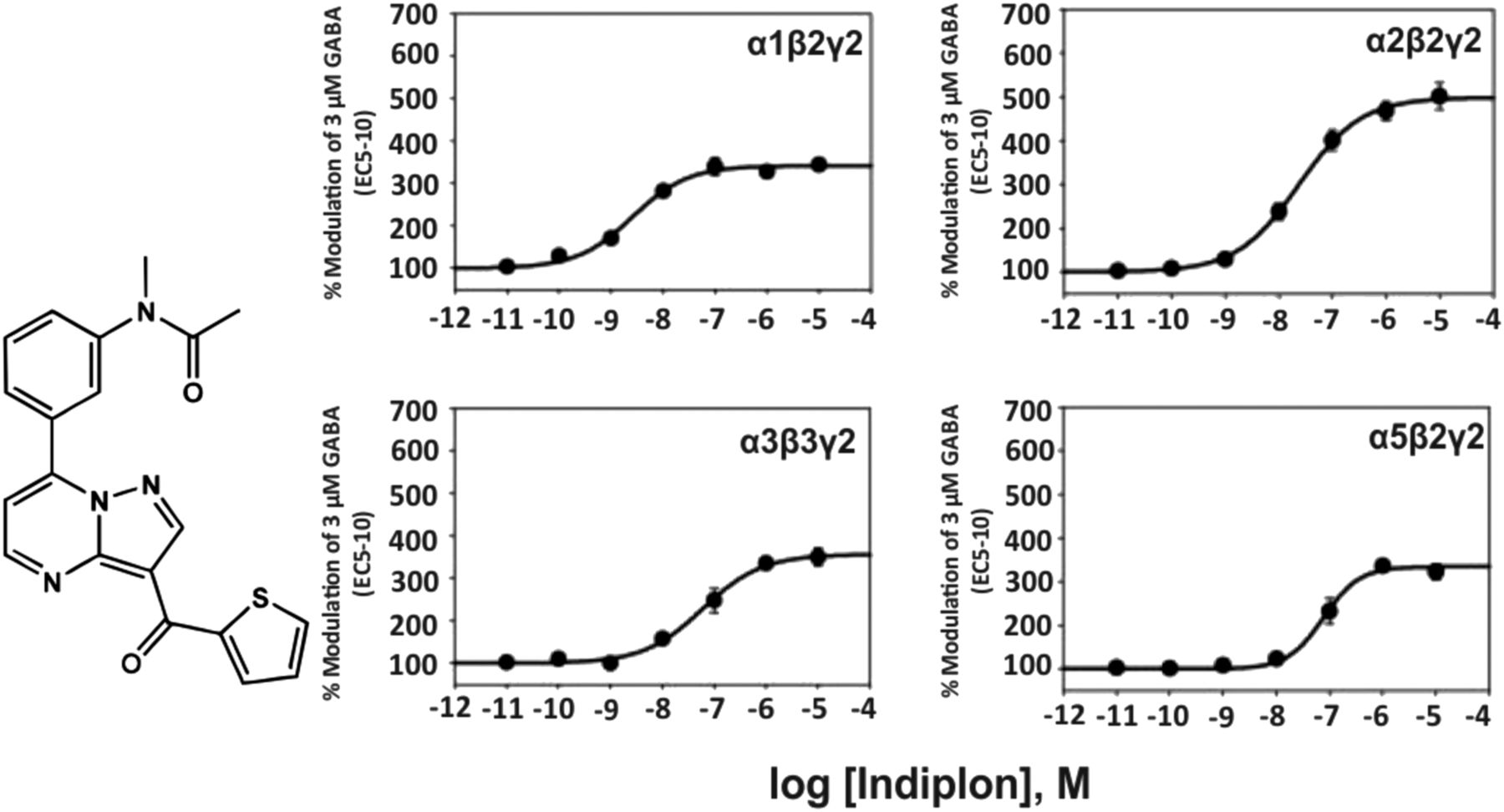
Chemical structure and concentration-response curves of indiplon at various recombinant GABAA receptor subtypes from the rat expressed in HEK 293 cells. GABA concentration was 3 μM (GABA EC5–10) for all recombinant receptors, and data were measured by patch-clamp recording. The EC50 values for allosteric modulation of α1β2γ2, α2β2γ2, α3β3γ2, α5β2γ2 receptors by indiplon were 2.6, 24, 60, and 77 nM, respectively. Figure modified from (Petroski et al., 2006).
3. Zaleplon, Zopiclone
The structurally related pyrazolopyrimidine zaleplon (Fig. 3) exhibits a much lower affinity for the benzodiazepine binding site of recombinant α1β2γ2, α2β1γ2, α3β3γ2, α2β3γ2 receptors from rat (Ki of 66, 830, 710, 1780 nM, respectively) (Dämgen and Lüddens, 1999) and, due to its lower potency in electrophysiological experiments, seems to be α1-selective up to 100 nM, while at 1 µM all receptor subtypes are modulated by zaleplon (Petroski et al., 2006). At 10 μM concentration it is a full benzodiazepine site agonist at α1-3βγ2 receptors and a partial agonist at α5βγ2 receptors. Similar to zolpidem and indiplon, zaleplon exhibits sedative-hypnotic properties in rodents and humans.
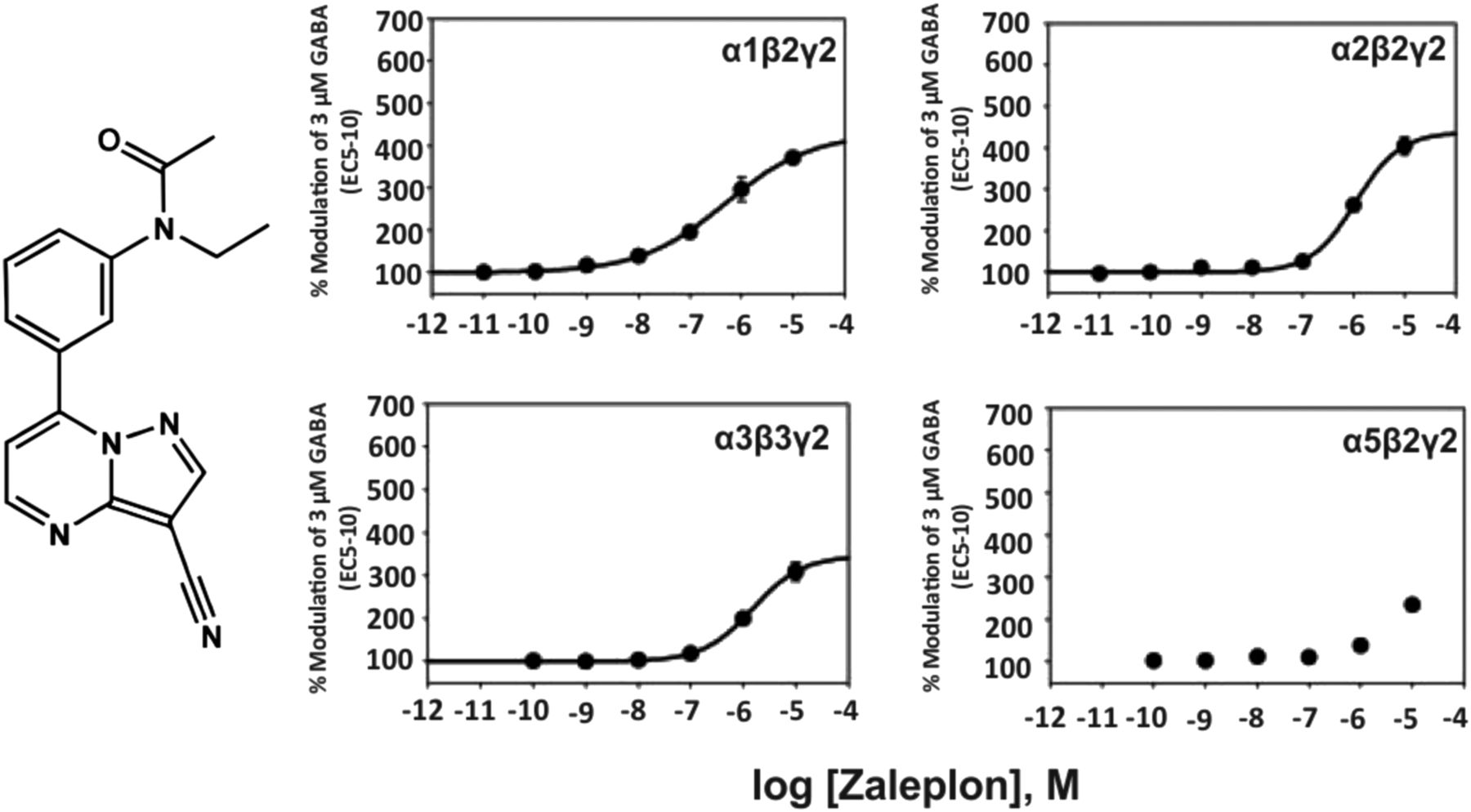
Chemical structure and concentration-response curves of zaleplon at various recombinant GABAA receptor subtypes from the rat expressed in HEK 293 cells. GABA concentration was 3 μM (GABA EC5–10) for all recombinant receptors, and data were measured by patch-clamp recording. The EC50 values for allosteric modulation of α1β2γ2, α2β2γ2, α3β3γ2, or α5β2γ2 receptors by zaleplon were 499, 1098, 1514, or >3000 nM, respectively. Figure modified from (Petroski et al., 2006).
The structurally dissimilar cyclopyrrolone derivative zopiclone, the third so-called “Z” drug after zolpidem and zaleplon, apparently does not exert significant effects up to 10 nM concentrations at recombinant α1β2γ2, α2β2γ2, α3β3γ2, α5β2γ2 receptors from rat (Petroski et al., 2006). At 100 nM concentration, however, it modulates α1β2γ2 receptors to about 300% of GABA EC5–10, and α5β2γ2 to about 250%. The allosteric modulation by 100 nM of zopiclone of recombinant α2β2γ2 or α3β3γ2 receptors, however, was below 200% of GABA EC5–10. The EC50 values for the allosteric modulation of α1β2γ2, α2β2γ2, α3β3γ2, α5β2γ2 receptors by zopiclone, were 158, 598, 1187, and 146 nM, respectively (Petroski et al., 2006). Zopiclone thus is not GABAA receptor subtype-selective at all. It only preferentially modulates α1β2γ2 and α5β2γ2 receptors but also significantly modulates the other GABAA receptor subtypes at a similar concentration. Similar to the other “Z” drugs, zopiclone exhibits sedative-hypnotic properties in rodents and humans. But this compound also exhibits anxiolytic, anticonvulsant, and muscle relaxant properties.
4. β-CCt
The β-carboline-3-carboxylate-t-butyl ester (Fig. 4) was described as a mixed benzodiazepine agonist-antagonist ligand with >10-fold binding selectivity for α1 over α2, and α3 receptors and >110-fold selectivity for α1- over α5-containing receptors (June et al., 2003; Yin et al., 2010). To the best of our knowledge, no concentration-response curves have been published for β-CCt, but from the bar graphs in Fig. 4, it can be deduced that this compound, even at high concentrations, acts more or less as a null modulator (antagonist) at α1- and α2-containing receptors, as a weak positive modulator at α3 and α4 receptors, and a negative modulator at α5 receptors. Thus, despite its at least 10-fold selectivity for α1 receptors in benzodiazepine binding studies, part of the effects of β-CCt are presumably exerted via other GABAA receptor subtypes. Looking at these data, conclusions that the anti-ethanol actions of this compound are primarily mediated via α1-containing receptors (June et al., 2003; Yin et al., 2010) seem not to be sufficiently supported. Nevertheless, in behavioral studies, β-CCt demonstrated clear differences when directly compared with flumazenil (Savić et al., 2004), and it is still used as one of the rare “selective” antagonists at the benzodiazepine binding site (see section V.H).

Chemical structure of β-CCt and bar graph showing the maximal efficacy of this compound as well as that of flumazenil or the β-carboline ZK93426 in modulating rat recombinant α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2, and α5β3γ2 GABAA receptor subtypes expressed in Xenopus laevis oocytes. A saturating concentration (1–10 μM) of β-CCt, flumazenil, or ZK93426 was coapplied over voltage-clamped oocytes along with an EC50 of GABA. Modified from Yin et al. (2010), Bioorg Med Chem, 18,7548-7564, Elsevier, 2010.
5. Ro15-1788 (Flumazenil)
As can be seen in Fig. 4, the imidazobenzodiazepine flumazenil (Ro15-1788) (Fig. 5), which originally was assumed to be a general antagonist at the benzodiazepine site of all GABAA receptor subtypes, acts as an antagonist only at α1β3γ2, but not at other GABAA receptor subtypes. This conclusion is supported by the concentration-response curves of Fig. 5, indicating that flumazenil, like other imidazobenzodiazepines, is also a positive allosteric modulator at α4β3γ2 and α6β3γ2 receptors (Ramerstorfer et al., 2010). The relatively weak positive or negative allosteric modulation of this compound at these other receptors as well as changes in GABAA receptor expression and activity during benzodiazepine treatment or disease states can at least partially (see also section V.H) explain a variety of surprising observations when flumazenil was used in humans as an antagonist of benzodiazepine actions (Nutt, 1983; Hulse et al., 2015).

Chemical structure and concentration-response curves of Ro15-1788 (flumazenil) at various recombinant GABAA receptor subtypes from the rat expressed in Xenopus laevis oocytes and measured at GABA EC3. Modified from Ramerstorfer et al., Eur J Pharmacol, 636,18-27, Elsevier, 2010.
C. Compounds Claimed to Selectively Modulate α2βγ2, α3βγ2, and α5βγ2 Receptors
Since GABAA receptors composed of α1βγ2 subunits seemed to mediate the sedative effects of benzodiazepine site ligands, the development of drugs with reduced or eliminated modulation of these receptors became a high priority of pharmaceutical industry.
1. L-838,417
The triazolopyridazine L-838,417 (Fig. 6) has a similar low nanomolar affinity for the benzodiazepine binding site of recombinant human GABAA receptors composed of α1,2,3,5β3γ2 subunits and expressed in Ltk cells. The affinity for α4β3γ2 or α6β3γ2 receptors was 267 or 2183 nM, respectively. Concentration-response curves indicate that L-838,417 is a positive allosteric modulator with nanomolar potency at α2β3γ2, α3β3γ2, and α5β3γ2 receptors, but is devoid of modulatory actions at α1β3γ2 receptors (Fig. 6). Due to the low affinity at α4- and α6-containing receptors, physiologic effects of this compound possibly mediated through these subtypes were not investigated (McKernan et al., 2000). In contrast to diazepam, L-838,417 did not impair the motor performance of wild-type mice on the rotarod, but it enhanced locomotor activity. This is a sign of behavioral disinhibition that might have been caused by a positive allosteric modulation of α2-containing GABAA receptors under conditions where it is not opposed by a simultaneous modulation of α1 receptors (Ralvenius et al., 2015). At doses that occupied less than 50% of the benzodiazepine binding sites, L-838,417 retained anticonvulsant activity in mice and anxiolytic-like activity in rat. The data indicated that, in rodents, a compound with no efficacy at the α1 subtype does not produce sedation, but retains its anxiolytic-like properties (McKernan et al., 2000).

Chemical structure and concentration-response curves of L838,417 at various human recombinant GABAA receptor subtypes at GABA EC20 expressed in Ltk cells. Figure modified from (McKernan et al., 2000), with permission by Springer Nature.
2. TPA-023B
The imidazotriazine TPA-023B (Fig. 7) exhibits a similar low nanomolar affinity for the benzodiazepine binding site of human GABAA receptor subtypes composed of α1,2,3,5β3γ2 subunits and expressed in Ltk cells. The affinity for α4β3γ2 or α6β3γ2 receptors was 3300 or 4700 nM, respectively. In electrophysiological experiments, it is a comparably strong modulator of GABA EC20 at α2β3γ2, α3β3γ2, and α5β3γ2 receptors and only weakly modulates α1β3γ2 receptors (Atack et al., 2011a). The high receptor occupancy in the rat at a dose that generates significant anxiolytic-like effects (87% at 1 mg/kg) indicates that all these receptors are fully modulated in vivo at a low dose of the drug already. Rotarod performance of rats was not significantly impaired by TPA-023B, even at a dose (10 mg/kg) that gave essentially complete receptor occupancy. Other studies indicated that TPA-023B is a nonsedating anxiolytic in primates (Atack et al., 2011a). The inset in Fig. 7 indicates that TPA-023B exhibits less than half of the maximal efficacy of chlordiazepoxide at α2, α3, and α5 receptors and about 3% of the maximal efficacy of chlordiazepoxide at α1 receptors.

Chemical structure and concentration-response curves of TPA-023B at various human recombinant GABAA receptor subtypes stably expressed in Ltk cells and measured at GABA EC20. Inset shows the maximum efficacy at each receptor subtype relative to that produced by 3 μM of the nonselective full agonist chlordiazepoxide (CDP). Three micromolars chlordiazepoxide potentiated GABA EC20 currents by 105% ± 6%. Figure modified from Atack et al. (2011a), with permission by SAGE Publishing.
3. NS11394, NS16085
The [3′-[5-(1-hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile] NS11394 (Fig. 8) exhibited a subnanomolar affinity for the benzodiazepine site of human α1-, α2-, α3-, and α5β3γ2 receptors, whereas the affinity for α4β3γ2 and α6β3γ2 receptors was weak (Ki of 324 and 1009 nM, respectively). Based on oocyte electrophysiology with human GABAA receptors (GABA EC5–25) relative to 0.5 μM diazepam, NS11394 was claimed to exhibit a functional selectivity profile at GABAA receptors of the α5 > α3 > α2 > α1 order (Mirza et al., 2008). Nevertheless, the concentration-response curves indicate that, despite its distinct efficacy for the individual receptor subtypes, this compound cannot selectively modulate a single GABAA receptor subtype at any concentration. NS11394 behaved as an anxiolytic-like compound with a reduced side-effect profile in rat and mouse, even at full receptor occupancy (Mirza et al., 2008).

Chemical structure and concentration-response curves of NS11394 at various human recombinant GABAA receptor subtypes expressed in Xenopus laevis oocytes and measured at GABA EC5–25. Data are expressed relative to the effects of 0.5 μM diazepam on the same oocyte (Christian et al., 2015) (rel. DZ). Figure modified from Mirza et al. (2008).
The structurally related compound NS16085 (de Lucas et al., 2015), is α2/α3 selective up to a concentration of 3 nM, where it exerts about 10% of the actions of 0.5 μM diazepam at these receptors. In addition, it is a weak negative allosteric modulator at α1-containing receptors and a marginally positive allosteric modulator at α5-containing receptors. Both NS16085 and NS11394 were demonstrated to exert an analgesic action by depressing activity-dependent spinal sensitization after inflammatory injury. These data indicate that potentiation of α2- and α3-GABAA receptors is sufficient and that positive modulation at α5-containing GABAA receptors as with NS11394 is not necessary for inducing this pharmacological effect (de Lucas et al., 2015).
D. Compounds Claimed to Selectively Modulate α2βγ2 and α3βγ2 Receptors
Molecular genetic and pharmacological evidence indicated that α2βγ2 receptors predominantly mediate the anxiolytic effects of diazepam (Löw et al., 2000; Behlke et al., 2016; Engin et al., 2016). In addition, α2βγ2 and α3βγ2 receptors also exhibit antihyperalgesic actions (Knabl et al., 2008; Ralvenius et al., 2015) and at higher concentrations also seem to mediate the muscle relaxant effects of diazepam. Because α1βγ2 receptors seemed to mediate the sedative effects (Rudolph et al., 1999; McKernan et al., 2000) and α5βγ2 receptors the cognition-impairing effects of classic benzodiazepines (Collinson et al., 2002; Crestani et al., 2002), avoiding modulation of both of these receptor types was another major goal of pharmaceutical industry.
1. Compound 4
The first compound claimed to be α2 selective was compound 4, a modified quinolone antibiotic that exhibited anxiolytic-like properties but did not cause sedation. It produced stronger effects than L-838,417 at α2-GABAA receptors but did not act via the benzodiazepine binding site (Johnstone et al., 2004). Unfortunately, however, only α1β2γ2 and α2β2γ2 receptors were investigated in this study, and to the best of our knowledge, no further information on this compound is available in the literature.
2. Pyridazine Series of α2βγ2- and α3βγ2-selective Compounds
In addition to the compounds mentioned in the previous section, some anxiolytic-like pyridazine compounds with no overt signs of ataxia or sedation have been identified that depending on their exact structure exhibit α2βγ2, α3βγ2, and α5βγ2 selectivity (compound 15); α2βγ2 and α3βγ2 selectivity (compound 14, Fig. 9); or α3βγ2 selectivity (compound 16). These ligands exhibit no modulatory activity at the α1 subtype, a good central nervous system penetration and receptor occupancy, and excellent oral bioavailability (Lewis et al., 2006). To the best of our knowledge, no additional studies have been reported on these compounds.

Chemical structure and concentration-response curves of compound 14 at various recombinant human GABAA receptor subtypes stably expressed in L(tk-) cells measured at GABA EC20. Figure modified from Lewis et al. (2006), with permission of The American Chemical Society.
3. SL-651,498
The pyridoindole derivative SL-651,498 (Fig. 10) exhibited a differential affinity for the benzodiazepine site of rat α1-, α2-, α3-, or α5β3γ2 receptors (Ki of 17, 73, 80, or 215 nM, respectively) and based on electrophysiological measurements was claimed to be an α2-, α3-selective positive allosteric modulator (Griebel et al., 2001). But the functional concentration-response curves at various GABAA receptor subtypes, relative to zolpidem (α1β2γ2) or diazepam (α2β2γ2, α3β2γ2, and α5β3γ2), provide evidence that this compound is only α2,α3 selective up to a 10 nM concentration. In rats, this compound administered at 1–10 mg/kg elicited anxiolytic-like activity similar to that of diazepam and induced muscle weakness, ataxia, or sedation at substantially higher doses (≥30 mg/kg) (Griebel et al., 2001).
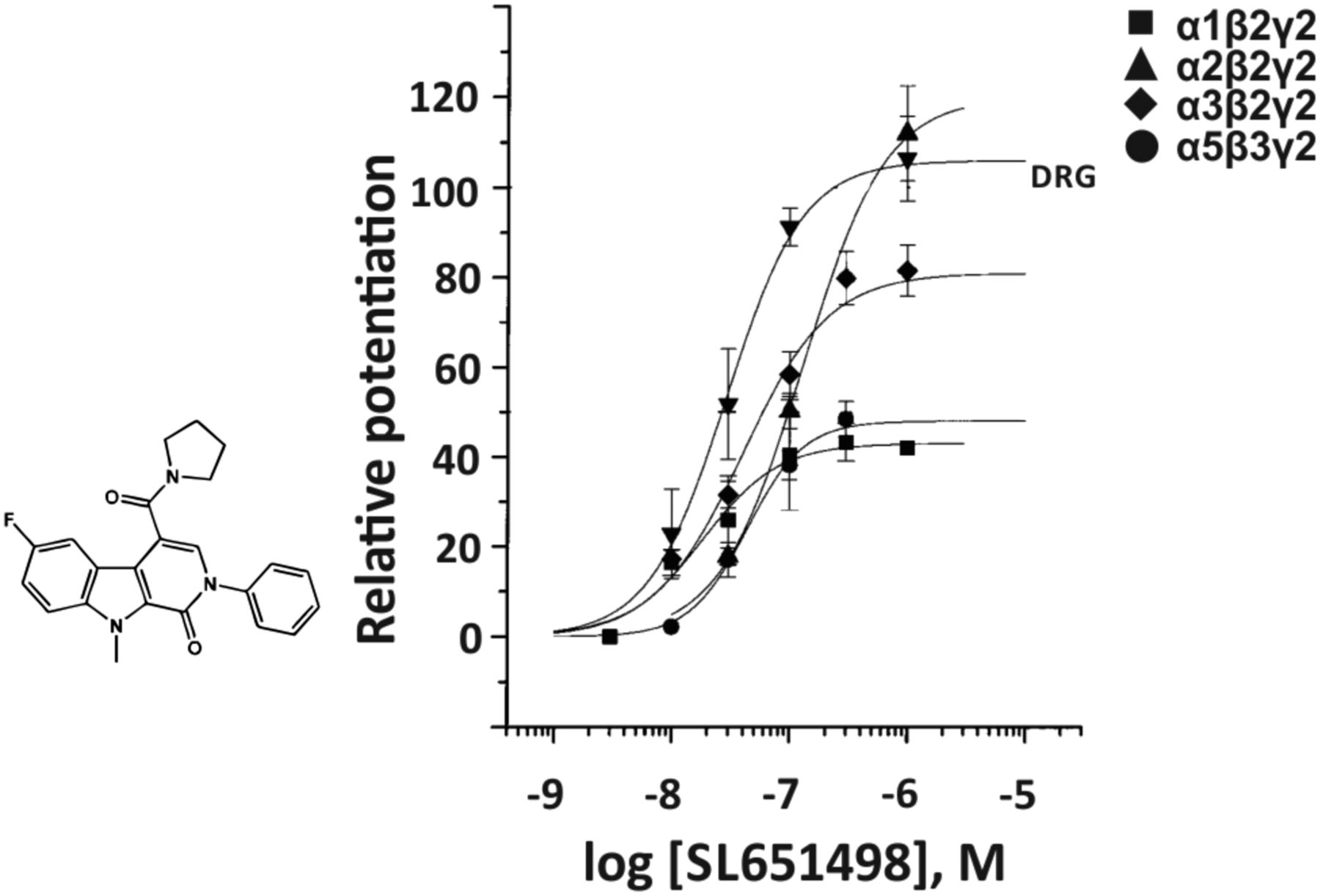
Chemical structure and concentration-response curves of SL-651,498 at various rat recombinant GABAA receptor subtypes stably expressed in HEK 293 cells measured using whole cell patch-clamp at GABA EC5–10 and presented relative to zolpidem (α1β2γ2) or diazepam (α2β2γ2, α3β2γ2, and α5β3γ2). DRG indicates the concentration-response curve of SL-651,498 at dorsal root ganglia neurons in culture that contain exclusively native α2-containing GABAA receptors. Figure modified from Griebel et al. (2001).
4. TPA023
The triazolopyridazine TPA023 (Fig. 11) has a subnanomolar affinity for the benzodiazepine binding site of α1-, α2-, α3-, or α5β3γ2 receptors and a reduced affinity for α4β3γ2 or α6β3γ2 receptors (Ki of 60 or 418 nM, respectively). It was claimed to be selective for α2β3γ2 and α3β3γ2 receptors (Atack et al., 2006b). However, this compound already produces a subnanomolar subtle positive modulation at α5β3γ2 receptors. At 1 nM it starts to positively modulate α3β3γ2 and at 10 nM concentrations also α2β3γ2 receptors. Thus there is no concentration where this compound exhibits pure α2/α3 selectivity. This conclusion is supported by other researchers (Christian et al., 2015; de Lucas et al., 2015), who identified even stronger interactions of this compound with other receptor subtypes and indicated that in their hands TPA023 exhibited only marginal selectivity; see Fig. 11B (de Lucas et al., 2015) or Fig. 11, C and D (Christian et al., 2015). In rats, a 50% occupancy of TPA023 corresponded to an oral dose of 0.42 mg/kg. TPA023 produced anxiolytic-like effects in rodents at minimal effective doses of 1–3 mg/kg corresponding to 70%–88% occupancy, while there was no appreciable sedation up to 30 mg/kg (Atack et al., 2006b). The inset in Fig. 11A indicates that the maximal efficacy elicited by TPA023 relative to 3 μM chlordiazepoxide is quite weak at the various GABAA receptor subtypes. This conclusion is confirmed by Fig. 11B, which presents modulation of GABA EC10 by TPA023 relative to the modulation by 0.5 μM diazepam, or by Fig. 11D, which presents modulation of GABA EC10 by TPA023 relative to 1 μM diazepam.

Chemical structure and concentration-response curves of TPA023. (A) Figure modified from Atack et al. (2006b), generated at various recombinant human GABAA receptor subtypes (containing β3 subunits) stably expressed in L(tk-) cells measured at GABA EC20. Inset shows the maximum efficacy of TPA023 at each subtype relative to that produced by 3 μM of the nonselective full agonist chlordiazepoxide (CDP). (B) Figure modified from de Lucas et al. (2015), Biochemical Pharmacology, 93, 370-379, Elsevier, 2015, generated at various recombinant human GABAA receptor subtypes (containing β2 subunits) expressed in Xenopus laevis oocytes measured at GABA EC10 relative to 0.5 μM diazepam in the same oocyte. Inset shows % of modulation of GABA EC10 by 0.5 μM diazepam at the individual receptors. The percentages given at the right of the curves represent the Emax elicited by TPA023 as % of the effect of 0.5 μM diazepam (de Lucas et al., 2015). (C and D) Figures modified from Christian et al. (2015) with permission by the American Physiological Society, generated with TPA023 at various recombinant human GABAA receptor subtypes (containing β3 subunits) expressed in Xenopus laevis oocytes and measured at GABA EC10. (C) Data represent % modulation of GABA EC10 by TPA023. (D) Data represent % modulation of GABA EC10 by TPA023, and normalized to the current potentiation produced by 1 μM diazepam that elicited a near maximal response.
5. MRK-409
The structurally related triazolopyridazine MRK-409 (Fig. 12) has a comparable subnanomolar affinity for the benzodiazepine binding site of recombinant human α1β3γ2, α2β3γ2, α3β3γ2, α5β3γ2 GABAA receptor subtypes and a lower affinity for α4β3γ2 or α6β3γ2 receptors (Ki = 78 or 980 nM, respectively). In electrophysiological studies it is relatively selective for α3 receptors compared with α1β3γ2, α2β3γ2, and α5β3γ2 receptors at subnanomolar but not at higher concentrations (Fig. 12). MRK-409 produced anxiolytic-like activity in rodents and primates, with minimum effective doses corresponding to occupancies from 35% to 65%, depending on the particular model used, and first overt signs of sedation at occupancies greater than 90% (Atack et al., 2011b). The inset in Fig. 12 indicates that MRK-409, relative to 3 μM chlordiazepoxide, behaves as a weak to moderate partial agonist at the benzodiazepine binding site of diazepam-sensitive GABAA receptor subtypes.

Chemical structure and concentration-response curves of MRK-409 at various recombinant human GABAA receptor subtypes stably expressed in mouse L(tk-) cells, measured using whole cell patch-clamp electrophysiology at GABA EC20. Inset shows maximal potentiation at each cell type expressed relative to that produced by 3 μM of the nonselective full benzodiazepine site agonist chlordiazepoxide. Figure modified from Atack et al. (2011b) with permission by SAGE Publishing.
6. SH-053-2′-N (HZ166), MP-III-024, KRM-II-81
The imidazobenzodiazepine SH-053-2′-N (Fig. 13) exhibits a moderate affinity for the benzodiazepine site of GABAA receptor subtypes (Ki of 300, 160, 527, or 82 nM for α1β3γ2, α2β3γ2, α3β3γ2, or α5β3γ2 receptors, respectively) (Fischer et al., 2010) and was claimed to be an α2/α3 GABAA receptor-selective benzodiazepine. MP-III-024, the methyl ester analog of the ethyl ester HZ166, exhibited a slightly lower efficacy than HZ166, but a similar preferential activity at α2β3γ2 and α3β3γ2 receptors (Fischer et al., 2017). KRM-II-81, a derivative of HZ166 carrying an oxazole ring instead of the ethyl ester of HZ166, exhibited a higher potency and efficacy for α2β3γ2 and α3β3γ2 receptors (Lewter et al., 2017). However, the respective concentration-response curves indicate that all three of these compounds already at 100 nM concentrations significantly modulate α1 and α5 receptors in addition to α2/α3 receptors (Rivas et al., 2009; Fischer et al., 2010, 2017; Lewter et al., 2017) (Fig. 13). MP-III-024 and KRM-II-81 exhibited significant antinociceptive effects (Fischer et al., 2017; Lewter et al., 2017). HZ166 produced some anticonvulsive (Rivas et al., 2009), anxiolytic-like (Savić et al., 2010), and antihyperalgesic effects (Di Lio et al., 2011), while it was devoid of sedation and motor impairment in rodents in some (Rivas et al., 2009; Di Lio et al., 2011), but not all, studies (Savić et al., 2010). In the latter study, the magnitude of the hypolocomotor effect of 30 mg/kg SH-053-2′-N in rats was somewhere in the middle between the effects of two tested doses of diazepam (1.25 and 2.5 mg/kg) (Savić et al., 2010).
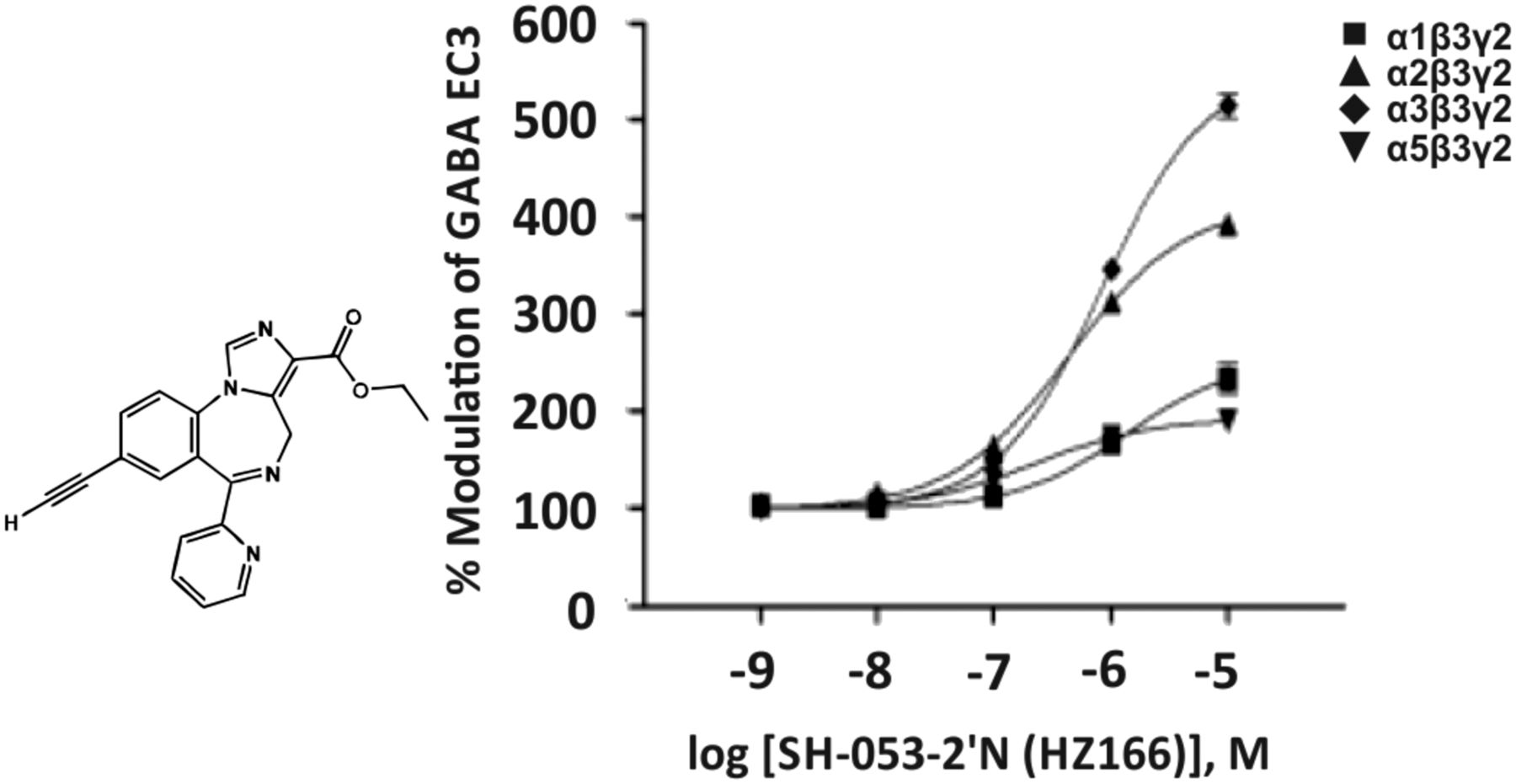
Chemical structure and concentration-response curves of SH-053-2′-N (HZ166) at various rat recombinant GABAA receptor subtypes expressed in Xenopus laevis oocytes and measured at GABA EC3. Figure modified from Rivas et al. (2009) with permission of The American Chemical Society.
7. Baicalin
A variety of flavonoids have been demonstrated to interact with the benzodiazepine site of GABAA receptors and some of them also have some preferences for certain GABAA receptor subtypes (Furtmueller et al., 2008; Wang et al., 2008; Karim et al., 2012a). The flavonoid baicalin (Fig. 14) has been isolated from the traditional Chinese herb Huangqin, the dry root of Scutellaria baicalensis Georgi, and interacts with the benzodiazepine binding site of GABAA receptors with a poor Ki value of 77.1 μM. It produced anxiolytic-like effects in a Vogel conflict test and elevated plus maze test, and was devoid of sedation, myorelaxation, anticonvulsant, amnesic, and motor incoordination effects. In whole cell patch-clamp studies at 1 μM concentrations, baicalin showed significant preference for α2- and α3-containing compared with α1- and α5-containing GABAA receptor subtypes (Wang et al., 2008). However, baicalin in addition also is a known prolyl endopeptidase inhibitor (Tarragó et al., 2008) and induces apoptosis in pancreatic cancer cells (Takahashi et al., 2011).

Chemical structure of baicalin and concentration-response curves of diazepam (DZ, open circles) and baicalin (filled triangles) on various human recombinant GABAA receptor subtypes expressed in HEK 293T cells under whole cell patch-clamp at GABA EC20. Modified from Wang et al., Neuropharmacology, 55:1231–1237, Elsevier, 2008.
8. 6-Hydroxyflavone
The flavonoid 6-hydroxyflavone (Fig. 15) has a quite moderate affinity of 1.3–4.9 μM for the benzodiazepine binding site of human diazepam-sensitive GABAA receptor subtypes expressed in HEK 293T cells (Ren et al., 2010) and preferentially modulates α2- and α3-containing GABAA receptors in electrophysiological experiments. This compound produced anxiolytic-like effects in mice without the side effects observed with classic benzodiazepines (Ren et al., 2010). It should be kept in mind, however, that flavonoids can mediate their effects via multiple binding sites at GABAA receptors (Hanrahan et al., 2015) and that the actions of flavonoids are not necessarily limited to GABAA receptors. Depending on their structure they also can interact with nicotinic acetylcholine receptors, serotonin type 3A receptors, glutamate AMPA/kainate receptors and others (Johnston and Beart, 2004).
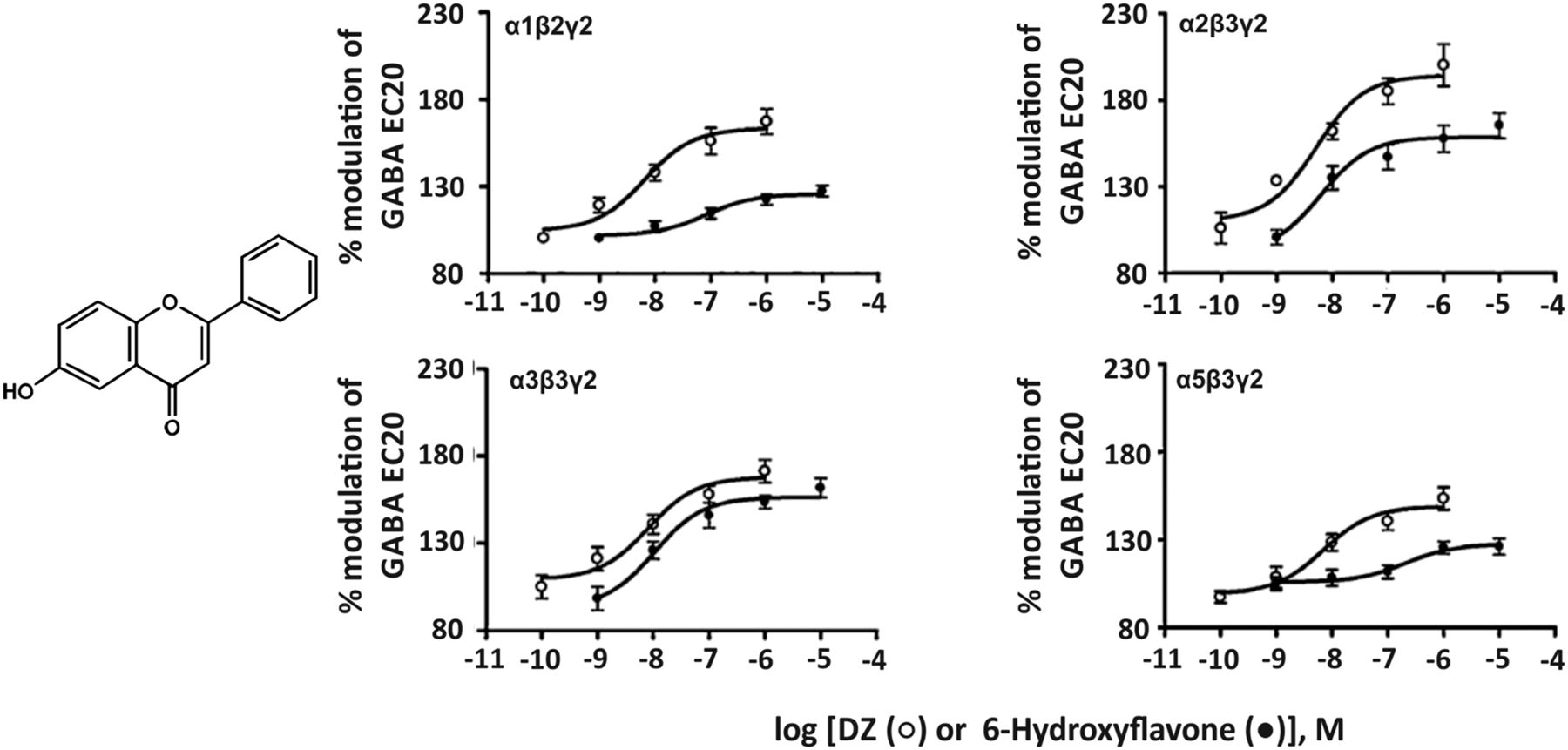
Chemical structure of 6-hydroxyflavone and concentration-effect curves of diazepam (DZ, open circles) and 6-hydroxyflavone (filled circles) on human recombinant GABAA receptor subtypes expressed in HEK 293T cells under whole cell patch-clamp at GABA EC20. Modified from Ren et al., Biochemical Pharmacology 79: 1337–1344, Elsevier, 2010.
9. Fa131
The flavan-3-ol derivative Fa131 (Fig. 16) does not inhibit [3H]flunitrazepam binding to rat cortical membranes. However, at low micromolar concentrations it is a positive allosteric modulator of human recombinant α1,2,3,5β2γ2L and α1β2 receptors expressed in Xenopus laevis oocytes, and this enhancement is not mediated via the benzodiazepine site, as it could not be blocked by the benzodiazepine site antagonist flumazenil (Fernandez et al., 2008). Fa131 preferentially modulates α2 receptors below 3 μM concentrations and induces anxiolytic-like but no sedative effects in rodents. This compound highlights the potential of targeting nonbenzodiazepine allosteric sites in the search for new anxioselective drugs.

Chemical structure and concentration-response curves of Fa131 at various human recombinant GABAA receptor subtypes expressed in Xenopus laevis oocytes and measured at GABA EC5. Modified from Fernandez et al., Neuropharmacology, 55: 900–907, Elsevier, 2008.
10. AZD7325
The cinnoline-carboxamide AZD7325 (Fig. 17) was claimed to be an α2/α3 GABAA receptor-selective partial modulator acting via the benzodiazepine site (Christian et al., 2015; Jucaite et al., 2017). Concentration-response curves at GABA EC10 and human αxβ3γ2 receptors expressed in Xenopus laevis oocytes indicate, however, that α5 receptors also are modulated to a small extent already at nanomolar concentrations (Christian et al., 2015). Positron emission tomography studies demonstrated that up to 80% receptor occupancy could be reached in the human brain by AZD7325 for its anxiolytic effects without overt sedation or cognitive impairment. The declared lack of side effects in humans can be explained by an insufficient modulation of α1- and α5-containing receptors under these conditions (Jucaite et al., 2017).

Chemical structure and concentration-response curves of AZD7325 at various human recombinant GABAA receptor subtypes expressed in Xenopus laevis oocytes and measured at GABA EC10. (A) % Modulation of GABA EC10. (B) The same data relative to the current potentiation produced by 1 μM diazepam, which elicited near maximal response at all receptor subtypes investigated. Figure modified from Christian et al. (2015) with permission by the The American Physiological Society.
E. Compounds Claimed to Selectively Modulate α3βγ2 Receptors
So far, no reasonably investigated compound has been identified that modulates only α2βγ2 GABAA receptors with a certain selectivity, but several compounds have been identified that were claimed to selectively modulate α3β3γ2 receptors.
1. TP003
The imidazopyridin-3-yl-biphenyl-2-carbonitrile TP003 (Fig. 18) has a comparable subnanomolar affinity for the benzodiazepine binding site of recombinant human α1-, α2-, α3-, and α5β3γ2 GABAA receptors (Ki between 0.3 and 0.5 nM), and a low affinity for α4- or α6β3γ2 receptors (Ki of 2.4 or 1.8 μM, respectively) and was declared to be an α3β3γ2-selective positive allosteric modulator (Dias et al., 2005). The complete functional concentration-response curves (at GABA EC20, referred to the potentiation of 3 μM chlordiazepoxide) indicate, however, that due to the exceptionally high potency of this compound this at best holds true only at concentrations up to 1 nM (Fig. 18A); even before 10 nM, TP003 additionally modulates α2β3γ2 and α5β3γ2 receptors. More recently, stronger effects of TP003 at various receptor subtypes were demonstrated (de Lucas et al., 2015) (Fig. 18B) when GABA EC10 was used and it was concluded that TP003 exhibits only marginal receptor subtype selectivity. In Table 1 of Christian et al. (2015), data are presented indicating that TP003 exhibits no selectivity at all for α3-containing GABAA receptor subtypes.

Chemical structure and concentration-response curves of TP003. (A) Figure modified from Dias et al. (2005) with permission of the Society of Neuroscience, generated at various human αxβ3γ2 recombinant GABAA receptor subtypes stably expressed in L(tk-) cells and measured at GABA EC20. Data presented are referred to the potentiation of 3 μM chlordiazepoxide. The inset histogram shows % modulation of GABA EC20 by 3 μM chlordiazepoxide (CDP) at these receptor subtypes. (B) Figure modified from de Lucas et al., Biochemical Pharmacology 93, 370–379, Elsevier, 2015, generated at various human recombinant GABAA receptor subtypes (containing β2 subunits in this case) expressed in Xenopus laevis oocytes and measured at GABA EC10 relative to 0.5 μM diazepam in the same oocyte. Inset shows % of modulation of GABA EC10 by 0.5 μM diazepam at the individual receptors. The percentages given at the right of the curves represent the Emax elicited by TP003 as % of the effect of 0.5 μM diazepam.
Nevertheless, based on the apparent selectivity of TP003 for α3-containing receptors and on experiments indicating that TP003 generated anxiolytic-like effects even in mice with a point mutation that renders α2-containing receptors benzodiazepine insensitive (Dias et al., 2005), it was claimed that α3-containing receptors have a significant role in mediating the anxiolytic effects of benzodiazepines. However, in contrast to diazepam, which not only is inactive at the αβγ2 GABAA receptor subtypes carrying the point mutation α2H101R but also at receptors carrying the mutations α1H101R, α3H126R, or α5H105R that are also used for demonstrating the function of the respective receptor subtypes in the brain (Rudolph and Knoflach, 2011; Rudolph and Möhler, 2014), other ligands of the benzodiazepine site, such as bretazenil or Ro15-4513, are even more active at all these point-mutated receptors (Benson et al., 1998). The anxiolytic effects of TP003 in α2-point-mutated mice thus cannot be interpreted in the absence of evidence that TP003 is really inactive at recombinant α2H101Rβγ2 receptors. Moreover, due to the only marginal selectivity of TP003, the involvement of other receptor subtypes such as α5-containing receptors in anxiolytic-like activity (Behlke et al., 2016), as well as the absence of molecular genetic evidence indicating an involvement of α3β2γ2 receptors in the anxiolytic-like action (Löw et al., 2000), it has to be concluded that an anxiolytic-like role of α3-containing GABAA receptors is not supported by the available data.
2. YT-III-31
The imidazobenzodiazepine YT-III-31 (Fig. 19) preferentially modulates the recombinant α3β3γ2 GABAA receptor subtype (Namjoshi et al., 2013). Although the concentration-response curves at first sight seem to point to a high selectivity for α3 receptors, this compound also significantly modulates most investigated receptor subtypes at 10 nM concentrations. In rats, YT-III-31 produced anxiolytic-like actions in a narrow dose range (<10 mg/kg) but sedation at higher doses (Batinić et al., 2018). The concentration-response curves of Fig. 19 predict moderate potentiation of α3 as well as α2 and α5 receptors before sedative effects mediated via α1 receptors are activated.

Chemical structure and concentration-response curves of YT-III-31 at various rat recombinant GABAA receptor subtypes expressed in Xenopus laevis oocytes and measured at GABA EC3. Modified from Namjoshi et al., Bioorg Med Chem, 21, 93–101, Elsevier, 2013.
3. α3IA
The pyridone α3IA (Fig. 20) has a modest affinity for the benzodiazepine binding sites of α1-, α2-, α3-, or α5β3γ2 receptors (Ki = 1029, 323, 82, or 410 nM, respectively, and a very weak affinity for that of α4- or α6β3γ2 receptors (Ki > 10,000 nM) and was claimed to be an α3β3γ2 receptor-selective negative allosteric modulator (Atack et al., 2005). Concentration-response curves measured at GABA EC20, however, indicate that this only holds true up to a concentration of 30 nM, as at 100 nM concentrations, receptors composed of α1β3γ2 and α2β3γ2 subunits also become negatively modulated by this drug. At doses that produce relatively low levels of occupancy (12%) in the rat cerebellum, a brain region that contains predominantly α1- and α6-containing GABAA receptors and only 2% of α3- and 7% of α2-containing receptors (Pöltl et al., 2003), this compound elicited an anxiogenic-like effect similar to FG7142, and this effect could be blocked by the benzodiazepine site antagonist flumazenil (Atack et al., 2005). From the concentration-response curves in Fig. 20 it can be concluded that the concentration eliciting a 12% occupancy of α1 receptors amounts to >100 nM α3IA. At that concentration, α3IA elicits a similar negative allosteric modulation at α3β3γ2 receptors as the anxiogenic compound FG7142.

Chemical structure and concentration-response curves of α3IA at various human recombinant αxβ3γ2 GABAA receptor subtypes stably expressed in L(tk-) mouse fibroblast cells and measured at GABA EC20. The shaded areas represent the range of inverse agonist efficacies across subtypes for the nonselective partial inverse agonists FG7142 or the nonselective full inverse agonist DMCM. Figure modified from Atack et al. (2005). Reprinted with permission of John Wiley & Sons, Inc.
F. Compounds Claimed to Selectively Modulate α5βγ2 Receptors
A combination of molecular genetic and pharmacological approaches indicated that α5βγ2 receptors mediate the unwanted cognitive effects of diazepam and that a negative modulation of these receptors enhances learning and memory (Collinson et al., 2002; Crestani et al., 2002). Other experiments indicated that reducing excessive tonic inhibition by negative allosteric modulators at α5βγ2 receptors may promote functional recovery from stroke (Clarkson et al., 2010). Therefore, the main effort of researchers was directed to the development of negative allosteric modulators of α5βγ2 receptors. However, recent evidence indicated beneficial effects of positive allosteric modulators at α5-containing GABAA receptors on cognition in the aging brain (Koh et al., 2013), in schizophrenia (Gill and Grace, 2014), or in neuropsychiatric disorders characterized by cognitive deficits due to impaired memory interference management (Engin et al., 2015). These observations point to a more complex bidirectional modulation of cognition by α5-containing GABAA receptors and provide new impetus also to develop selective positive allosteric modulators at α5βγ2 receptors. This impetus is further enhanced by recent findings that positive allosteric modulators at α5-containing GABAA receptors have beneficial effects in the treatment of asthma (Gallos et al., 2015) or medulloblastomas (Jonas et al., 2016).
1. SH-053-R-CH3-2′F, MP-III-022
One of the first relatively α5β3γ2-selective positive allosteric modulators reported with concentration-response curves was the imidazobenzodiazepine SH-053-R-CH3-2′F (Savić et al., 2010) (Fig. 21). This compound has a moderate affinity for the benzodiazepine binding site of α5 receptors (Ki = 95.2 nM) and a low affinity for α1-, α2-, or α3β3γ2 receptors (Ki = 759, 948, or 768 nM, respectively) (Fischer et al., 2010). In electrophysiological experiments, SH-053-R-CH3-2′F is selective for α5 receptors up to a 30 nM concentration. At 100 nM concentration, this compound significantly enhances GABA EC3 currents at α1β3γ2, α2β3γ2, α3β3γ2, and α5β3g2 receptors from 100% to 111%, 124%, 125%, and 183%, respectively (Fischer et al., 2010; Savić et al., 2010). At doses up to 30 mg/kg, SH-053-R-CH3-2′F depressed locomotion but did not induce cognitive impairment or anxiolytic-like activity (Savić et al., 2010). In addition, it was demonstrated to relax precontracted intact airway smooth muscle cells (Gallos et al., 2015).

Chemical structure and concentration-response curves of SH-053-R-CH3-2′F at various rat recombinant GABAA receptor subtypes expressed in Xenopus laevis oocytes and measured at GABA EC3. Modified from Savic et al., Prog Neuropsychopharmacol Biol Psychiatry, 34:376–386, Elsevier, 2010.
Ester to amide substitution in SH-053-R-CH3-2′F led to MP-III-022, with improved selectivity, efficacy, and kinetic behavior as a positive modulator of GABAA receptors containing the α5 subunit (Stamenić et al., 2016). While at doses 1–10 mg/kg it was devoid of ataxia, sedation, or an influence on the extent of anxiety-related behavior in rats, at the dose of 10 mg/kg MP-III-022 caused a strong positive modulation of α5β3γ2 receptors and mild, but significant, muscle relaxation (Stamenić et al., 2016).
2. α5IA
The triazolopyridazine α5IA (Fig. 22) was demonstrated to bind with equivalent subnanomolar affinity (0.5–0.9 nM) to the benzodiazepine binding site of recombinant human GABAA receptors containing α1-, α2-, α3-, or α5β3γ2 subunits and possessed much lower affinity (Ki = 60 or 418 nM, respectively) for receptors containing α4 or α6β3γ2 subunits. α5IA was declared to be an α5 receptor-selective negative allosteric modulator (Dawson et al., 2006), but the data presented for human recombinant αβ3γ2 GABAA receptors indicate that there was no concentration where this compound acted exclusively via α5 receptors (Fig. 22). Interestingly, however, data obtained from different expression systems [Xenopus oocytes or L(tk-) cells stably expressing the same human receptors), depending on the receptor subtype investigated were found to be significantly different (Dawson et al., 2006). A possible additional action of this compound via α4β3γ2 receptors was not investigated. Other studies indicated that with oral doses of 0.1, 1.0, and 10 mg/kg, GABAA receptor occupancies in Sprague-Dawley rat brain were 27%, 79%, and 87%, respectively, 2 hours after oral administration (Atack et al., 2009a). This compound significantly enhanced performance in a rat hippocampal-dependent test of learning and memory with a minimum effective oral dose of 0.3 mg/kg. It was not convulsant or anxiogenic in rodents and did not impair performance in the mouse rotarod test (Dawson et al., 2006).
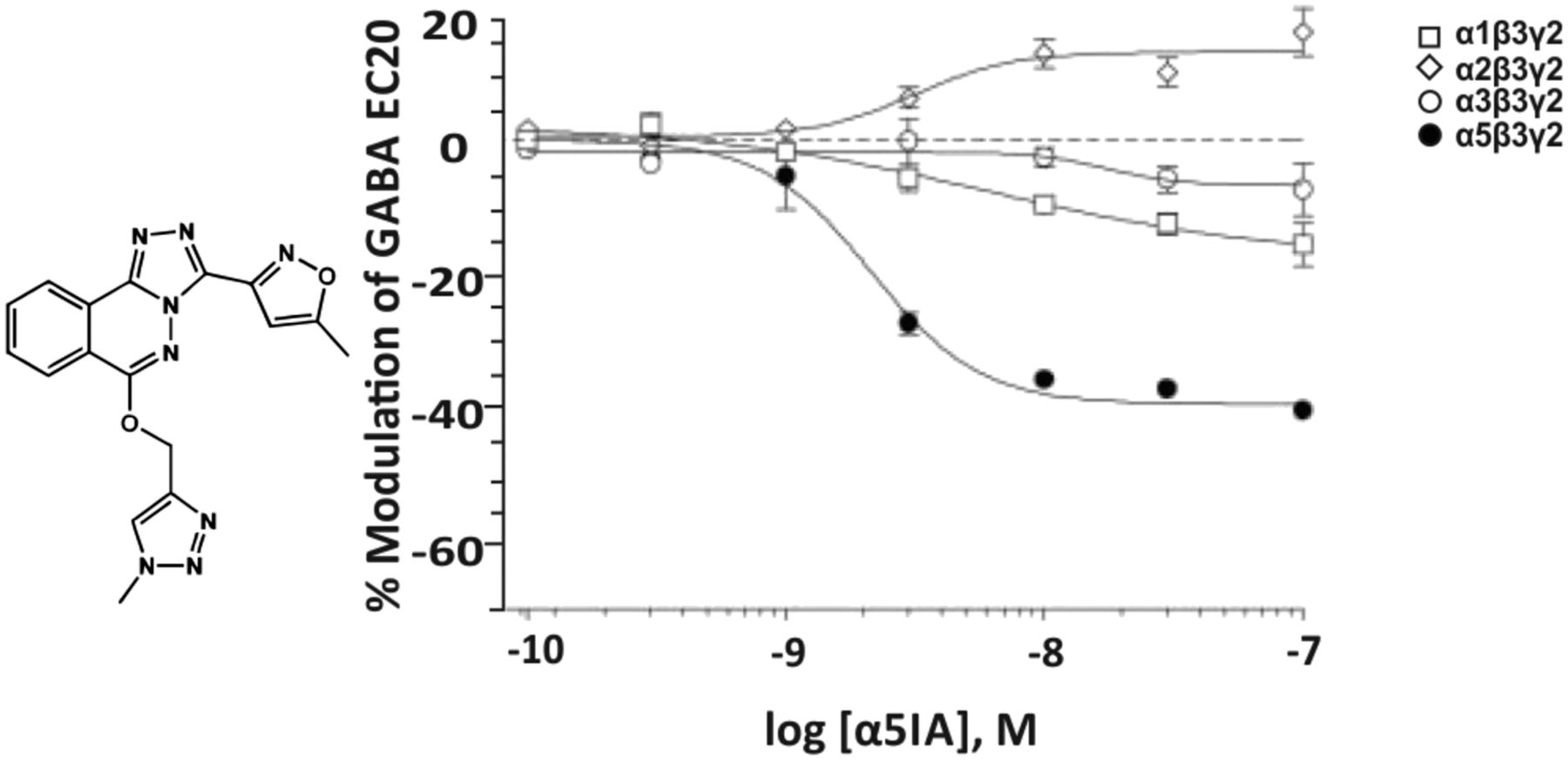
Chemical structure and concentration-response curves of α5IA at various human recombinant αxβ3γ2 GABAA receptor subtypes stably expressed in L(tk-) mouse fibroblast cells and measured at GABA EC20. Figure modified from Dawson et al. (2006).
3. α5IA-II
The structurally related triazolopyridazine α5IA-II (Fig. 23) exhibited a comparably high affinity for the benzodiazepine site of α1-, α2-, α3-, or α5β3γ2 receptors (Ki = 1.4, 2.7, 1.4, or 0.8 nM, respectively). The affinity for α4- or α6β3γ2 receptors was not investigated. α5IA-II was claimed to be a selective negative allosteric modulator at α5βγ2 receptors (Collinson et al., 2006). Nevertheless, there is no concentration where this compound modulates only α5βγ2 receptors. Occupancy studies indicated that a dose of 1 mg/kg α5IA-II produced sustained and high level occupancy in rats, with maximum occupancy (80%) being achieved within 15 minutes of dosing. Assuming that receptor occupation parallels receptor modulation, this indicates that an in vivo application of this drug at 1 mg/kg probably is able to modulate not only α5β3γ2 receptors. Behavioral studies indicated that α5IA-II induced an enhancement of cognitive performance in the Morris water maze, affecting encoding and recall but not the consolidation phases of performance (Collinson et al., 2006).
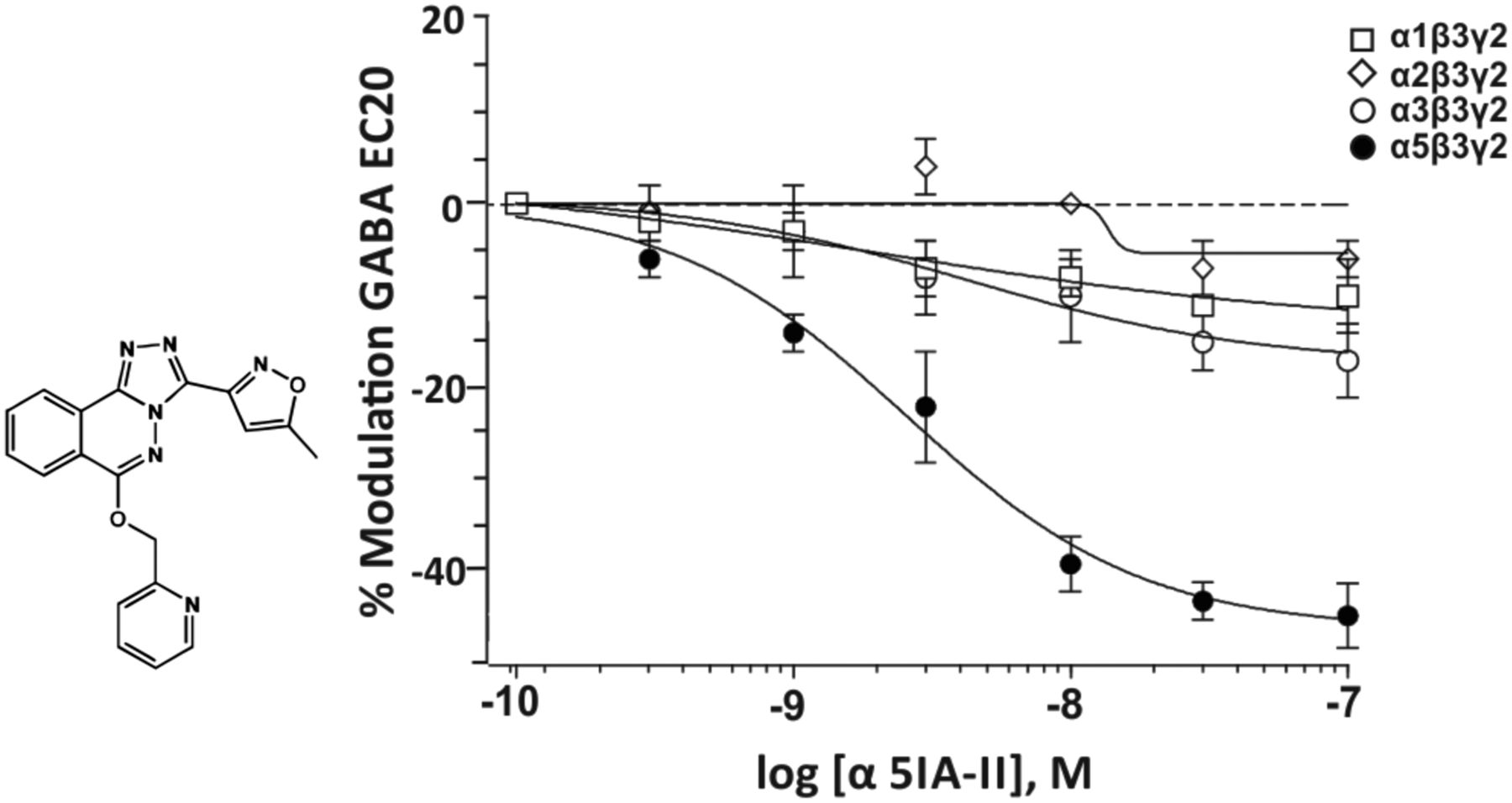
Chemical structure and concentration-response curves of a5IA-II at various human recombinant GABAA receptor subtypes stably expressed in L(tk-) mouse fibroblast cells and measured at GABA EC20. Adapted with permission, Springer, Psychopharmacology (Berl), Collinson et al. (2006).
4. MRK-016
The pyrazolotriazine MRK-016 (Fig. 24) exhibited a high and essentially equivalent affinity for the benzodiazepine binding site of human recombinant GABAA receptors composed of α1-, α2-, α3-, or α5β3γ2 subunits (Ki value range, 0.83–1.4 nM). The affinity was much lower for α4- or α6β3γ2 receptors (Ki = 400 or 4100 nM, respectively). MRK-016 was declared to be a selective negative modulator at α5 receptors (Atack et al., 2009b). However, there is no concentration where this compound only modulates α5β3γ2 receptors (Fig. 24). Other experiments indicated that the maximal occupancies of GABAA receptors in the rat brain after 1, 3, or 10 mg/kg orally, measured 0.5 hour after dosing, were 79%, 81%, and 91%, respectively, again suggesting that α5β3γ2 receptors were not exclusively modulated in vivo under these conditions (Atack et al., 2009b). MRK-016 increased long-term potentiation in mouse hippocampal slices to a greater extent than α5IA, consistent with its greater α5-inverse agonism, and enhanced cognitive performance in the Morris water maze. In mice, it was not anxiogenic, not proconvulsant, and did not produce kindling. Nevertheless, it was poorly tolerated in elderly subjects, and this precluded its further development (Atack et al., 2009b). Given the recent evidence that negative allosteric modulators at α5-containing GABAA receptors improve cognitive function in young, but not in aged, rats (Koh et al., 2013), it is not surprising that this extremely strong negative allosteric modulator at α5 receptors was poorly tolerated in elderly subjects.

Chemical structure and concentration-response curves of MRK-016 at various human recombinant GABAA receptor subtypes stably expressed in L(tk-) mouse fibroblast cells and measured at GABA EC20. Figure modified from Atack et al. (2009b).
5. L-655,708
The imidazobenzodiazepine L-655,708 (Fig. 25) exhibited a 30–70-fold selectivity for α5- compared with α1-, α2-, and α3β3γ2 receptors (Ki of 1, 70, 48, and 31 nM, respectively) in benzodiazepine binding studies and was described as a highly selective weak negative allosteric modulator at α5β3γ2 receptors as indicated by its concentration-response curves (Atack et al., 2006a). However, due to the exceptionally high potency of this compound at α5β3γ2 receptors, this is true up to only a 10 nM concentration (Fig. 25A). L-655,708 enhanced long-term potentiation in a mouse hippocampal slice model. Under conditions where it achieved 75% occupancy of α5 and 22% occupancy of α1, α2, and α3 receptors, L-655,708 enhanced cognition in the Morris water maze, but was devoid of proconvulsant activity.
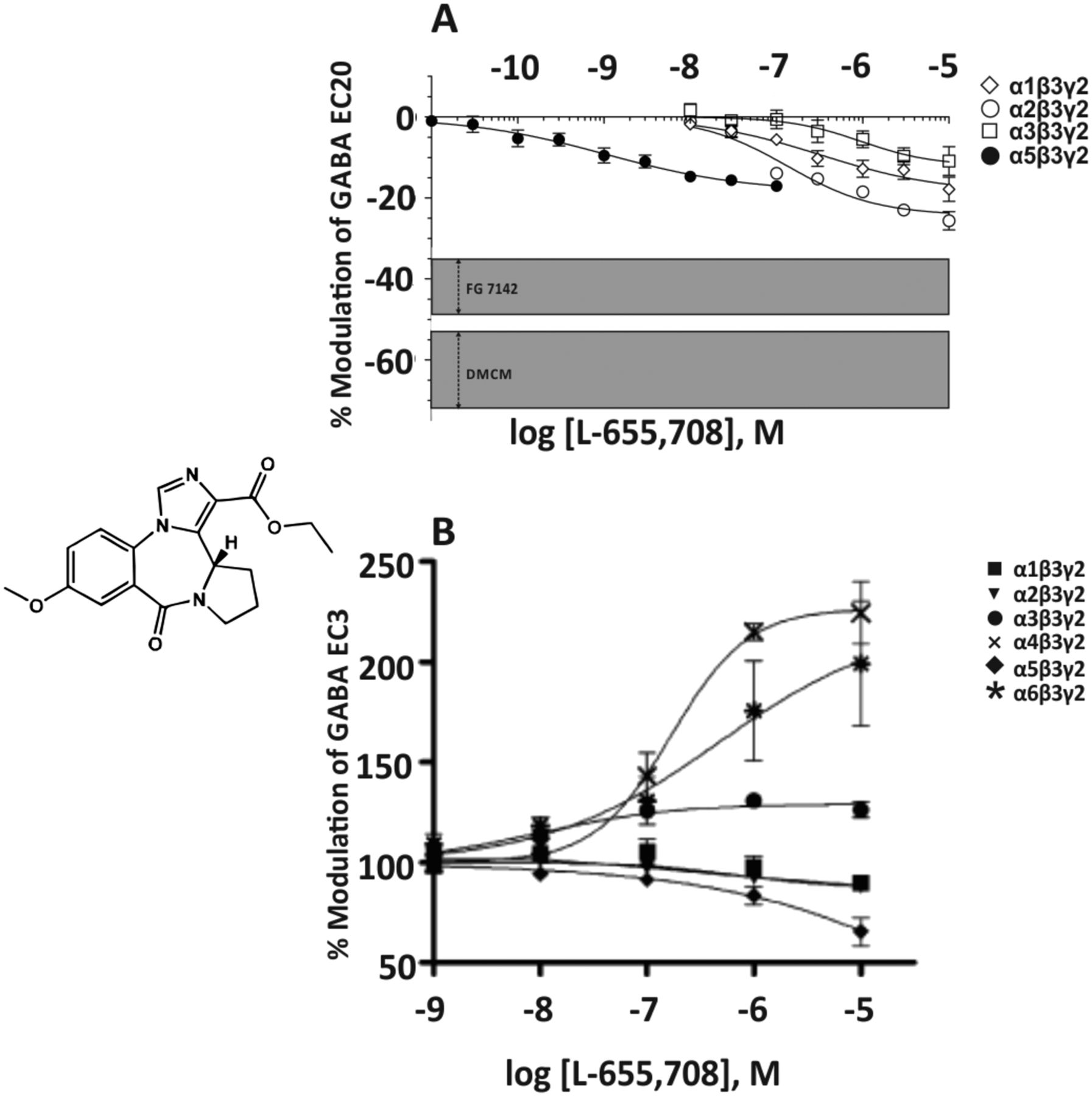
Chemical structure and concentration-response curves of L-655,708. (A) Modified from Atack et al., Neuropharmacology, 51:1023–1029, Elsevier, 2006a. Data were generated at various human recombinant GABAA receptor subtypes stably expressed in L(tk-) mouse fibroblast cells and measured at GABA EC20. The shaded areas represent the range of inverse agonist efficacies across subtypes for the nonselective partial inverse agonists FG7142 or the nonselective full inverse agonist DMCM. (B) Figure modified from Ramerstorfer et al., Eur J Pharmacol, 636,18–27, Elsevier, 2010. Data were generated at various rat recombinant GABAA receptor subtypes expressed in Xenopus laevis oocytes and measured at GABA EC3. The concentration-response curve of α2β3γ2 receptors is overlapping with that of α1β3γ2 receptors.
The data presented, however, are different from our own data presented in Fig. 25B (Ramerstorfer et al., 2010). The opposite direction of α3β3γ2 receptor-modulation by this compound in the two data sets is especially puzzling. Presumably, the concentration of applied GABA (EC20 vs. EC3) or the exact experimental conditions affected the final results. It can be assumed that at low GABA concentrations, predominantly one of the two GABA binding sites of GABAA receptors is occupied. At higher GABA concentrations, increasingly both sites at the receptors become occupied by GABA. Interaction of a compound with a receptor containing two occupied GABA sites might cause a conformational change different from that with only one site occupied, opening the possibility that extent and direction of the allosteric modulation by some compounds, depending on the receptor subtype investigated, could also depend on the GABA concentration used. However, so far, this possibility has not been systematically investigated for any compound. In any case, this compound, similar to other imidazobenzodiazepines, additionally exhibits significant positive modulatory effects at α4β3γ2 and α6β3γ2 receptors (Fig. 25B) that were not investigated in previous studies and could have contributed to the behavioral effects observed.
6. PWZ-029
The imidazobenzodiazepine PWZ-029 (Fig. 26) exhibited a moderate affinity for the benzodiazepine binding site of α5β3γ2 receptors (Ki of 38.8 nM) and Ki values of >300 nM for all other GABAA receptors investigated. In electrophysiological studies it was demonstrated to be a relatively selective negative allosteric modulator at α5β3γ2 receptors up to a concentration of 100 nM (Harris et al., 2008; Savić et al., 2008a), while this compound modulates other receptor subtypes by less than 10%. At concentrations >100 nM this compound exhibits weak positive modulatory effects at α1β3γ2, α2β3γ2, α3β3γ2, α4β3γ2 or α6β3γ2 receptors (Harris et al., 2008). This compound was able to attenuate scopolamine-induced contextual memory impairment in mice (Harris et al., 2008) and improved passive avoidance learning in rats (Savić et al., 2008a), indicating that it facilitates some aspects of cognitive performance.
Chemical structure and concentration-response curves of PWZ-029 at various rat recombinant GABAA receptors expressed in Xenopus laevis oocytes and measured at GABA EC3. The concentration-response curves at α4β3γ2 and α6β3γ2 receptors were measured at GABA EC20. Figure modified from Harris et al. (2008) with permission of The American Chemical Society.
7. RY-024
The imidazobenzodiazepine RY-024 (Fig. 27) exhibits nanomolar affinities for the benzodiazepine site of α1-, α2-, α3-, α5-, α6β3γ2 receptors (Ki values of 26.9, 26.3, 18.7, 0.4, 5.1 nM, respectively) and a unique spectrum of actions at these receptor subtypes (Harris et al., 2008). It is a negative allosteric modulator at α5β3γ2, α1β3γ2, and α2β3γ2; has no action via α3β3γ2; and is a positive allosteric modulator at α4β3γ2 and α6β3γ2 receptors. Due to its high potency for α5 receptors, this compound only up to a 10 nM concentration can be considered a more or less selective α5β3γ2-negative modulator. Nevertheless, based on receptor binding studies only, which indicated an about 70-fold selectivity of this compound for α5 over α1 receptors, in experiments with RY024 it was claimed that α5-containing receptors play an important role in regulating the reinforcing, motor-impairing, and sedative effects of alcohol in outbred rats (McKay et al., 2004).
Chemical structure and concentration-response curves of RY024 at various rat recombinant GABAA receptors expressed in Xenopus laevis oocytes and measured at GABA EC20. Figure modified from Harris et al. (2008) with permission of The American Chemical Society.
8. RO4938581
The imidazobenzodiazepine RO4938581 (Fig. 28) has a high affinity (Ki of 4.6 nM) for the benzodiazepine site of rat α5β3γ2 receptors and a much lower affinity for that of α1β3γ2, α2β3γ2, or α3β3γ2 receptors (Ki values of 174, 185, or 80 nM, respectively) and was also suggested to be an α5-selective negative allosteric modulator (Ballard et al., 2009). The incomplete presentation of the concentration-response curves at the remaining receptor subtypes as well as the missing data for α4β3γ2 and α6β3γ2 receptors cast doubt on that claim. The degree of α5 receptor occupancy in the rat hippocampus produced by RO4938581 at oral doses of 0.1, 1.0, and 10 mg/kg was 30%, 74%, and 90%, respectively (Ballard et al., 2009). In experiments with mice, rats, and monkeys, this compound enhanced hippocampal long-term potentiation and was cognition enhancing at 30% receptor occupancy in the rat, while being devoid of anxiety-like and proconvulsant actions (Ballard et al., 2009).

Chemical structure and concentration-response curves of RO4938581 at various rat recombinant GABAA receptor subtypes stably expressed in HEK 293 cells and measured using the whole cell patch-clamp technique at GABA EC20. Adapted with permission, Springer, Psychopharmacology (Berl), Ballard et al. (2009).
9. TB21007
The 6,6-dimethyl-3-(2-hydroxyethyl)thio-1-(thiazol-2-yl)-6,7-dihydro-2-benzothiophen-4(5H)-one (TB21007) (Fig. 29, compound 43 in Chambers et al. (2003)) exhibits a 10- to 13-times selectivity for α5β3γ2 receptors in benzodiazepine binding studies (Ki of 20, 16, 20, and 1.6 nM, for human α1-, α2-, α3-, and α5β3γ2 receptors, respectively) and acts as a strong negative allosteric modulator at the α5β3γ2 receptor subtype (Chambers et al., 2003). Concentration-response curves, however, indicate that due to the exceptionally high potency of this compound at α5β3γ2 receptors and the only 10–13-fold difference of its affinity and potency for α5β3γ2 and α1β3γ2 receptors, this compound is α5-selective at only subnanomolar concentrations. TB21007 improved memory performance of young rats but not of rats exhibiting age-related memory impairment in a radial arm maze task (Koh et al., 2013). Interestingly, in rats with age-related memory impairment some poorly characterized positive allosteric modulators at α5-containing receptors were effective in improving memory performance [compound 6 in van Niel et al. (2005) and compound 44 in Chambers et al. (2003)].

Chemical structure and concentration-response curves of TB21007 at various human recombinant GABAA receptor subtypes stably expressed in L(tk-) mouse fibroblast cells and measured at GABA EC20. Figure modified from Chambers et al. (2003) with permission of The American Chemical Society.
10. S44819
Recently, a substituted 8-methyl-5-[1-benzothiophen-2-yl]-1,9-dihydro-2H-[1,3]oxazolo[4,5-h][2,3]benzodiazepin-2-one (S44819) was claimed to be an α5-GABAA receptor-selective antagonist competitively inhibiting the action of GABA at these receptors (Ling et al., 2015; Etherington et al., 2017) (Fig. 30). This compound enhanced object recognition memory, long-term potentiation, blocked tonic current mediated by extrasynaptic α5-GABAA receptors, but had no effect on synaptic GABAA receptors and reversed scopolamine-induced impairment of spatial working memory in the eight-arm radial maze. Data presented indicate, however, that S44819 exhibited only marginal receptor subtype selectivity. It preferentially inhibited GABA-induced currents at α5β2γ2 receptors in the concentration range of 1 nM to 10 μM, with a binding constant of 221 nM. It required about threefold higher concentrations to inhibit α1β2γ2 or α3β2γ2 receptors to the same extent, with few effects on α2β2γ2 receptors up to 10 μM concentrations (Etherington et al., 2017). This compound thus selectively inhibits α5β2γ2 receptors up to only 100 nM concentration in recombinant receptors. Nevertheless, it is the first in class compound that uniquely acts as a relatively potent, competitive, and selective antagonist of recombinant as well as native α5-GABAA receptors. Its apparent selectivity in vivo cannot be explained by its only weak selectivity for α5β3γ2 receptors. Its selectivity presumably is enhanced by the low GABA concentration acting at extrasynaptic α5βγ2 receptors that can be more easily overcome by the competitive GABA-site antagonist than the much higher GABA concentrations acting at synaptic α1βγ2 or α3βγ2 receptors (Ling et al., 2015). In addition, the low potency of this compound also might contribute to its selectivity, because concentrations sufficient to inhibit significantly synaptic receptors might not be easily achieved in the brain.

Chemical structure and concentration-response curves of S44819 at various recombinant GABAA receptor subtypes, stably expressed in HEK 293 cells and investigated by use of the fluorometric imaging plate reader dye assay at 1.6 μM GABA (EC30–50). Recombinant receptors were composed of the human α-subunit isoforms, together with the rat β2S and rat γ2L subunits. Modified from Etherington et al., Neuropharmacology, 125,353–364, Elsevier, 2017.
11. XLi-093
The bivalent imidazobenzodiazepine XLi-093 (Fig. 31) has been identified as a selective antagonist at the benzodiazepine site of α5β3γ2 GABAA receptors. XLi-093 exhibits an affinity of 15 nM for the benzodiazepine site of α5β3γ2 GABAA receptors and a 60-fold to >100-fold lower affinity for α1-, α2-, α3-, α4-, or α6β3γ2 receptors. In electrophysiological experiments, it has been identified as a potent antagonist of the effects of diazepam at rat α5β3γ2 GABAA receptors but not at α1β3γ2, α2β3γ2, or α3β3γ2 GABAA receptors (Li et al., 2003). This compound could be a valuable tool for validating effects of compounds mediated via α5β3γ2 GABAA receptors.

Chemical structure of XLi-093 and its inhibition of the effects of diazepam at the diazepam-sensitive GABAA receptor subtypes. The concentration-response curves show the effects of diazepam in the absence or presence of 1 μM XLi-093. Inhibition is shown by the right-shift of the diazepam concentration-response curve. Figure modified from Li et al. (2003) with permission of The American Chemical Society.
G. Compounds Preferentially Modulating α4βγ2 and/or α6βγ2 Receptors
GABAA receptor subtypes composed of α6βγ2 (Nusser et al., 1998) and presumably also those composed of α4βγ2 are located synaptically as well as extrasynaptically and thereby contribute to phasic and tonic inhibition in the central nervous system, respectively. Whereas α4βγ2 receptors are located predominantly in the forebrain, hippocampus, and thalamus (Benke et al., 1997; Bencsits et al., 1999), GABAA receptors composed of α6βγ2 are predominantly located in the cerebellar granule cells (Nusser et al., 1998; Pirker et al., 2000). Currently, not much is known about the function of α4βγ2 or α6βγ2 receptors.
1. CMD-45
The imidazobenzodiazepine CMD-45 (Fig. 32) preferentially enhances the action of GABA at α4β3γ2- and α6β3γ2-containing recombinant GABAA receptors, presumably by acting via the benzodiazepine binding site. But this compound also modulates α1β3γ2, α2β3γ2, α3β3γ2, and α5β3γ2 receptors to a smaller extent. It was recently demonstrated that human airway smooth muscle cells among GABAA receptor α subunits express only α4 and α5 subunits (Gallos et al., 2015) and that CMD-45 is able to significantly relax precontracted human airway smooth muscle cells ex vivo (Yocum et al., 2016). This seems to indicate that selective targeting of α4-containing GABAA receptors with inhaled ligands may be a novel therapeutic pathway to treat bronchoconstriction while avoiding sedative effects in the central nervous system, which are largely mediated by α1- and possibly also α3-containing GABAA receptors (Ralvenius et al., 2015; Behlke et al., 2016; Yocum et al., 2016).

Chemical structure of CMD-45 and concentration-response curves at various recombinant GABAA receptor subtypes from the rat expressed in Xenopus laevis oocytes and measured at GABA EC3. *Data for α4β3γ2 and α6β3γ2 are significantly different from those of α1β3γ2 receptors. Figure modified from Yocum et al. (2016) with permission of The American Thoracic Society. Copyright © 2016.
2. XHe-III-74
The structurally similar imidazobenzodiazepine XHe-III-74 (Fig. 33) exhibits a comparable preferential efficacy for the modulation of α4β3γ2 and α6β3γ2, but exhibits a weaker effect at α1β3γ2 receptors than CMD-45. Similar to CMD-45, it significantly relaxed precontracted human airway smooth muscle cells ex vivo and reduced respiratory system resistance in an asthmatic mouse model in vivo (Yocum et al., 2016). These results were confirmed and extended by using the structurally similar XHe-III-74 ethyl ester and XHe-III-74 acid (Forkuo et al., 2016), but the respective concentration-response curves of the two latter compounds were only incompletely published.

Chemical structure and concentration-response curves of XHe-III-74 at various recombinant GABAA receptor subtypes from the rat expressed in Xenopus laevis oocytes and measured at GABA EC3. *Data for α4β3γ2 and α6β3γ2 are significantly different from those of α1β3γ2 receptors. Figure modified from Yocum et al. (2016) with permission of The American Thoracic Society. Copyright © 2016.
3. PZ-II-029 (Compound 6), LAU159, LAU463
The pyrazoloquinolinone PZ-II-029 (compound 6) (Fig. 34), as well as other structurally related compounds, are high affinity null modulators at the benzodiazepine binding sites of various GABAA receptor subtypes but in addition also positively modulate α6β3γ2 receptors with low potency and exceptionally high selectivity via the α6+β3− interface (Varagic et al., 2013a). Recently, the structurally related LAU159 (8-chloro-2-(3-methoxyphenyl)-2H-pyrazolo[4,3-c]quinolin-3(5H)-one) has been demonstrated to show the highest functional selectivity for positive modulation at α6β3γ2 receptors with nearly no residual activity at the other α1−5β3γ2 receptors up to 10 μM concentrations (Treven et al., 2018). PZ-II-029 and LAU159, together with LAU463 (7-bromo-2-(4-methoxyphenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]-quinolin-3-one), another α6β3γ2 receptor-selective compound, were recently used as lead compounds for the development of a variety of deuterated α6β3γ2 receptor-selective compounds with increased metabolic stability and bioavailability (Knutson et al., 2018). These compounds achieve 100–300 nM concentrations in the rat brain in in vivo studies at 10 mg/kg (Knutson et al., 2018), and due to their low potency, this is sufficient to selectively modulate α6β3γ2 receptors. The recent demonstration that α6β3γ2 receptors not only occur in the cerebellar granule cells, the cochlea nucleus, olfactory bulb, spinal cord, and retina (Gutiérrez et al., 1996) but also in the trigeminal ganglia (Hayasaki et al., 2006), striatum (Leggio et al., 2015) and in the hippocampus (Yang et al., 2016), suggests that PZ-II-029, LAU159, and its congeners might have interesting applications in diseases in which this receptor subtype plays a role. This conclusion is supported by recent reports suggesting that α6-containing GABAA receptors may play a role in neuropsychiatric disorders with sensorimotor gating deficits, such as tic disorders, certain symptoms of schizophrenia, obsessive compulsive disorders, attention deficit disorders, and Huntington’s chorea (Liao et al., 2016; Chiou et al., 2018), as well as in depression (Yang et al., 2016), trigeminal orofacial pain (Puri et al., 2012; Kramer and Bellinger, 2013), trigeminal neuropathy, and migraine (PCT/US2016/035761).

Chemical structure and concentration-response curves of PZ-II-029 (compound 6) at a variety of recombinant GABAA receptor subtypes from the rat expressed in Xenopus laevis oocytes and measured at GABA EC3. Figure modified from Varagic et al. (2013a). Reprinted with permission of John Wiley & Sons, Inc.
4. (+)ROD188
Bicuculline is a competitive antagonist at the GABA binding site of GABAA receptors. The bicuculline derivative (+)ROD188 (Fig. 35) is an allosteric modulator of GABAA receptors (Thomet et al., 2000). Surprisingly, this compound does not interact with the GABA binding site but competitively inhibits [3H]Ro15-1788 binding with an IC50 value of 33.1 μM. Electrophysiological experiments, however, indicated that the positive allosteric modulation of GABAA receptors by (+)ROD188 was inhibited only partially (29%) by Ro15-1788, indicating that most of the effects of this compound are mediated by a so far unidentified second site of action different from the benzodiazepine binding site. Concentration response curves shown in Fig. 35 indicate that this compound preferentially modulates α6β2γ2 receptors. The in vivo effects of this compound so far have not been investigated. In a subsequent study, some structural analogs of (+)ROD188 also induced a preferential modulation of α6β3γ2 receptors (Ramerstorfer et al., 2015). The absence of any direct effects at GABAA receptors, as well as their potential selectivity for receptor subtypes, make this compound class suitable for drug discovery programs.

Chemical structure and concentration-response curves of (+)ROD188 at various recombinant GABAA receptor subtypes from the rat expressed in Xenopus laevis oocytes and measured at GABA EC5. Figure modified from Thomet et al. (2000). Reprinted with permission of John Wiley & Sons, Inc.
5. Amiloride
The diuretic amiloride (Fisher, 2002) is an antagonist at α6β3γ2 receptors, with no actions at α1–5β3γ2 receptors up to 30 μM concentrations (Fig. 36). It seems to act competitively via the GABA binding site and in addition seems to be an open channel blocker, and the α6-subunit seems to confer higher affinity for both sites. Amiloride inhibition was only dependent on the α6 subunit and not influenced by the type of β or γ subunit or the presence of a δ subunit in GABAA receptors (Fisher, 2002). However, its in vivo application is limited by the fact that amiloride does not readily cross the blood-brain barrier, by its inhibitory action at Na+ channels and transporters, and its additional modulation of other transmitter receptors (Fisher, 2002).

Chemical structure and concentration-response curves of amiloride showing its inhibition of the currents elicited by 1 (α6), 3 (α4,α5), or 10 μM (α1, α2, α3) GABA at various rat GABAA receptor subtypes recombinantly expressed in mouse fibroblast cell line L929. Figure modified from Fisher (2002).
6. Furosemide
The diuretic furosemide (Fig. 37) is another relatively selective antagonist at α6β2/3γ2, α6β2/3, and α6β2/3δ receptors, with no interaction with receptor subtypes containing other α subunits up to 3 μM concentrations. But furosemide does not interact with β1-containing receptors (Korpi et al., 1995; Wafford et al., 1996; Korpi and Lüddens, 1997) and presumably interacts with a binding site within the transmembrane domain of α6 subunits (Thompson et al., 1999), which is different from that of amiloride (Fisher, 2002). As with amiloride, its in vivo application is limited due to its diuretic properties.

Chemical structure and concentration-response curves of furosemide showing its inhibition of a GABA EC50 response at human GABAA receptors expressed in Xenopus laevis oocytes. Figure modified from Thompson et al. (1999).
H. Benzodiazepine Site Antagonist Actions
Currently, there are only a few compounds that are antagonists (null modulators) at the benzodiazepine site of GABAA receptor subtypes. β-CCt (Fig. 4, see section V.B.4) is an antagonist at α1, and less avidly, at α2 receptors, but is a weak positive modulator at α3 and α4 and a negative modulator at α5 receptors (June et al., 2003; Yin et al., 2010). Flumazenil (Ro15-1788) (Figs. 4 and 5, see section V.B.5), is an antagonist at α1 receptors only, but a positive allosteric modulator at α2-, α3-, α4-, and α6-containing receptors and a weakly negative modulator of α5β3γ2 receptors (Ramerstorfer et al., 2010). The bivalent imidazobenzodiazepine XLi-093 (Fig. 31, see section V. F. 11) is a selective antagonist at the benzodiazepine site of α5β3γ2 GABAA receptors (Li et al., 2003), but unfortunately its actions at α4βγ2 and α6βγ2 receptors have not been investigated. Since imidazobenzodiazepines usually can bind to the benzodiazepine site of α4βγ2 and α6βγ2 receptors, some actions of XLi-093 at these receptors can be expected. PZ-II-029 (Fig. 34, see section V.G.3) or LAU159 is a high affinity antagonist at the benzodiazepine site of α1–6βγ2 receptors, but low potency positive modulators of α6βγ2 receptors by an additional action via the α+β− interface of GABAA receptors (Varagic et al., 2013a).
It could be concluded that these compounds on systemic or local application are only silent at receptors for which they are null modulators, but in addition modulate other receptor subtypes according to their respective potency and efficacy. In addition, all these compounds are able to antagonize the actions of strongly positive or negative allosteric modulators at the benzodiazepine site, at all receptor subtypes at which they exhibit a null modulatory, weakly positive, or weakly negative allosteric modulation.
But null modulators at the benzodiazepine site also may exert additional actions in the brain. More than 30 years ago, in addition to other compounds, a peptide was identified in the brain that is able to inhibit the binding of diazepam to brain membranes (diazepam binding inhibitor, DBI (Costa and Guidotti, 1985). DBI and other compounds with similar actions might thus be endogenous modulators of GABAA receptors acting via the benzodiazepine site. Recently, new evidence for the existence and function of endogenous ligands for the benzodiazepine binding site has been accumulated (Möhler, 2014), and it seems clear now that endozepines, such as DBI or octadecaneuropeptide, might negatively or positively modulate the function of GABAA receptors via the benzodiazepine site and by that modulate, for instance, neurogenesis or thalamic oscillations, respectively. Flumazenil seems to be able to inhibit the actions of these endogenous modulators (Möhler, 2014), and similar effects can also be expected from other benzodiazepine site null modulators. Thus antagonists at the benzodiazepine site are not necessarily silent but might interfere with the actions of endogenous modulators in the brain, and this could contribute to their spectrum of action in vivo (Hulse et al., 2015). Because the receptor subtype selectivity of endogenous modulators of the benzodiazepine site of GABAA receptors currently is not known, the receptor subtype(s) mediating the behavioral action of a benzodiazepine site antagonist via blockade of endogenous modulators cannot be delineated. In addition, it is not possible to distinguish between effects of benzodiazepine site antagonists elicited by their direct modulation of some GABAA receptor subtypes and effects elicited by blockade of endogenous ligands.
I. Compounds Claimed to Selectively Modulate β1-containing GABAA Receptors
The majority of GABAA receptors is composed of two α, two β, and one γ or one δ subunit (Barnard et al., 1998; Olsen and Sieghart, 2008), and there is also evidence for the existence of receptors composed of αβ subunits in the brain (Mortensen and Smart, 2006; Olsen and Sieghart, 2008). The latter receptors might be composed of two α and three β subunits (Tretter et al., 1997; Baumann et al., 2001), but different stoichiometries seem to be possible (Im et al., 1995; Boileau et al., 2005; Che Has et al., 2016). Receptors containing ε, θ, or π subunits also seem to contain β subunits (Olsen and Sieghart, 2008). Depending on the type of β subunit present in receptors, their regional, cellular, and subcellular distribution, as well as their function in the brain, might be different. Drugs that are able to selectively modulate receptors containing a specific β subunit only will thus exhibit more specific properties and less unwanted effects than those that cannot differentiate between receptors containing different β subunits. In addition, there is ample evidence for the existence of two different α and/or two different β subunits in native GABAA receptors (Jechlinger et al., 1998; Benke et al., 2004; Olsen and Sieghart, 2008). Depending on the types of α and β subunits within the receptors, their stoichiometry, and their subunit arrangement, the receptors contain distinct subunit interfaces and thus, exhibit distinct pharmacological properties that might allow for selectively targeting the respective receptor subtypes (Che Has et al., 2016). However, the pharmacology of receptor subtypes containing two different α or β subunits so far has not been sufficiently investigated. Depending on their regional, cellular, and subcellular location in the brain, they contribute to different types of behavior (Ralvenius et al., 2015). It thus can be expected that the behavioral selectivity of compounds can be increased if they not only can distinguish between different α but also between different β subunits or even between receptors containing different α and β subunit combinations. First evidence for this assumption is provided by the in vivo actions of compounds that can distinguish between receptors containing different types of β subunits.
1. Salicylidene Salicylhydrazide
By screening approximately 10,000 compounds from a structurally diverse screening library for their activity on α2β1γ1θ, α3β3γ2s, and α4β3γ2s GABAA receptors at a single concentration (8 μM) and using a high-throughput voltage/ion probe reader assay, salicylidene salicylhydrazide (Fig. 38), was identified as a potent selective inhibitor of α2β1γ1θ, with a maximum inhibition of 56% and an IC50 of about 32 nM (Thompson et al., 2004). By using patch-clamp electrophysiological techniques, it was then demonstrated that the compound in a limited set of receptor subtypes was selective for the β1 subunit. The extent of inhibition was modulated by α1 and α2, γ1, and γ2, but was not dependent on the presence of θ subunits. Salicylidene salicylhydrazide produced incomplete inhibition on all β1-containing subtypes investigated and, hence, seemed not to act within the ion channel but via a so far unidentified binding site (Thompson et al., 2004). Although there are only limited biologic data with salicylidene salicylhydrazide, it is a known chelator of metal ions and in vitro has comparable cytotoxicity to cisplatin. In addition, this compound also exhibited poor in vivo pharmacokinetics and thus seems to be of limited use for the investigation of the function of β1 subunit-containing receptors (Thompson et al., 2004).
Compounds selective for β1- or β2/3-containing GABAA receptors.
2. Fragrant Dioxane Derivatives
By screening several libraries of odorants, fragrant (1,3)-dioxane derivatives were identified (Fig. 38) that enhance the action of GABA with six times higher potency at β1 subunit-containing compared with the β2 or β3 subunit-containing GABAA receptors (Sergeeva et al., 2010). In the limited set of recombinant receptors investigated, the effects of the fragrant dioxane derivatives depended only on the type of β subunit, were independent of the γ subunit, and obviously were similar in α1- or α2-containing receptors. These compounds act via a so far unidentified binding site at GABAA receptors and up to a 10 μM concentration selectively modulated α1β1γ2 receptors. With the help of these compounds, β1 subunits were identified in synapses that modulate wake-promoting histaminergic neurons in the posterior hypothalamus, indicating that β1-containing receptors might regulate wakefulness and sleep (Sergeeva et al., 2010).
3. Pyrazoloquinolinones
Pyrazoloquinolinones, such as CGS 9895 or PZ-II-029 (compound 6; see section V.G.3) in many cases are high-affinity antagonists (null modulators) at the benzodiazepine binding site (α+γ2− interface) of GABAΑ receptors and exert their low-potency allosteric modulatory action of GABAA receptors via an additional binding site at the α+β− interface (Ramerstorfer et al., 2011; Varagic et al., 2013b). Since both α and β subunits contribute to the latter interface, the action of these compounds is strongly dependent on the α as well as the β subunit type. Whereas CGS 9895 and PZ-II-029 preferentially modulate GABAA receptors containing β2 or β3 subunits, some other pyrazoloquinolinones, such as PZ-II-028 (compound 11 of Varagic et al., 2013b), as well as some structural analogs thereof, preferentially or exclusively (DCBS96, Fig. 38) modulate GABAA receptors containing β1 subunits (Simeone et al., 2017). Unfortunately, however, the so far investigated compounds show only limited α selectivity and, thus, can only be used for investigating the effects of β1 subunit-containing receptors irrespective of the associated α subunit type.
J. Compounds Claimed to Selectively Modulate β2/3-containing GABAA Receptors
The majority of GABAA receptors contain β2 and/or β3 subunits (Sieghart and Sperk, 2002), and so far, no compounds could be identified that can distinguish between receptors containing one or the other β subunit type, with the possible exception of Thio-THIP (Fig. 39; see section V.K.1.c). However, molecular genetic techniques provided evidence that β2- or β3-containing GABAA receptors contribute to distinct behavioral actions of anesthetics.

GABA-site ligands. (A) GABA-site agonists at αβγ receptors. (B) GABA-site antagonists at αβγ receptors. (C) GABA-site agonists and antagonists at ρ-containing receptors.
1. Loreclezole, Etomidate, and Others
A variety of compounds have been claimed to be β2/3 selective (Fig. 38), such as the anticonvulsant loreclezole (Wafford et al., 1994; Wingrove et al., 1994), the intravenous general anesthetics etomidate (Belelli et al., 1997; Hill-Venning et al., 1997) or E-6375 (Pau et al., 2003), the inhalation anesthetic isoflurane (Li et al., 2010), some γ-butyrolactones (El Hadri et al., 2002), mefenamic acid (Halliwell et al., 1999) and certain other nonsteroidal anti-inflammatory agents (Smith et al., 2004), the anxiolytic and anticonvulsant tracazolate (Thompson et al., 2002b; Smith et al., 2004), the anticonvulsant sesquiterpenoid valerenic acid and its derivatives (Khom et al., 2007, 2016), furosemide (Fig. 37) (Thompson et al., 1999), or the anxiolytic-like flavanoid Fa131 (Fig. 16) (Fernandez et al., 2012). All of these compounds, with the exception of etomidate, for which binding sites have been identified in the two β+α− transmembrane interfaces of α1β2/3γ2 receptors (Feng and Forman, 2018), are allosteric modulators at GABAA receptors acting via so far unidentified binding sites. But actually they are not β2/3 selective at all, and only preferentially modulate β2/3-containing GABAA receptors. In addition, they also modulate β1-containing receptors and sometimes to a quite significant extent (Gee et al., 2010).
Loreclezole was the first one of these compounds identified, and it was demonstrated that the preferential interaction of loreclezole with β2- or β3-containing GABAA receptors depended on a single asparagine residue in the second transmembrane domain of β subunits (β2Asn289 or β3Asn290, when numbered including the signal peptide, or Asn265 in the β2 and β3 subunit, numbering of the mature subunits) that was different from the serine at the homologous position in β1 subunits (Wingrove et al., 1994). Since the β2/3 selectivity of all the above mentioned compounds depended on the same amino acid residues, all these compounds were claimed to act via this “loreclezole binding site.” These residues, however, are not necessarily near the respective binding sites but might be involved in the signal transduction of the drug effects to the ion channel.
Interestingly, the flavanoid Fa173 (Fig. 38) was demonstrated to be an antagonist of the positive modulatory actions of the flavanoid Fa131 (Fig. 16; see section V.D.9), as well as of etomidate (Fig. 38), loreclezole (Fig. 38), and the GABA-potentiating effects induced by high, but not low, concentrations of diazepam at α1β2 and α1β2γ2L receptors. The action of all these compounds could be reduced by the point mutation β2Asn265Ser (Fernandez et al., 2012). These results reinforce the idea that these compounds either share their binding pocket or activation domains. Fa173, however, did not block the modulatory actions of the neuroactive steroid 5α-pregnan-3α-ol-20-one, the barbiturate thiopental, and the anesthetic propofol at GABA-induced currents, suggesting that these compounds act via a binding site different from that of loreclezole and etomidate (Fernandez et al., 2012). Further investigations using this antagonistic compound might answer the question whether the effects of Fa173 occur through competition for the loreclezole, etomidate, flavanoid site (loreclezole site), or via another allosteric site, blocking the signal transduction of these compounds. In any case, Fa173 represents a lead compound in the development of novel antagonists at GABAA receptors (Fernandez et al., 2012).
The convulsant β-carboline DMCM (Fig. 38) is a benzodiazepine site ligand with negative allosteric modulatory actions at most GABAA receptor subtypes below 1 μM concentration. At higher concentrations, this compound turns into a positive allosteric modulator, suggesting interaction with a second binding site at GABAA receptors (Ramerstorfer et al., 2010). At α6β3γ2 receptors, however, DMCM does not exhibit a negative modulation of GABA-induced currents and is a positive allosteric modulator above 100 nM concentrations (Ramerstorfer et al., 2010). This positive allosteric modulation by >1 μM DMCM at all receptor subtypes investigated was not inhibited by the benzodiazepine site antagonist Ro15-1788, was only observed at β2- or β3-containing receptors and was dependent on the presence of β3Asn290 (numbering including signal peptide) (Stevenson et al., 1995). Therefore, DMCM not only interacts with the benzodiazepine site at nanomolar concentrations but also with the “loreclezole site” at >1 μM concentrations.
2. Etifoxine and Polyacetylene Compounds
In contrast to the compounds mentioned above, the anxiolytic-like and anticonvulsant etifoxine (Fig. 38) (Schlichter et al., 2000; Hamon et al., 2003) or some polyacetylene compounds (Baur et al., 2005) might mediate their preferential modulation of β2- or β3-containing receptors via an unidentified site different from the “loreclezole site.” Interestingly, etifoxine might elicit its anxiolytic-like action at least partially via an additional interaction with the so called “peripheral benzodiazepine receptor,” now called the 18-kDa translocator protein (TSPO) that seems to be involved in the regulation of neurosteroid synthesis and is also discussed as a target for anxiolytic drugs (Costa et al., 2012; Nothdurfter et al., 2012).
3. β2/3-Selective Enaminones (Compound 2-261)
Empirical observations of the β2/3 selective compounds loreclezole, mefenamic acid, tracazolate, and etifoxine (section V.J.1) in both animals and humans provided anecdotal evidence for the possibility that the degree of activation of β1 subunit-containing GABAA receptors may contribute to their sedative/ataxic properties (Gee et al., 2010). Based on this hypothesis, several enaminones were investigated for their positive allosteric modulation of α1β1γ2 or α1β2γ2 GABAA receptors. These compounds allosterically modulate GABAA receptors via a so far unidentified binding site and compared with other β2/3-selective compounds, such as loreclezole, tracazolate, or etomidate, exert a dramatically reduced maximal efficacy at α1β1γ2 over α1β2γ2 GABAA receptors (Gee et al., 2010). GABA-dependent modulation by the prototypic compound 2-261 (Fig. 38) at β2/3-containing receptors did not appear to be strongly dependent on the type of α subunits. This compound showed equivalent maximal modulation at α1-, α2-, or α3β2/3γ2 receptors. Compound 2-261 produced anxiolytic-like actions with reduced ataxic or sleep-inducing effects (Gee et al., 2010; Yanovsky et al., 2012), and the degree of sedation/ataxia induced by different enaminones or other β2/3-selective compounds (section V.J.1) correlated with their activity at α1β1γ2 receptors. Compounds with high efficacy at β1-containing GABAA receptors induced rotarod failures, whereas those with low activity at these receptors did not. In addition, it was demonstrated that compounds that reach brain levels associated with >47% stimulation of the α1β1γ2 receptor elicit rotarod deficits regardless of the potency of the compound (Gee et al., 2010).
4. Function of β2 or β3 Subunit-containing GABAA Receptors
Given the relatively low selectivity of the β2/3-selective compounds, their inability to distinguish between β2- or β3-containing receptors, their differential interaction with various GABAA receptor subtypes, and their widely different effects in vivo, these compounds could not be used to delineate a possible function of β2- or β3-containing receptors in the brain. Nevertheless, using mice harboring point mutated β2 or β3 subunits that drastically reduced the effects of etomidate, loreclezole, or of the inhalation anesthetic isoflurane, either in β2 (β2Asn265Ser)- (Reynolds et al., 2003; Groves et al., 2006) or β3 (β3Asn265Meth)- (Jurd et al., 2003; Lambert et al., 2005) containing receptors, some attribution of the in vivo functions of β2 or β3 subunit-containing receptors was still possible. The anticonvulsant effects of loreclezole seem at least partially to be mediated via β2-containing receptors (Reynolds et al., 2003; Belelli et al., 2005; Zeller et al., 2005; Groves et al., 2006). The hypothermic and cardiac depressant effects of etomidate seem to be predominantly mediated by β2-containing receptors (Zeller et al., 2005), suggesting that avoiding β2-containing receptor modulation should lead to an improved recovery after anesthesia (Cirone et al., 2004). The sedative action of etomidate (action on locomotor activity) is mediated by β2-containing GABAA receptors (Reynolds et al., 2003). The hypnotic actions of etomidate and propofol (loss of righting reflex) seem to be mediated via both β3 and β2 receptors (Jurd et al., 2003; Reynolds et al., 2003; Zeller et al., 2005). In contrast, both the immobilizing action of etomidate and of propofol and their induction of respiratory depression seem to be mediated via β3-containing receptors (Jurd et al., 2003; Reynolds et al., 2003; Zeller et al., 2005, 2007). The anterograde amnesic action of propofol is independent of β3-containing receptors (Zeller et al., 2007). The type(s) of α subunits present in receptors mediating the individual effects of anesthetics, however, could not be identified by these experiments. In addition, these experiments could not answer the question whether the respective receptors were of the αβ, αβγ, αβδ, αβε type or whether they contained γ1, γ2, or γ3 subunits.
K. Compounds Claimed To Modulate Selectively δ-Containing GABAA Receptors
GABAA receptors containing δ subunits seem to be exclusively located extrasynaptically (Nusser et al., 1998) and contribute to the tonic inhibition of neurons in the brain that modulates both cell and network behavior (Farrant and Nusser, 2005). They seem to be dynamically expressed during ovarian cycle, stress, and puberty (Shen et al., 2017), and evidence has been accumulated that drugs selectively modulating δ-containing GABAA receptors might be beneficial for a variety of human disorders (Brickley and Mody, 2012). However, studies with concatenated α1β3δ GABAA receptors have indicated that there might be several possibilities for the incorporation of a δ subunit in recombinant receptors (Kaur et al., 2009; Eaton et al., 2014; Botzolakis et al., 2016; Wongsamitkul et al., 2016). Depending on the subunit stoichiometry and arrangement, these receptors exhibit distinct properties. This conclusion is supported by the finding that nonconcatenated α6βδ (Hadley and Amin, 2007) and α4β1/3δ (Karim et al., 2012b) receptors can be activated by GABA with a nanomolar and a micromolar potency and that the latter receptor might form a novel GABA-binding site at the δ subunit interface (Karim et al., 2012b). The mixture of recombinant receptors formed will thus determine the pharmacological properties measured (Hartiadi et al., 2016). If receptors with distinct stoichiometry and subunit arrangement are also present in the brain, they represent novel receptor subtypes that, depending on their regional, cellular, and subcellular distribution, will modulate distinct brain functions.
1. γ-Aminobutyric Acid Site Ligands
GABA and orthosteric GABA-site agonists, such as muscimol, isoguvacine, or THIP, exhibit up to 100-fold difference in their potency and efficacy for activation of various GABAA receptor subtypes (Fig. 39A) (Ebert et al., 1994; Ducić et al., 1995; Frølund et al., 2002; Mortensen et al., 2012; Karim et al., 2013).
a. THIP (gaboxadol)
The GABA agonist THIP (gaboxadol, Fig. 39A) has been shown to be devoid of the neurotoxic properties of muscimol and, in contrast to muscimol, is metabolically stable. It is less potent than GABA but approximately 10 times more potent at δ-containing receptors than at γ2S-containing receptors (Brown et al., 2002; Frølund et al., 2002) and in contrast to GABA that is a partial agonist at δ-containing receptors (Bianchi and Macdonald, 2003; Meera et al., 2011), it is a full agonist at these receptors and thus elicits a markedly larger response than GABA. This view, however, was recently challenged by demonstrating that THIP at α6β3δ receptors exhibits a high potency but low efficacy, whereas at the contaminating α6β3 receptors it exhibits a low potency high efficacy modulation (Meera et al., 2011). The previously observed high efficacy modulation by THIP of α6β3δ receptors might thus have been mediated by simultaneously formed α6β3 receptors, which not only might be present in recombinant expression systems but also in the brain. Due to its sleep-inducing and analgesic action (Krogsgaard-Larsen et al., 2004; Wafford and Ebert, 2006), gaboxadol was developed as a hypnotic drug with no tolerance to sleep EEG and sedative effects after repeated daily dosing (Ebert et al., 2008), but its clinical development was abandoned due to safety concerns. In any case, this compound provided some insight on the role of δ-containing GABAA receptors in sedation and sleep (Krogsgaard-Larsen et al., 2004; Herd et al., 2009). It has to be kept in mind, however, that THIP is also an antagonist at ρ-containing GABAA receptors (Chebib, 2004). The interpretation of the in vivo results of this compound may therefore be more equivocal than previously assumed.
b. Thio-4-PIOL
The GABA site agonist Thio-4-PIOL (Fig. 39A) displayed substantial partial agonist activity of up to 30% relative to the effect of GABA at the human extrasynaptic GABAA receptor subtypes composed of α5β3γ2s, α4β3δ, and α6β3δ, and somewhat lower efficacies (4%–12%) at the corresponding α5β2γ2s, α4β2δ, and α6β2δ subtypes (Hoestgaard-Jensen et al., 2013). In contrast, it was an antagonist at the synaptic GABAA receptors composed of α1β2,3γ2S, α2β2,3γ2S, and α3β2,3γ2S (maximal responses of 0%–4% of the GABA current). Thio-4-PIOL thus possibly could be used for exploring the physiologic roles of native synaptic and extrasynaptic GABAA receptors (Hoestgaard-Jensen et al., 2013).
c. Thio-THIP
The GABA site agonist Thio-THIP (Fig. 39A) displayed weak antagonistic activity at α1,2,5β2,3γ2s and ρ1 receptors and partial agonism at α6β2,3δ receptors. It also exhibited a pronounced agonism at α4β1δ and α4β3δ and a negligible activity at α4β2δ receptors. Thio-THIP is thus the first published ligand capable of discriminating between β2- and β3- containing receptor subtypes and could be a valuable tool for the exploration of native α4βδ GABAA receptors (Hoestgaard-Jensen et al., 2014).
d. GABA site antagonists
While the potency of GABA site agonists and modulators of GABAA receptors varies with subunit composition, the potency of GABA site antagonists such as bicuculline, SR95531 (Fig. 39B), and others is largely independent of receptor subunit composition (Lüddens and Korpi, 1995; Ebert et al., 1997; Frølund et al., 2002; Johnston, 2013).
Recently, however, the compound S44819 was claimed to be an orthosteric, competitive, α5-GABAA receptor-selective antagonist (Ling et al., 2015; Etherington et al., 2017; see Fig. 30, section V.F.10). Although this compound exhibited only an about threefold selectivity for α5β3γ2 over α1β3γ2 or α3β3γ2 receptors when recombinantly expressed, its selectivity in vivo is enhanced due to the low extrasynaptic GABA concentrations at α5β3γ2 receptors, which can be more easily overcome by this antagonist than the high synaptic GABA concentrations at α1β3γ2 or α3β3γ2 receptors (Ling et al., 2015; see section V.F.10). In addition, the low potency of this compound also might have contributed to its selectivity in vivo, because concentrations sufficient to inhibit significantly fully saturated synaptic receptors might not be easily achieved in the brain. So far the action of S44819 at δ-containing receptors has not been investigated. It can be assumed, however, that it also will preferentially inhibit α5βδ over other δ-containing receptors.
In addition, the diuretic amiloride (Fig. 36; see section V.G.5) is a relative selective antagonist at α6β1−3γ1−3, or α6β3δ receptors and seems to mediate its action primarily as a competitive antagonist of the GABA binding site of these receptors (Fisher, 2002).
2. Neurosteroids and Tracazolate
The anxiolytic, sedative, and anticonvulsant neurosteroids seem to act via several neurosteroid binding sites in the transmembrane domain of GABAA receptors that still have not been unequivocally identified and might differ in different receptor subtypes (Seljeset et al., 2015; Laverty et al., 2017). They are able to modulate δ-containing receptors to a much stronger extent than other GABAA receptors presumably by shifting the physiologic GABA-induced partial agonist activation of δ-containing GABAA receptors from low-to high-efficacy gating patterns (Belelli et al., 2002; Bianchi and Macdonald, 2003; but see also Meera et al., 2011). The same seems to hold true for the nonsedative anxiolytic-like and anticonvulsant pyrazolopyridine tracazolate (Fig. 38) that seems to act via a so far unidentified binding site at GABAA receptors (Thompson et al., 2002b; Zheleznova et al., 2008). This compound, however, is also interacting with adenosine receptors and phosphodiesterases (Thompson et al., 2002b).
3. Ketamine
The chiral arylcyclohexylamine (Fig. 40) is a dissociative anesthetic capable of inducing analgesia, psychomimetic behavior, and a catatonic state of unconsciousness. It noncompetitively inhibits NMDA receptors, and at anesthetically relevant concentrations is also a positive allosteric modulator at GABAA receptors. Up to concentrations of 20 μM ketamine exhibits some selectivity for α6β2δ- and α6β3δ-containing receptors compared with a limited set of other GABAA receptor subtypes (α1β2γ2, α1β2, α1β2δ, α4β2γ2, α4β2δ, α6β2γ2) (Hevers et al., 2008). At higher concentrations, ketamine directly activates both α6β2δ- and α6β3δ-receptor subtypes.

Chemical structure of ketamine and bar graphs representing the average of ketamine-dependent potentiation (at 10, 20, 50, and 100 μM ketamine) of the indicated rat recombinant receptors at GABA EC3. Figure modified from Hevers et al. (2008) with permission of the Society of Neuroscience.
4. DS2
The imidazopyridine DS2 (Fig. 41) is a relatively selective positive modulator of δ-containing receptors (Wafford et al., 2009). It exhibits weak effects at α1β2γ2, α2β2γ2, α3β2γ2, and α5β2γ2 receptors, but strongly modulates α4β1δ, α4β2δ, α4β3δ, and α6β2δ receptors (Jensen et al., 2013). It interacts with a novel binding site at GABAA receptors that so far has not been identified. The bar graph in Fig. 41 shows the extremely strong potentiation of GABA-induced currents by this compound at δ-containing GABAA receptors. Unfortunately, however, in vivo experiments with this compound are hampered because of its very poor brain penetration. Nevertheless, this compound can be used in slice experiments to clarify the function of δ-containing GABAA receptors (Ye et al., 2013).

Chemical structure of DS2 and bar graphs indicating the modulation by 10 μM DS2 of the indicated human recombinant receptor subtypes expressed in Xenopus laevis oocytes and stimulated by GABA EC5–20 (for α1–5β2γ2) or GABA EC20–50 for α1β2δ and α4β1–3δ and α6β2δ relative to the action of the agonist GABA (rel.AG). In addition, EC50 values of DS2 were determined from full concentration-response curves and given below the indicated receptor subtypes. Unfortunately, most of these full concentration-response curves were not published. Figure modified from Jensen et al. (2013). Reprinted with permission of John Wiley & Sons, Inc.
5. Methaqualone
The sedative-hypnotic and recreational drug methaqualone (Fig. 42) was demonstrated to be a positive allosteric modulator at human α1,2,3,5β2,3γ2s GABAA receptors, whereas it displayed highly diverse functionalities at the α4,6β1,2,3δ GABAA receptor subtypes, ranging from inactivity (α4β1δ) through negative (α6β1δ) or positive allosteric modulation (α4β2δ, α6β2,3δ) to extremely strong effects at α4β3δ (Hammer et al., 2015). Methaqualone was proposed to interact with the transmembrane β+α− interface, possibly targeting a site overlapping with that of the general anesthetic etomidate. Methaqualone exhibited negligible activities at numerous neurotransmitter receptors and transporters, suggesting that it is a selective GABAA receptor modulator. In addition, the doses producing significant in vivo effects in assays for locomotion and anticonvulsant activity correlated fairly well with its potencies as a modulator of recombinant GABAA receptors (Hammer et al., 2015).

Chemical structure and concentration-response curves of methaqualone at various human GABAA receptors expressed in Xenopus oocytes, measured at GABA EC10. Data were normalized to the maximal response elicited by GABA on each oocyte. Figure modified from Hammer et al. (2015).
L. Compounds Modulating γ1- or γ3-Containing GABAA Receptors
GABAA receptors containing γ2 subunits are the major GABAA receptor subtypes in the brain. Sixty to seventy percent of all GABAA receptors contain this subunit (Sieghart and Sperk, 2002), and most of the compounds discussed so far exert their actions at least partially via these receptors. GABAA receptors containing γ1 or γ3 subunits have not been investigated as extensively as those containing γ2 subunits, presumably because of their low abundance in the brain. Three to eleven percent or 3%–14% of all GABAA receptors contain γ1 or γ3 subunits, respectively (Pirker et al., 2000; Sieghart and Sperk, 2002). The γ1 subunit is absent or only weakly expressed in most rat brain regions but is relatively enriched in the basal ganglia, the septal and basal forebrain region, the amygdala, in some thalamic and hypothalamic areas, cerebellum, pons, and medulla. The γ3 subunit is weakly expressed and diffusely distributed all over the rat brain (Wisden et al., 1992; Pirker et al., 2000). Nevertheless, despite their relatively low abundance, these receptors might have important regulatory functions in the brain. In the absence of any information on their abundance and regional and cellular distribution in the human brain, their possible importance for regulating the function of the human central nervous system cannot be estimated. Since these receptors contain most (if not all) drug binding sites that are also present on γ2-containing receptors, they can also be modulated by most, if not all the compounds modulating receptors containing γ2 subunits. In any case, γ1-containing (Ymer et al., 1990; Puia et al., 1991; Wafford et al., 1993; Belelli et al., 2002; Khom et al., 2006) or γ3-containing receptors (Knoflach et al., 1991; Herb et al., 1992; Graham et al., 1996; Sur et al., 1998; Davies et al., 2000; Lippa et al., 2005) also have a benzodiazepine binding site and thus can be modulated by benzodiazepine site ligands (Sieghart, 1995), although in most cases with a lower potency and/or efficacy as judged by the limited number of compounds investigated. So far, no compounds have been identified that selectively modulate γ1 or γ3 subunit-containing receptors.
M. Compounds Modulating ε-, θ-, or π-Containing GABAA Receptors
Not much is known about ε-containing GABAA receptors (Davies et al., 1997a, 2001; Whiting et al., 1997; Neelands et al., 1999; Moragues et al., 2000, 2002, 2003; Thompson et al., 2002a; Maksay et al., 2003; Sergeeva et al., 2005; Bollan et al., 2008) and even less about GABAA receptors containing θ (Bonnert et al., 1999; Sinkkonen et al., 2000; Moragues et al., 2002; Ranna et al., 2006) or π subunits (Hedblom and Kirkness, 1997; Neelands and Macdonald, 1999; Jin et al., 2005). The overall abundance of these receptors in the brain so far has not been investigated. The cDNA sequences of the ε and θ subunits were divergent in mouse, rat, and human tissues (Sinkkonen et al., 2000; Davies et al., 2002; Thompson et al., 2002a), and due to discrepant data in the literature, many researchers might have hesitated to investigate receptors containing these subunits. Receptors containing ε, θ, or π subunits are present in peripheral tissues as well as in the brain (Sieghart and Sperk, 2002), as indicated by in situ hybridization studies. The ε subunit has been demonstrated to be expressed by neurons located in septal and preoptic areas as well as in various hypothalamic nuclei, amygdala and thalamus, and the mRNA was also detected in major neuronal groups with broad-range influence, such as the cholinergic (basal nucleus), dopaminergic (substantia nigra compacta), serotonergic (raphe nuclei), and noradrenergic (locus coeruleus systems) neurons (Moragues et al., 2000, 2002, 2003). The θ subunit showed strikingly overlapping expression patterns with ε subunits throughout the brain. The π subunit was detected in several peripheral human tissues as well as in the brain (hippocampus and temporal cortex) (Hedblom and Kirkness, 1997; Neelands and Macdonald, 1999). But so far no study investigating the detailed regional distribution of π subunits in the brain has been published.
ε, θ, and π subunits can combine with other GABAA receptor subunits in recombinant expression systems, resulting in receptors with unique subunit composition and pharmacological properties. As with δ subunits, ε subunits might form receptors that differ in their subunit stoichiometry and arrangement (Wagner et al., 2005; Ranna et al., 2006; Bollan et al., 2008). However, the actual subunit composition of native GABAΑ receptors containing these subunits is not known and has not been extensively investigated due to a lack of subunit-specific antibodies for immunoprecipitation experiments. No compounds have been identified yet that selectively modulate these receptors.
N. Compounds Modulating ρ-Containing GABAA Receptors
ρ-Containing receptors form homo-oligomers or hetero-oligomers with other ρ subunits or possibly even with some other GABAA receptor or glycine receptor subunits (Qian and Ripps, 1999; Pan et al., 2000; Hartmann et al., 2004; Milligan et al., 2004; Frazao et al., 2007). Originally, ρ-containing receptors were named GABAC receptors because of some differences in their pharmacology compared with GABAA receptors (Chebib, 2004; Martínez-Delgado et al., 2010; Ng et al., 2011; Naffaa et al., 2017). Due to the sequence homology of ρ-subunits with other GABAA receptor subunits as well as the structural homology of ρ-containing receptors with αβγ GABAA receptors, the International Union of Pharmacology nomenclature commission decided that ρ-containing receptors belong to the GABAA receptor family and should be classified as such (Barnard et al., 1998; Olsen and Sieghart, 2008). This is even more justified considering homo-oligomeric β3 receptors. These receptors share many pharmacological properties with αβ or αβγ2 receptors (Slany et al., 1995; Zezula et al., 1996; Davies et al., 1997b; Wooltorton et al., 1997), but are activated by histamine and not by GABA or muscimol (Saras et al., 2008; Hoerbelt et al., 2016). A distinct pharmacology thus cannot be used as a criterion for a distinct receptor nomenclature (Barnard et al., 1998; Olsen and Sieghart, 2008). Although homo-oligomeric β3 receptors so far have not been identified in the brain due to the lack of selective ligands and the abundant expression of β3 subunits as constituents of αβ3γ2 GABAA receptors, their easy formation in recombinant expression systems that recently culminated in the first crystal structure of a GABAA receptor subtype (Miller and Aricescu, 2014), indicates that these receptors probably are expressed also in the brain.
GABAA receptors containing ρ subunits originally were identified in the retina (Cutting et al., 1991; Enz et al., 1996; Enz and Cutting, 1998), but later were also identified in many areas of the mammalian brain (Boue-Grabot et al., 1998; Wegelius et al., 1998; López-Chávez et al., 2005; Martínez-Delgado et al., 2010; Naffaa et al., 2017). However, the overall abundance of these subunits relative to other GABAA receptor subunits so far has not been investigated. The localization of the various ρ-containing GABAA receptors, as well as knockout and pharmacological studies, indicate that these receptors might play a role in visual processing and myopia development, olfactory senses, learning and memory, sleep patterns, nociception, and hormone secretion (Ng et al., 2011). Over time, a variety of GABA site agonists, such as CACA (cis-4-aminocrotonic acid; Fig. 39C), (+)-CAMP [(+)-cis-2-aminomethylcyclopropane carboxylic acid; Fig. 39C] or cis-3-ACPBuPA (cis-3-aminocyclopentanylbutyl-phosphinic acid; Fig. 39C), and antagonists, such as TPMPA (1,2,5,6-tetrahydropyridine-4-yl)methylphosphinic acid; Fig. 39C), were identified that selectively activated and inhibited, respectively, ρ-containing GABAA receptors compared with heteromeric GABAA receptors or metabotropic GABAB receptors (Chebib, 2004; Ng et al., 2011). In addition, some ligands with unique pharmacological profiles have been identified that show selectivity for one ρ subtype over others (Ng et al., 2011; Naffaa et al., 2017).
VI. Discrepancy between the In Vivo Effects of Drugs in Rodent and Human Studies
Subtype-selective drugs are important tools for in vitro cell culture or brain slice studies to clarify a possible contribution of a receptor subtype to the measured effect or in preclinical studies for the investigation of the role of receptor subtypes in the regulation of physiologic functions. But the main motivation for the development of such compounds is the hope to finally identify more selective drugs with fewer side effects and especially, anxiolytic drugs lacking sedation, hypnotic activity, and ataxia. One of the main obstacles in the development of such drugs, however, was recently addressed (Skolnick, 2012): compounds claimed to exhibit selectivity for α2/α3-containing GABAA receptor subtypes, such as MRK 409 (Atack et al., 2011b) or TPA023B (Atack et al., 2011a), and demonstrating a wide dose separation in their anxiolytic-like and sedative effects in rodents, in human studies exhibited actions more or less similar to those of the classic benzodiazepines, eliciting tiredness, drowsiness, and dizziness and at higher doses marked sedation in addition to their anxiolytic effects (Skolnick, 2012). Although the anxiolytic effects of TPA023 were significantly superior to placebo in three separate Phase II studies on generalized anxiety disorder without inducing sedation, in a study with 12 healthy volunteers this compound induced signs of sedation (including drowsiness and dizziness) in some volunteers and elicited a reduced saccadic peak velocity similar to a comparable dose of lorazepam as a possible additional indicator of a sedative action (de Haas et al., 2007; Skolnick, 2012). On the contrary, compounds such as ocinaplon or alpidem, which were not GABAA receptor subtype selective at all in recombinant receptor studies, reportedly behaved as anxiolytics with no sedative component in both rodents and humans (Musch et al., 1988; Lippa et al., 2005; Czobor et al., 2010; Skolnick, 2012). Based on these apparently paradoxical results, the concept of receptor subtype selectivity as a basis for generating drugs with reduced side effects was seriously questioned, and the enthusiasm of the pharmaceutical industry for the development of GABAA receptor subtype-selective drugs was dramatically reduced (Skolnick, 2012).
Given the discussion above, it is evident, however, that none of the drugs currently available is truly receptor subtype selective. By accepting the finding that even a subtle modulation of a receptor subtype can influence behavior, it has to be concluded that the effects of these “selective” drugs cannot be attributed exclusively to the receptor for which they possess the highest affinity or efficacy. In addition, evidence is accumulating that not only α2-containing receptor subtypes, but also α5- (Botta et al., 2015; Behlke et al., 2016)- or α4- and δ-containing receptor subtypes (Marowsky and Vogt, 2014) contribute to the modulation of anxiety. The anxiolytic but reduced sedative effects of the α2/α3-selective drugs in rodents might thus have been caused by an optimally balanced modulation of various receptor subtypes involved in the regulation of anxiety and sedation in these species. The comparable anxiolytic but relatively increased sedative properties of these drugs in humans could then suggest that GABAA receptor subtypes modulating anxiety might be similar, but receptors mediating sedation might be different in rodents and man (Skolnick, 2012).
The validity of animal models used in anxiety and depression research is not universally accepted. Especially scientists in the field of neurodevelopmental and psychiatric disorders often indicate that nonhuman animals can never express the full range of abilities and disabilities that characterize humans (Garner et al., 2009; Blanchard et al., 2013). While this may be and probably is true, it does not compromise the translational validity of at least some animal models used in anxiety studies. In recent years, fear conditioning models from non-human animal research have been substantiated and extended in humans, using neuropsychological and neuroimaging methodologies (Delgado et al., 2006). Recently, the open field test has been demonstrated to be related to human agoraphobic fear (Walz et al., 2016).
In addition, it is generally accepted that biology is similar in different mammalian species, although the complexity of their brains and the exact regulation of individual functions might be different. Apparently, the discrepancy in the behavioral effects of GABAA receptor subtype-preferring drugs in rodents and humans is observed rather in their sedative than anxiolytic-like effects, although animal models of sedation are much less disputed than those of anxiety research. It is thus more probable that additional neuronal systems are involved in the regulation of tiredness, drowsiness, dizziness, and sedation in the more complex and better controlled human brain, thus increasing the overall inhibition by the drugs applied. This possibility is supported by evidence indicating that the regional distribution of GABAA receptor subunits is at least partially different in rodent and human brains (Waldvogel et al., 1999; Loup et al., 2000; Stojanovic et al., 2016; Stefanits et al., 2018). In addition, it has been demonstrated that α1βγ2 receptors are expressed in human (Hellsten et al., 2010), but not reliably in rodent, noradrenergic locus coeruleus neurons (Luque et al., 1994; Chen et al., 1999). Since locus coeruleus neurons are involved in regulation of vigilance states, species differences in the expression profile of α1βγ2 receptors in these neurons might thus contribute to the increased sedative effects of α2/α3 preferring benzodiazepine site ligands in humans, if these compounds also exert a certain degree of activity at α1 receptors (Hellsten et al., 2010). A positive modulation of α5 receptors (Savić et al., 2008b) or α3 receptors (Behlke et al., 2016) might also contribute to sedation. Enhanced expression of such receptors in humans might thus also contribute to the enhanced adverse effects of insufficiently selective GABAA receptor ligands. Therefore, compounds that are really receptor subtype selective and that completely avoid the majority of receptors mediating sedation, might dramatically improve the situation and increase the separation between their anxiolytic and sedative action also in the human brain.
A. Receptors Containing β1-Subunits Might Contribute to the Sedative Action of Benzodiazepine Site Ligands
It is generally assumed that the type of β subunit in a receptor subtype does not significantly influence the action of benzodiazepine site ligands (Hadingham et al., 1993). However, this does not hold true for all benzodiazepine site ligands (Sieghart, 1995; Stevenson et al., 1995; Gee et al., 2010; Varagic et al., 2013a). A possible involvement of β1 subunit-containing GABAA receptors in the regulation of wakefulness was suggested by the use of the β1-preferring fragrant dioxane derivatives (section V.I.2) (Sergeeva et al., 2010). The importance of α1β1γ2 GABAA receptors for eliciting ataxia, sedation, and sleep was then further emphasized by studies using β2/3-selective enaminones (Gee et al., 2010) (section V.J.3). These compounds allosterically modulate GABAA receptors via a so far unidentified binding site and compared with other β2/3 selective compounds, such as loreclezole, tracazolate, or etomidate, exert a dramatically reduced maximal efficacy at α1β1γ2 over α1β2γ2 GABAA receptors (Gee et al., 2010). These enaminones produced anxiolytic-like actions with reduced ataxic or sleep-inducing effects (Gee et al., 2010; Yanovsky et al., 2012) and also reduced abuse liability or memory impairment (Yanovsky et al., 2012; Yoshimura et al., 2014).
The activity of loreclezole and other β2/3-selective compounds at β1-containing receptors might have been still too high (Gee et al., 2010) to avoid sedative side effects. In addition, β2- or β3-containing receptors might also contribute to the sedative actions of these compounds (Belelli et al., 2005; Groves et al., 2006). Nevertheless, the extent of modulation of α1β1γ2 receptors might have contributed to the extent of the sedative and ataxic effects of the currently available drugs. Ocinaplon exhibits a lower modulation of α1β1γ2 receptors than bretazenil or diazepam (Gee et al., 2010). It might thus be interesting to investigate whether the sedative and ataxic effects of ocinaplon, MRK 409, TPA 023, and TPA 023B or of any other drug for which preclinical as well as clinical data are available, correlate with their efficacy at β1-containing GABAA receptors.
GABAA receptors containing β1 subunits are expressed in the brain stem and arousal-related areas of the rat brain, for instance in the reticular thalamic nucleus, which is an important area for modulating sleep (Pirker et al., 2000; Gee et al., 2010). They are relatively minor receptor subtypes compared with those containing β2 or β3 subunits. Depending on the brain region investigated, about 20%–30%, 65%, or 55% of all GABAA receptors contain β1, β2 or β3 subunits, respectively (Sieghart and Sperk, 2002). The finding that these percentages add up to >100% indicates a significant colocalization of β1, β2, or β3 subunits in GABAA receptors (Jechlinger et al., 1998). A possible contribution of a minor GABAA receptor subtype to the sedative effects of MRK 409 in humans was suggested by the extremely low receptor occupancy (<10%) of this compound under conditions that caused sedation in young healthy male volunteers (Atack et al., 2011b). This might have been caused by an especially high potency or efficacy of this compound for a low abundance receptor subtype mediating sedation in man. To investigate a possible contribution of β1-containing receptors to the sedative effects of MRK 409 and to possibly identify the GABAA receptor subtype involved, the effects of MRK 409 at recombinant GABAA receptors composed of α1–6β1γ1−3 should be investigated and compared with the respective receptors containing β2 or β3 subunits.
B. Receptors Containing γ1 or γ3 Subunits, or Those Containing α4, α6, or δ Subunits, Might Contribute to the In Vivo Effects of Benzodiazepine Site Ligands
Benzodiazepine site ligands not only interact with GABAA receptors containing γ2 subunits, but also with those containing γ1 (Puia et al., 1991; Wafford et al., 1993; Sieghart, 1995; Khom et al., 2006) or γ3 subunits (Herb et al., 1992; Sieghart, 1995; Davies et al., 2000; Lippa et al., 2005). But so far, a possible contribution of such receptors to the spectrum of in vivo actions of benzodiazepine site ligands was not considered and no compounds have been identified that selectively modulate γ1 or γ3 receptors. Although γ1 or γ3 receptors are much less abundant in the mammalian brain than those containing γ2 subunits (Pirker et al., 2000; Sieghart and Sperk, 2002), they might have important regulatory functions. The presence of γ1 receptors in the amygdala (Pirker et al., 2000; Esmaeili et al., 2009) might suggest their possible involvement in the regulation of anxiety, and ligands interacting with such receptors might mediate some of their effects via this mechanism and this could enhance their anxiolytic relative to their sedative effects.
Imidazobenzodiazepines (Figs. 5, 25–29, 31–33), pyrazoloquinolinones (Fig. 34), β-carbolines, such as DMCM (Fig. 38) and other structural classes of benzodiazepine site ligands, not only modulate the diazepam-sensitive α1,α2,α3,α5βγ2 receptors, but also α4 and α6 subunit containing GABAA receptors (Sieghart, 1995; Stevenson et al., 1995; Ramerstorfer et al., 2010; Varagic et al., 2013a), and thus effects mediated by these drugs via these receptor subtypes cannot be ignored. The recent identification of a pyrazoloquinolinone modulating α1β3δ, α4β3δ, and α6β3δ receptors (Mirheydari et al., 2014) together with the observation that extrasynaptic receptors containing α4 and δ subunits seem to be involved in inducing sedation and sleep (Belelli and Lambert, 2005; Belelli et al., 2005) and possibly also anxiety (Marowsky and Vogt, 2014) indicate that modulation of such receptors can contribute to the overall action of such compounds. A possible modulation of such receptors cannot be excluded before appropriate experiments have been performed.
C. Additional Interaction with Targets Different from GABAA Receptors Could Also Contribute to the Separation of Anxiolytic and Sedative Properties of Ligands
Alpidem, similar to the structurally related zolpidem, potently and efficiently modulates α1-, α2-, and α3-containing receptors, and exhibits no effects at α5 receptors (Puia et al., 1991; Costa and Guidotti, 1996). Whereas alpidem has anxiolytic but low sedative effects, zolpidem is a sedative hypnotic drug in humans. Both of these drugs, however, exhibit a similar modulatory effect at GABAA receptors containing β1 subunits (Puia et al., 1991; Costa and Guidotti, 1996). But in contrast to zolpidem, alpidem also has a high affinity for TSPO, which seems to be involved in the regulation of neurosteroid synthesis and is also discussed as a target for anxiolytic drugs (Costa et al., 2012; Nothdurfter et al., 2012). Similarly, the anxioselective pyrazolopyridine CGS 20625 not only acts via GABAA receptors but additionally exhibits some interaction with TSPO, known previously as the “peripheral benzodiazepine receptor” (Williams et al., 1989). Therefore, the wider separation of the anxiolytic over the sedative action of CGS 20625 or of alpidem compared with zolpidem, might have been caused by an additional interaction of CGS 20625 or alpidem with TSPO.
D. A Low Potency and Efficacy of Compounds Might Enhance Their Receptor Subtype-selective Actions
But how can ocinaplon act as a selective anxiolytic in rodents and humans (Czobor et al., 2010), although it seems not to be receptor subtype selective at all (Lippa et al., 2005; Berezhnoy et al., 2008)? Ocinaplon is a low potency and low efficacy allosteric modulator. This is in contrast to the previously investigated high potency, low efficacy allosteric modulators such as bretazenil and abecarnil (Ramerstorfer et al., 2010), MRK 409 (Fig. 12; section V.D.5), TPA023 (Fig. 11; section V.D.4), or TPA023B (Fig. 7; section V.C.2), which lost their anxioselective action observed in rodents when applied in humans (Skolnick, 2012). Due to their high potency, the latter drugs probably achieved brain concentrations eliciting their maximal efficacy at all receptor subtypes under conditions of in vivo application and by that sufficiently activated also those receptors mediating sedation in humans. The potency of ocinaplon for modulating GABAA receptors, however, is about 100–1000-fold lower than that of diazepam, depending on the receptor subtype investigated, and in addition this compound exhibits lower efficacy (Berezhnoy et al., 2008). This indicates that an at least 100–1000-fold higher concentration of ocinaplon has to be reached at GABAA receptors to elicit a modulation similar to that of diazepam. It is quite possible that concentrations sufficient to modulate GABAA receptors mediating sedation cannot be achieved by this drug.
Therefore, the low potency and low efficacy of ocinaplon, possibly combined with an intrinsic inability to sufficiently interact with those receptor subtypes mediating sedation at the concentrations achieved, might have added up to the favorable profile of this compound in humans. If more compounds with low potency and low efficacy, but with receptor subtype selectivity, such as 6-hydroxyflavone (Fig. 15; section V.D.8 (Ren et al., 2010), baicalin (Fig. 14; section V.D.7) (Wang et al., 2008), AZD7325 (Fig. 17; section V.D.10) (Christian et al., 2015), PZ-II-029 (Fig. 34; section V.G.3) (Varagic et al., 2013a), LAU159 (Treven et al., 2018), ELB138 (imepitoin), or ELB139 (Rabe et al., 2007; Rundfeldt and Löscher, 2014), confirm this hypothesis in clinical investigations by demonstrating a wider separation of anxiolytic and sedative properties in humans, pharmaceutical companies might have to radically change their strategy for the development of more selective drugs: Instead of developing high potency, high efficacy drugs with receptor subtype selectivity, the development of low potency, low efficacy drugs with receptor subtype selectivity might be more promising for eliciting selective actions. Since drug efficacy plays a critical role in determining the degree of GABAA receptor uncoupling, and, perhaps in the development of tolerance and dependence (Primus et al., 1996), low efficacy drugs might have an added benefit in a long-term treatment of patients.
VII. Outlook
Given the data summarized above, it is evident that many of the compounds claimed to be selective for a specific α1-, α2-, α3-, or α5β3γ2 receptor subtypes are only marginally selective and that their limited selectivity is even further reduced by their high potency. Generating compounds with reduced potency and efficacy but high receptor subtype-selectivity seems to be a way out of this problem. Such compounds, in addition to their receptor subtype selectivity, might not achieve concentrations in the brain that sufficiently modulate receptor subtypes mediating sedative effects. Investigating such compounds in a clinical setting is also the only possibility to clarify whether it is their high receptor subtype selectivity that results in high functional selectivity or whether functional selectivity is achieved by an optimal balance of activity at various receptor subtypes.
In addition, the “receptor subtype selectivity” of benzodiazepine site ligands so far was predominantly investigated at the “diazepam-sensitive” α1β3γ2, α2β3γ2, α3β3γ2, and α5β3γ2 receptors only, although most if not all of these drugs additionally can modulate receptors containing the same α, but different β or γ, subunits. All these receptors exhibit a distinct regional, cellular, and subcellular distribution in the brain and can be assumed to exhibit distinct behavioral effects, but the investigation of their function so far has been neglected. The same holds true for receptors containing α4 or α6 subunits. Certain structural classes of benzodiazepine site ligands can also modulate these or even δ-containing receptors. Therefore, all of these receptor subtypes have to be considered and investigated when claiming a “receptor subtype selectivity” of a drug. Additional interaction with such so far not investigated receptors can shift the balance between anxiolytic and sedative effects. Although an investigation of all these additional receptor subtypes seems to be a tremendous task, it seems to be feasible using high-throughput electrophysiological measurements (Trumbull et al., 2003) or fluorometric imaging plate readers (FLIPR) using membrane potential red dye, which redistributes across the plasma membrane in a voltage-dependent manner (Nik et al., 2017). In addition, the already available compounds with limited receptor subtype selectivity and their congeners, together with structural models of individual GABAA receptor subtypes (Miller and Aricescu, 2014), receptor subtype-specific pharmacophore models (Clayton et al., 2007, 2015), and structure-based drug design (Richter et al., 2012), will facilitate the development of new and more selective drugs. Recent binding site mapping in models of GABAA receptors has identified additional binding sites in these receptors that possibly also can be exploited for the development of receptor subtype-selective drugs (Ernst and Sieghart, 2015; Puthenkalam et al., 2016).
Finally, to strengthen the conclusions from biologic datasets generated by available compounds with limited or unclear receptor subtype selectivity, compounds from the same structural class exhibiting a similar as well as dissimilar selectivity profile together with compounds selectively antagonizing the actions of the investigated drug at the receptor supposedly mediating its in vivo action, should be included into the protocol. In addition, positive controls (compounds from a different structural class that are also active at the respective receptor subtype) or negative controls (compounds exhibiting no activity at the respective receptor subtype) should also be included into the protocol, if available. The present review can serve as a guide for the selection of adequate positive or negative controls. Furthermore, the data obtained with compounds preferentially interacting with a certain receptor subtype have to be carefully interpreted by considering all the known interactions of the compound at the drug concentrations achieved in the brain. After all, a possible additional modulation by the drug of other GABAA receptor subtypes or binding sites or of different transmitter systems has to be acknowledged in the discussion of the data.
Acknowledgments
We thank the countless contributors to the work cited in this review, without whom the current understanding of the physiological and pharmacological role of GABAA receptor subtypes would not be possible. In addition, we want to thank Dr. Aleksandar Obradović, Dr. Bojan Marković, and Dr. Nebojša Cekić for their expert assistance in the preparation of the modified figures.
Authorship Contribution
Wrote or contributed to the writing of the manuscript: Sieghart, Savić.
Footnotes
In part, financial support was provided by the Ministry of Education, Science and Technological Development, R. Serbia – Grant No. 175076 (MMS).
Abbreviations
- DBI
- diazepam binding inhibitor
- GABA
- γ-aminobutyric acid
- GABAA-receptor
- GABA type A receptor
- GABAB-receptor
- GABA type B receptor
- KCC2
- K+-Cl− cotransporter 2
- NKCC1
- Na+-K+-2Cl− cotransporter 1
- TSPO
- translocator protein (18 kDa)
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
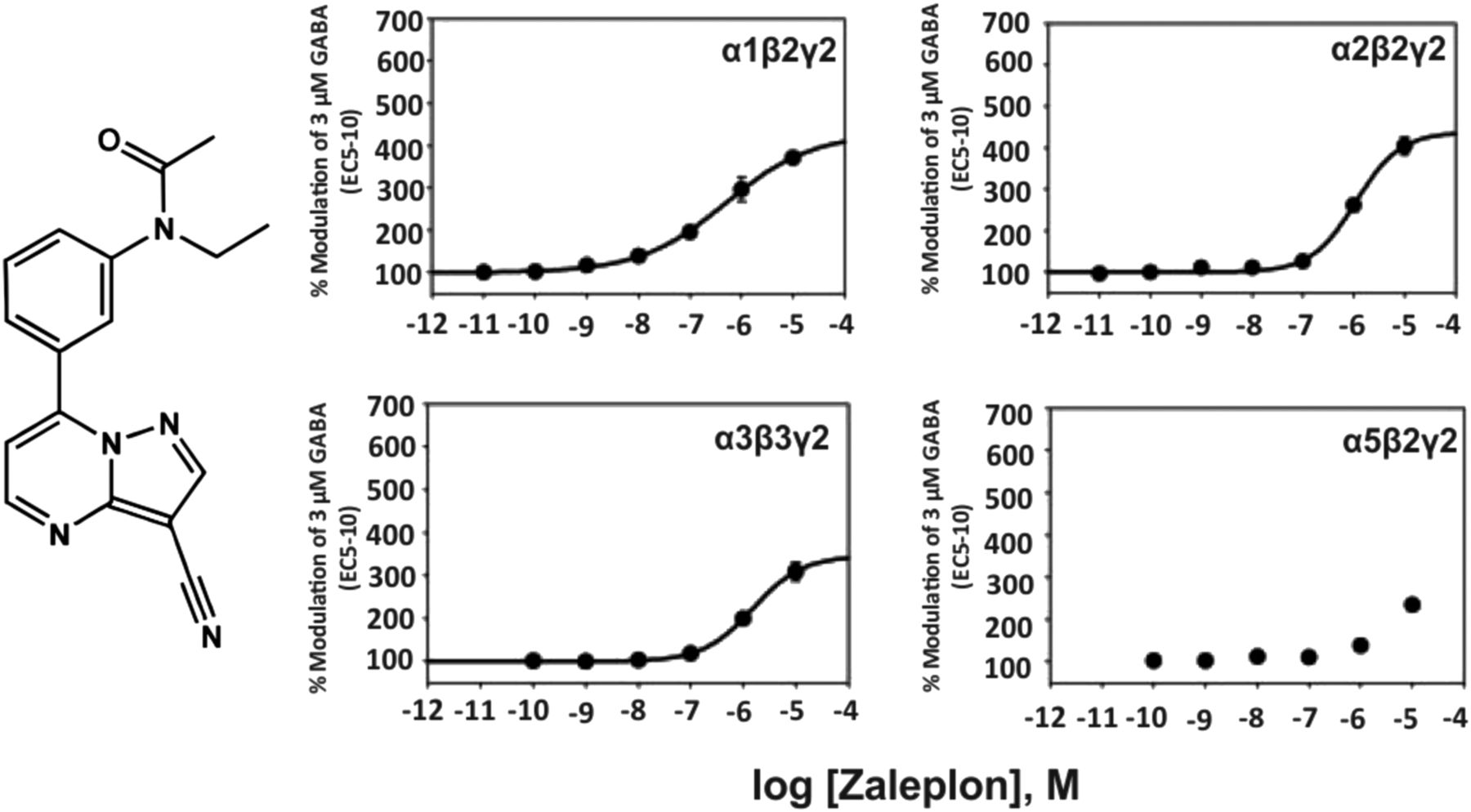{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
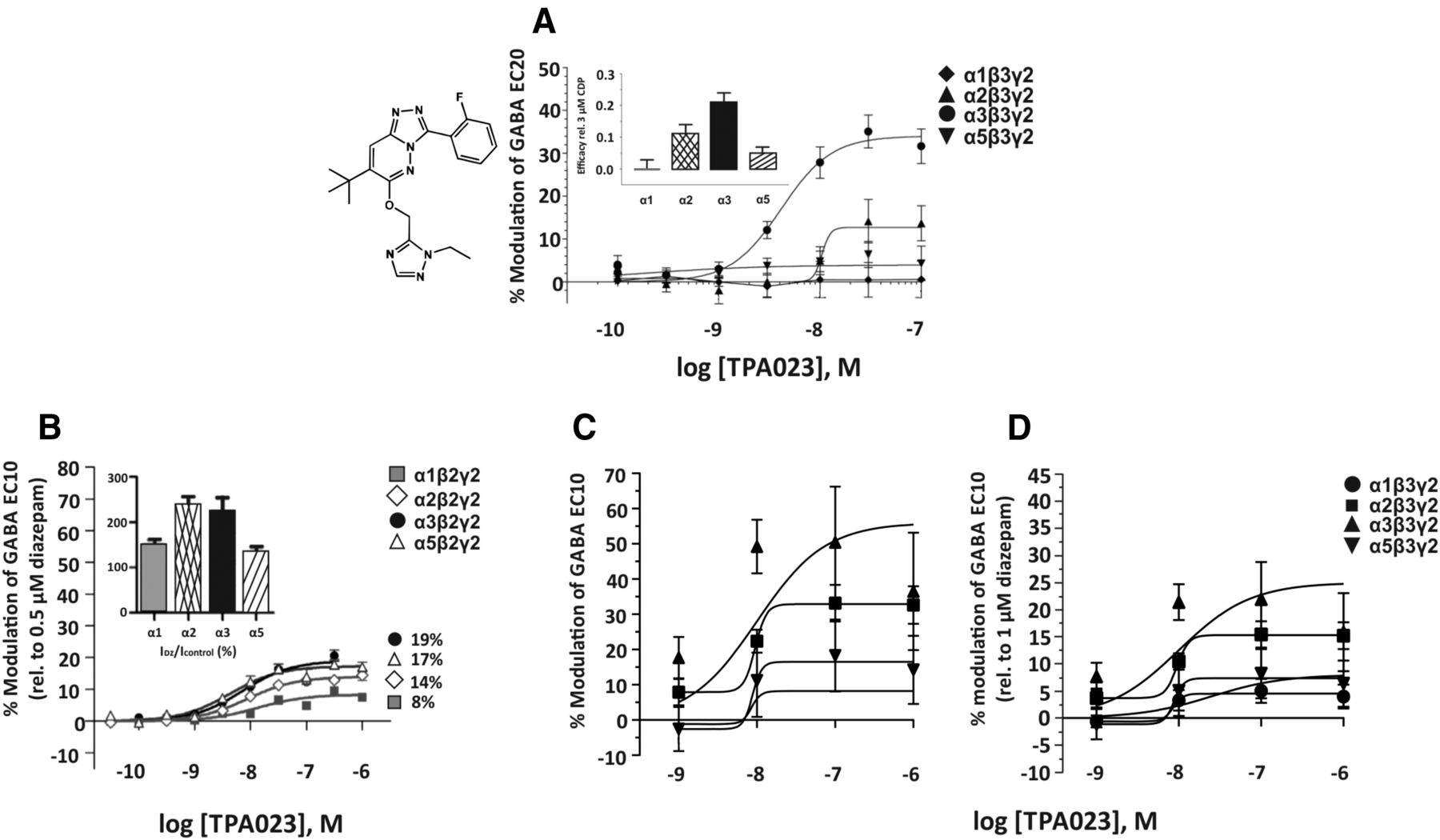{kind=link}
{kind=link}
{kind=link}
{kind=link}
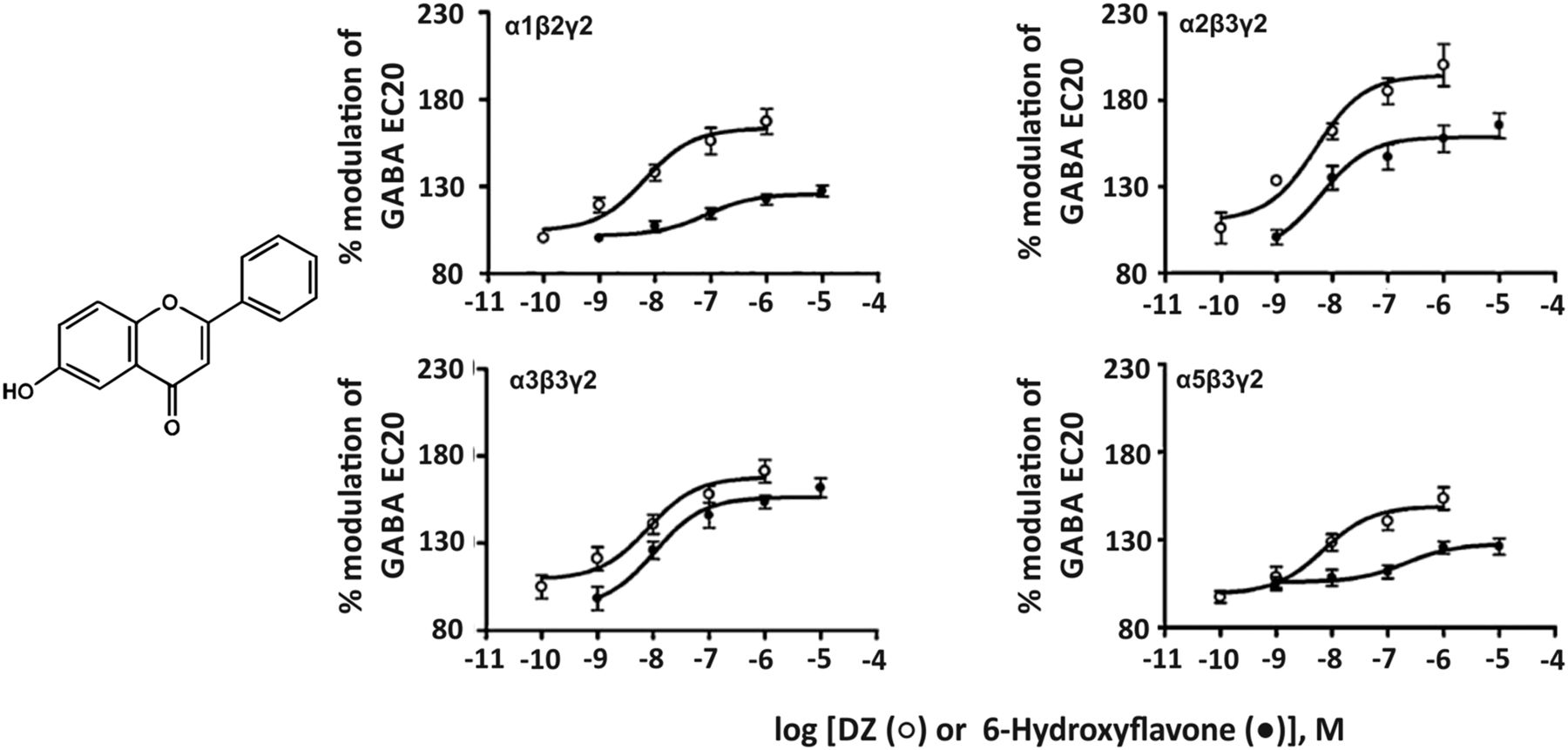{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
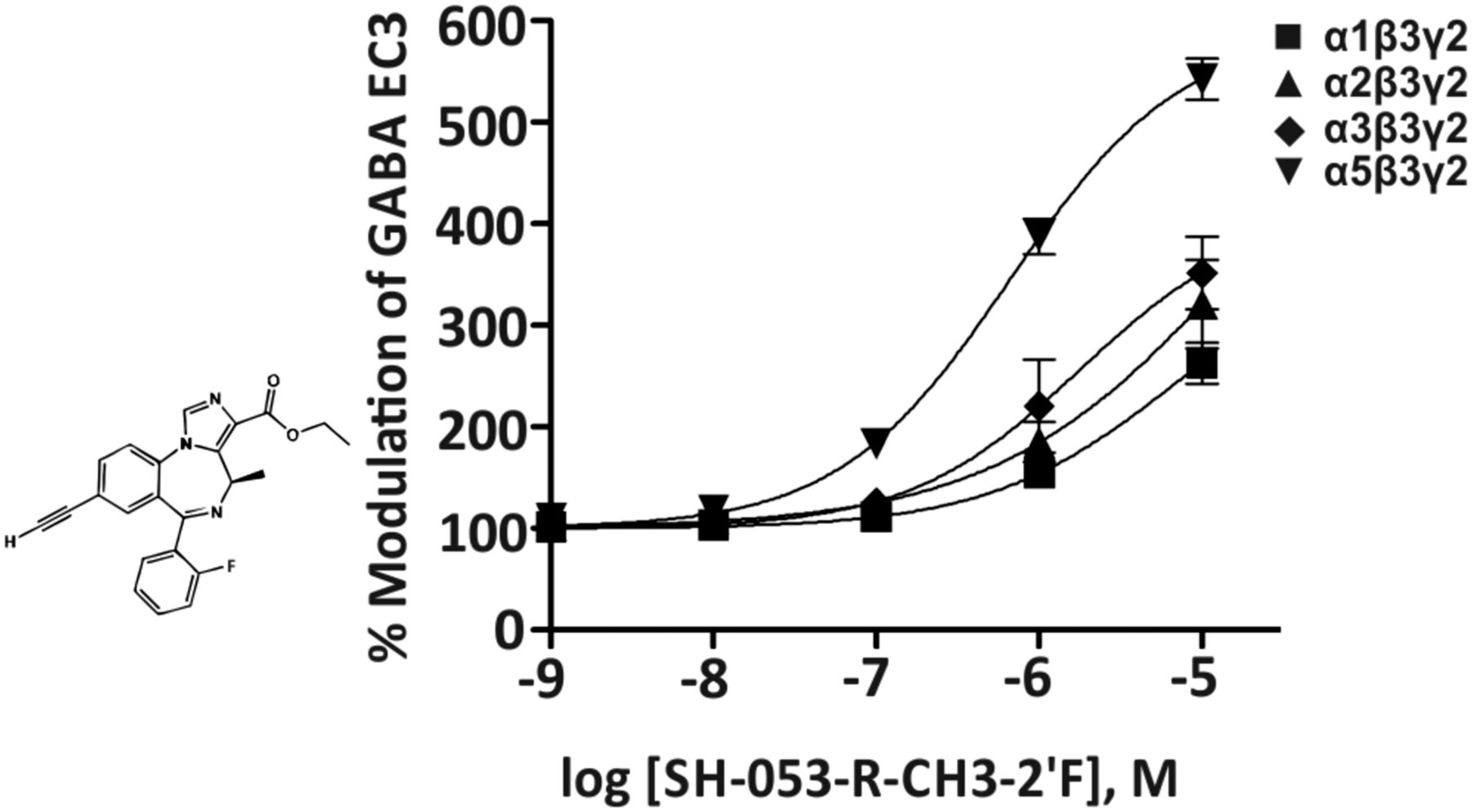{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
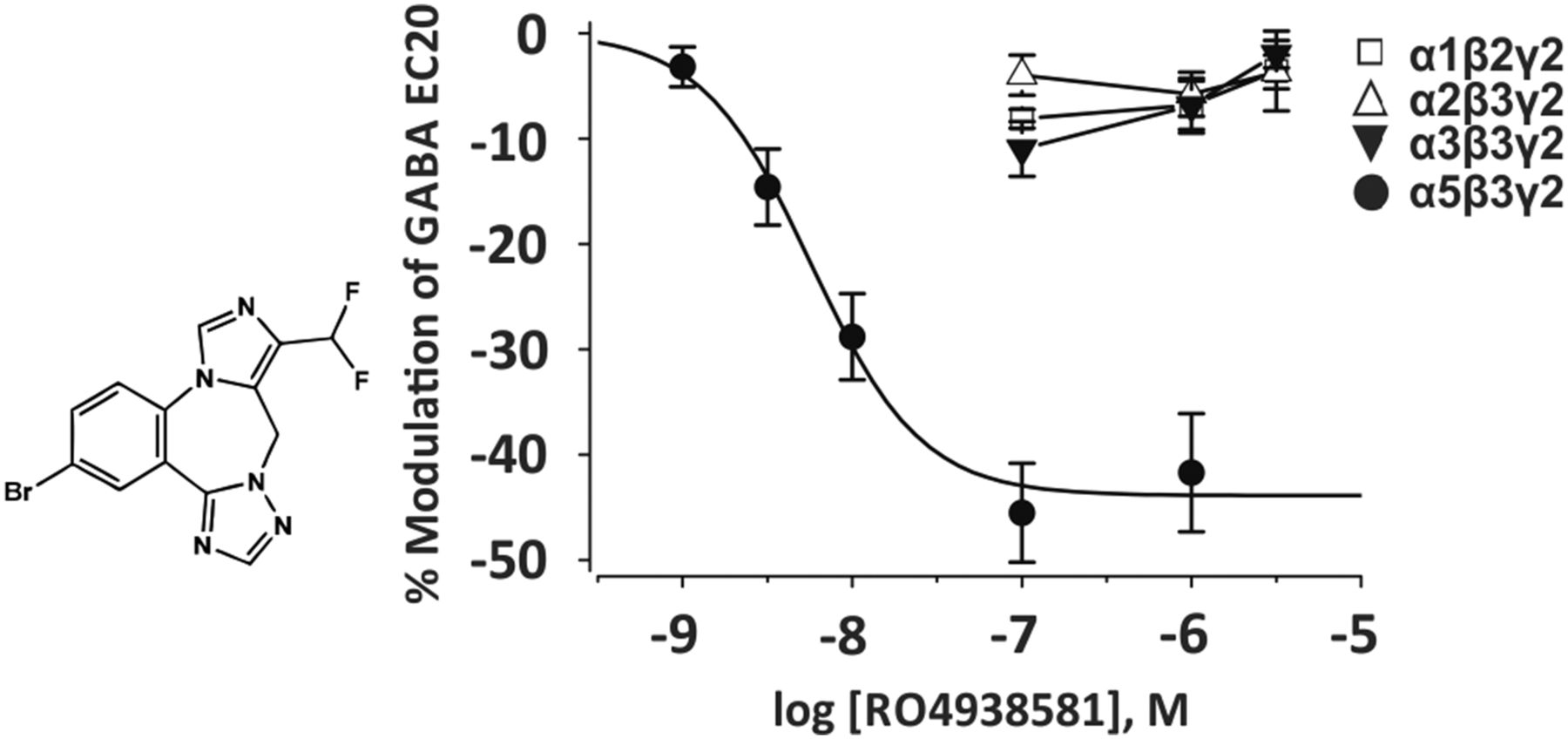{kind=link}
{kind=link}
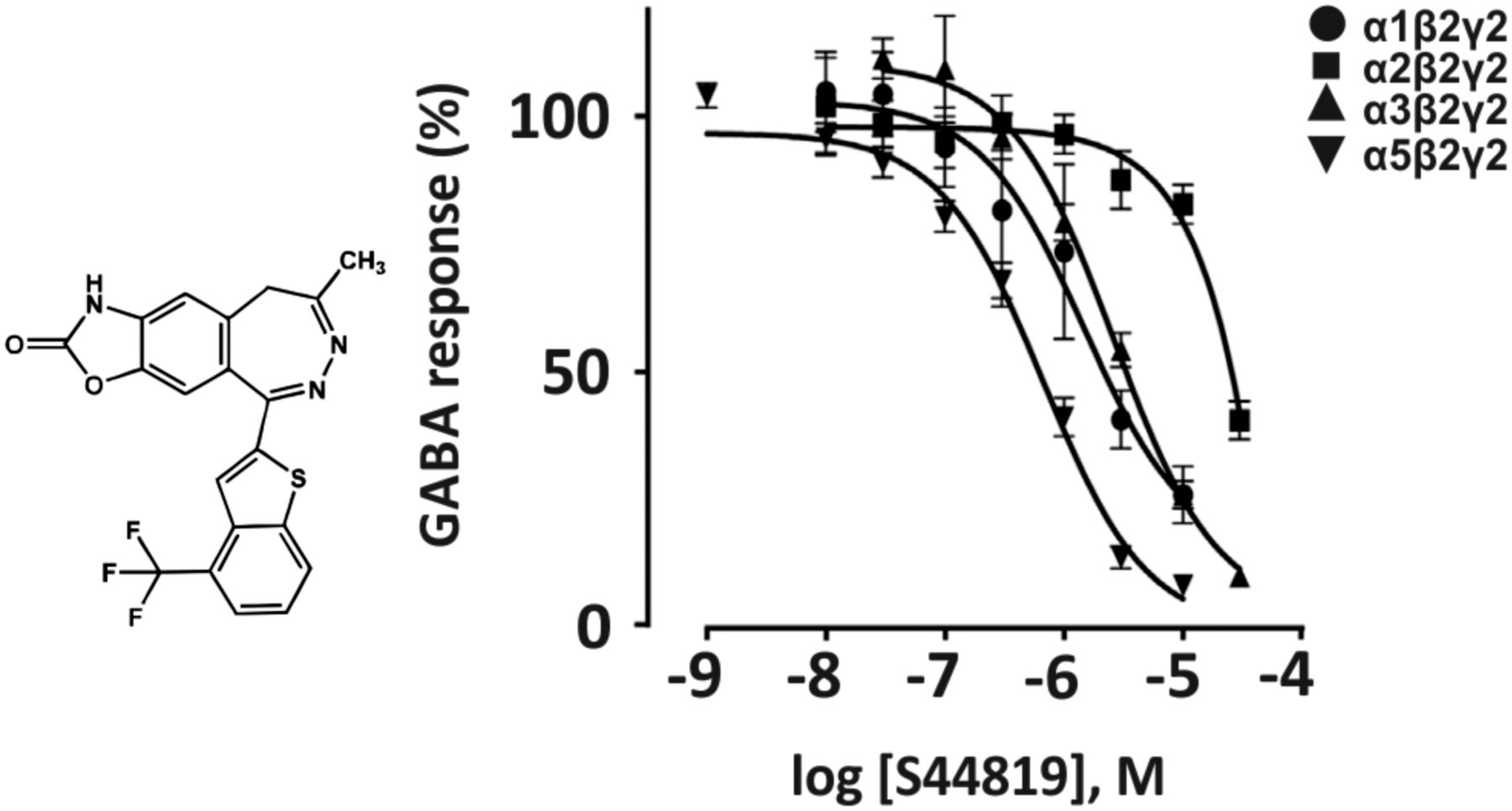{kind=link}
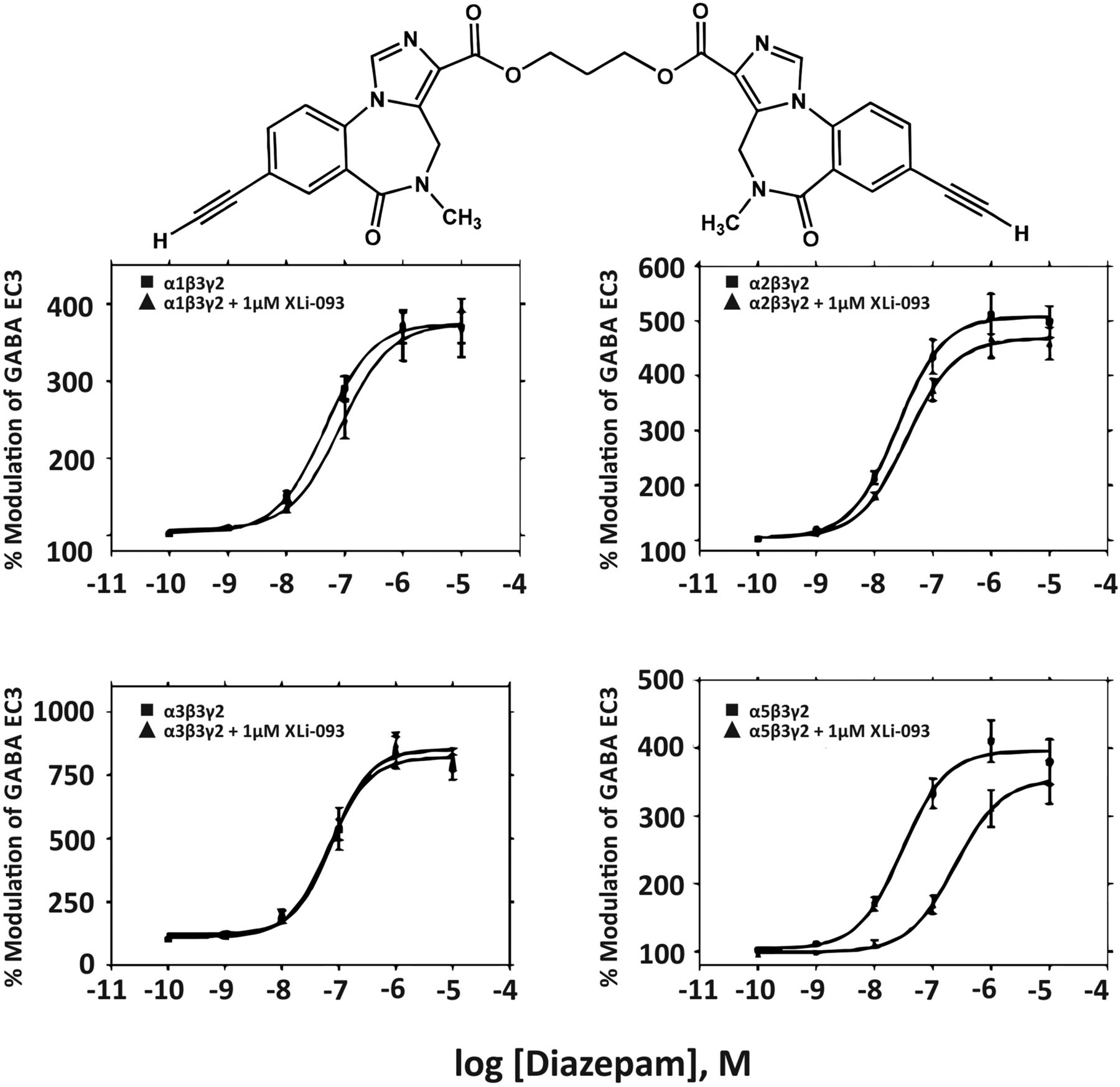{kind=link}
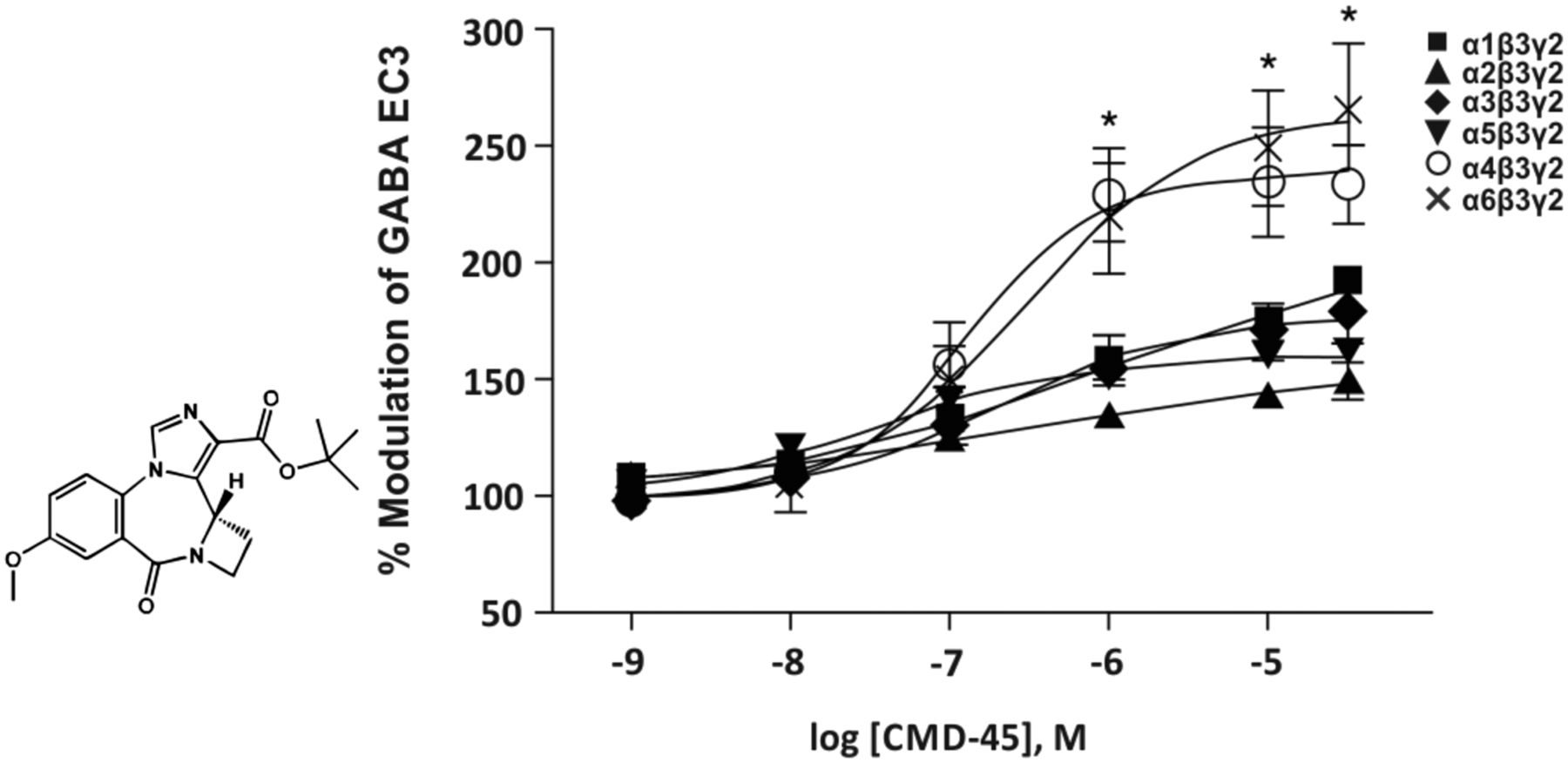{kind=link}
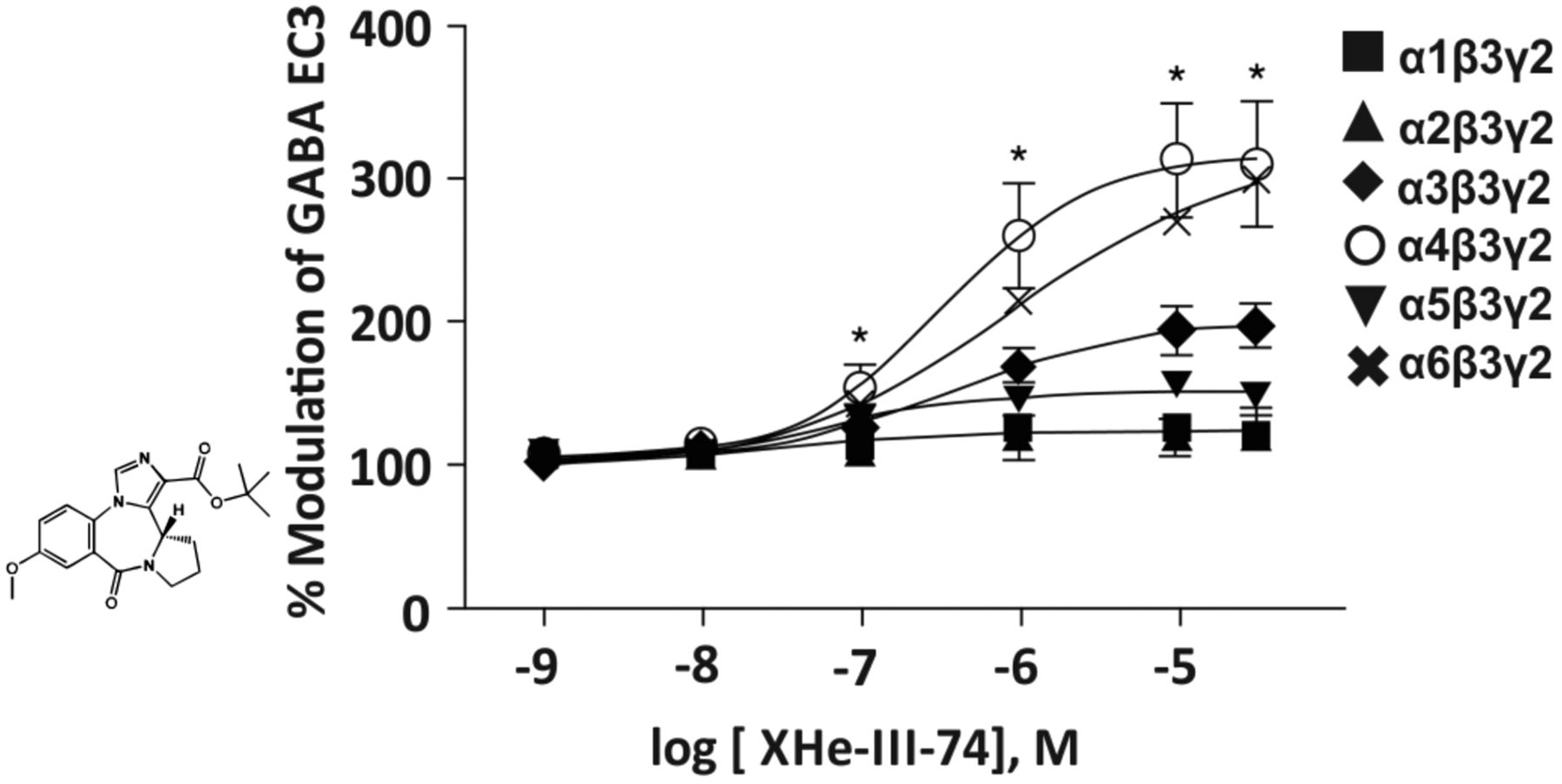{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
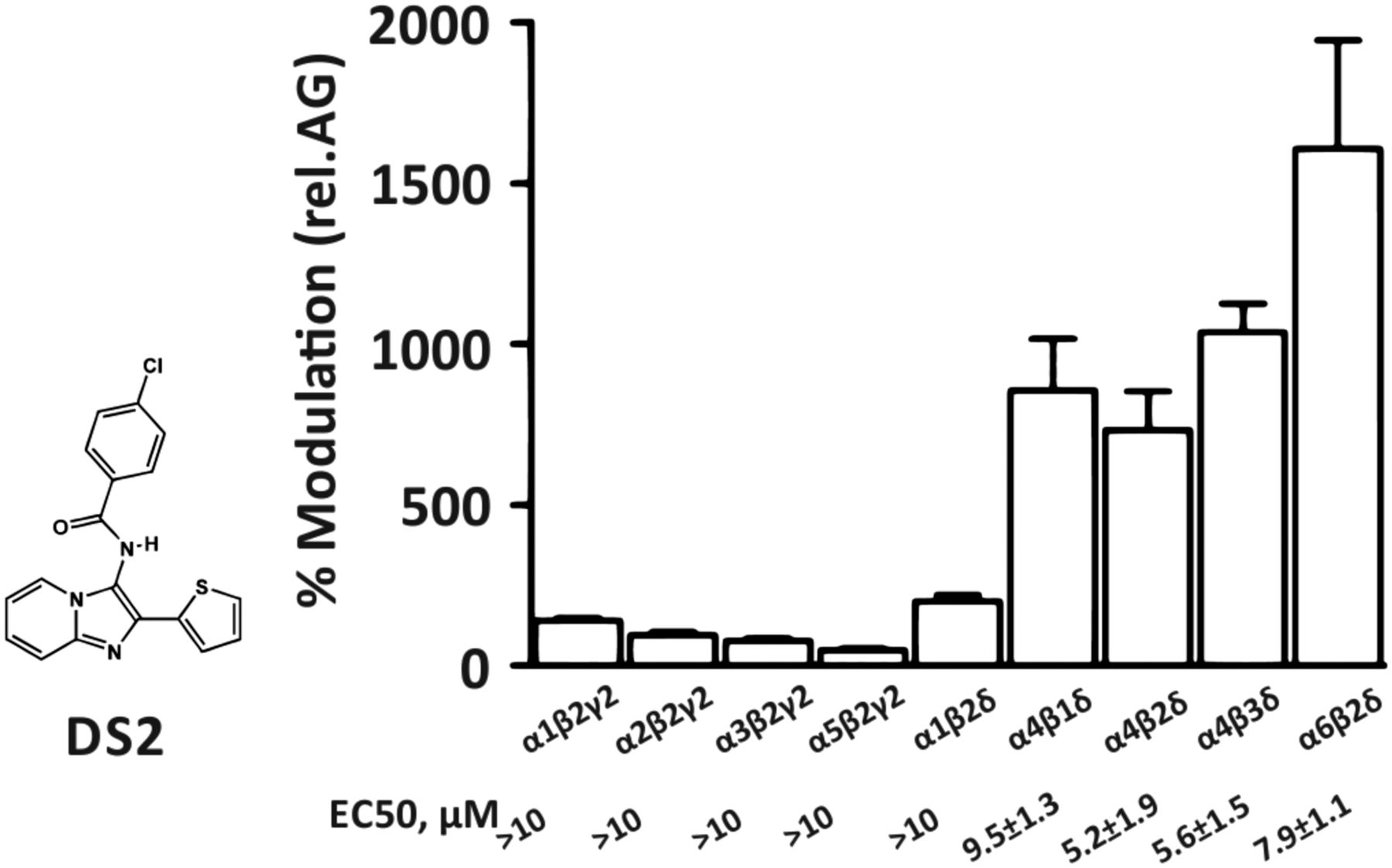{kind=link}
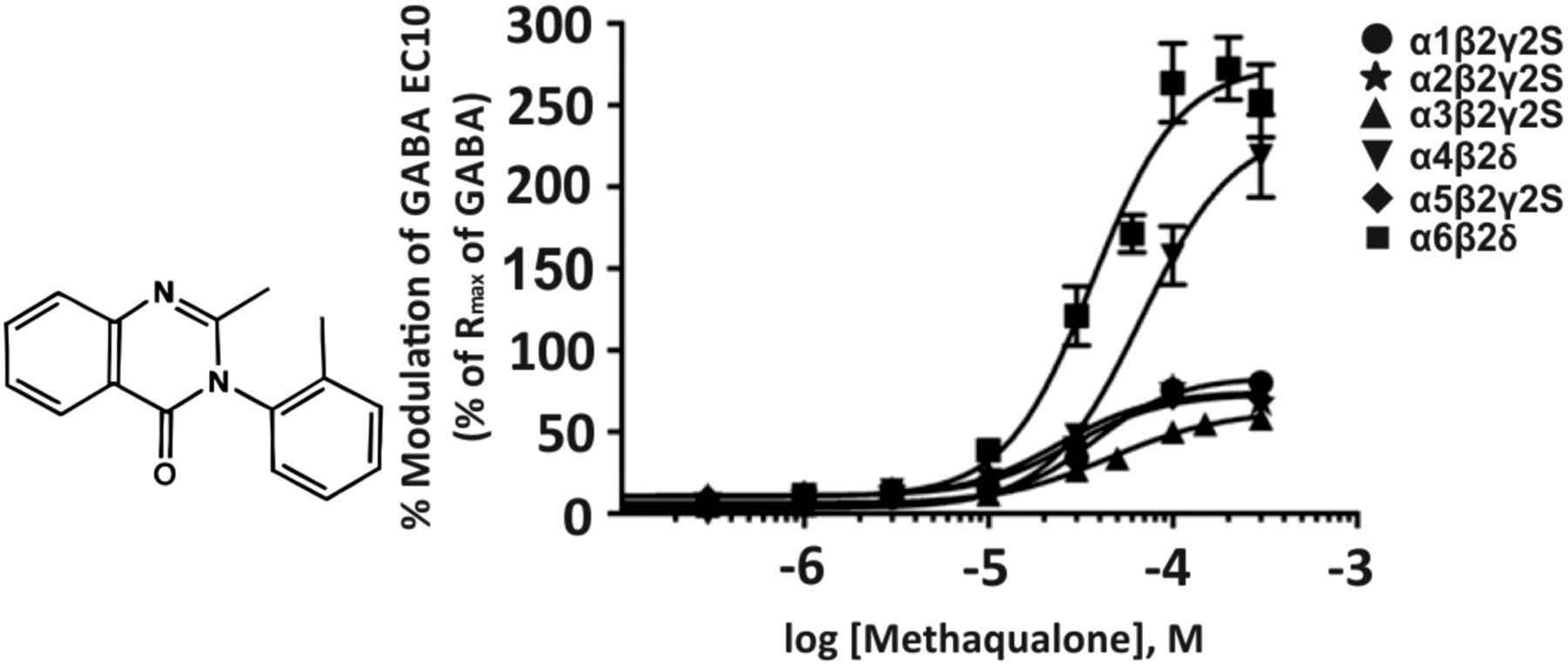{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Receptor Subtype-selective Binding Versus Subtype-selective Efficacy
- III. Receptor Subtype-selective Efficacy—Dependence on the Conditions of Measurements
- IV. Importance of Concentration-response Relationships of “Receptor Subtype-selective Ligands”
- V. Currently Available Compounds Claimed to Be γ-Aminobutyric Acid Type A Receptor Subtype-Selective
- VI. Discrepancy between the In Vivo Effects of Drugs in Rodent and Human Studies
- VII. Outlook
- Acknowledgments
- Authorship Contribution
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters