



Abstract
Many of the fundamental concepts of signal transduction and kinase activity are attributed to the discovery and crystallization of cAMP-dependent protein kinase, or protein kinase A. PKA is one of the best-studied kinases in human biology, with emphasis in biochemistry and biophysics, all the way to metabolism, hormone action, and gene expression regulation. It is surprising, however, that our understanding of PKA’s role in disease is largely underappreciated. Although genetic mutations in the PKA holoenzyme are known to cause diseases such as Carney complex, Cushing syndrome, and acrodysostosis, the story largely stops there. With the recent explosion of genomic medicine, we can finally appreciate the broader role of the Gαs-PKA pathway in disease, with contributions from aberrant functioning G proteins and G protein–coupled receptors, as well as multiple alterations in other pathway components and negative regulators. Together, these represent a broad family of diseases we term the Gαs-PKA pathway signalopathies. The Gαs-PKA pathway signalopathies encompass diseases caused by germline, postzygotic, and somatic mutations in the Gαs-PKA pathway, with largely endocrine and neoplastic phenotypes. Here, we present a signaling-centric review of Gαs-PKA–driven pathophysiology and integrate computational and structural analysis to identify mutational themes commonly exploited by the Gαs-PKA pathway signalopathies. Major mutational themes include hotspot activating mutations in Gαs, encoded by GNAS, and mutations that destabilize the PKA holoenzyme. With this review, we hope to incite further study and ultimately the development of new therapeutic strategies in the treatment of a wide range of human diseases.
Significance Statement Little recognition is given to the causative role of Gαs-PKA pathway dysregulation in disease, with effects ranging from infectious disease, endocrine syndromes, and many cancers, yet these disparate diseases can all be understood by common genetic themes and biochemical signaling connections. By highlighting these common pathogenic mechanisms and bridging multiple disciplines, important progress can be made toward therapeutic advances in treating Gαs-PKA pathway–driven disease.
I. Introduction
cAMP-dependent protein kinase, or protein kinase A, was one of the first kinases to be described as part of a signal transduction cascade and has served as the prototypical example ever since. As a holoenzyme, consisting of a regulatory (R) subunit dimer and two catalytic (C) subunits, PKA orchestrates complex protein phosphorylation networks by integrating upstream second messenger signals with spatial access to substrates; each layer is elegantly regulated to maintain homeostatic signaling across a diverse array of cell types. These signals manifest as a wide spectrum of physiologic functions, ranging from steroidogenesis in the adrenal cortex to stem cell maintenance in the hair follicle (Fig. 1A). Given this diversity and complex regulation, it is not surprising that mutations and dysregulation of PKA signaling can play a causative role in many human diseases. However, despite the vast amount of information surrounding PKA and its myriad of physiologic functions, the broad role of aberrant PKA signaling in disease is largely underappreciated. The study of signalopathies, or genetic disorders of signaling pathways, has emerged in recent years, including focuses on the Ras pathway (Rasopathies) (Tidyman and Rauen, 2009) and the transforming growth factor-β pathway (TGF-β signalopathies) (Cannaerts et al., 2015). Here, we define the newest member of the signalopathies, the Gαs-PKA pathway signalopathies. Gαs-PKA pathway signalopathies are defined as a family of diseases caused by germline, postzygotic, and somatic mutations in the Gαs-PKA pathway, with mutations commonly seen in GNAS, PRKACA, and PRKAR1A. In particular, we focus on endocrine and neoplastic diseases in which genetic data are strongly supported by mechanistic understanding of pathophysiology. With this review, we aim to bring together the existing body of knowledge surrounding aberrant pathway signaling in disease, bridging biochemistry, biology, physiology, and clinical practice under the umbrella of Gαs-PKA pathway signalopathies. By synthesizing the field, we hope to catalyze new efforts into the therapeutic targeting of a wide variety of human Gαs-PKA–driven diseases, ranging from endocrine and metabolic diseases to cancer.

(A) Protein kinase A is a central regulatory hub that mediates many physiologic processes, from hormonal growth and metabolism to transport and secretion. (B) Tables display the corresponding protein and gene names for each isoform of regulatory subunit and catalytic subunit. (C) Cartoon rendering of the PKA regulatory and catalytic subunit interactions. The interface of the catalytic subunit’s N-lobe (white) and C-lobe (olive) forms the active site of the kinase, helping to coordinate ATP and substrate. When the regulatory subunit is bound to the catalytic subunit, the inhibitory sequence (IS) occupies the active site to maintain the PKA holoenzyme in its inactive state. PKA exists as a holoenzyme composed of two regulatory and two catalytic subunits, that is coordinated through interactions with the D/D domains, which also bind to AKAPs (see Fig. 5D). When cAMP binds to and inactivates the two cAMP binding domains (CBD-A and CBD-B in teal) of the regulatory subunit, the catalytic subunit is free to phosphorylate its substrates.
II. Gαs–Protein Kinase A Pathway Basics
PKA is one of the best-characterized kinases and is a founding member of a large family of serine threonine kinases known as the ACG kinases (Hanks and Hunter, 1995). In 1991, PKA became the first kinase to have its crystal structure determined (Knighton et al., 1991a,b), and a similar architecture has now been characterized in over 550 structures to date. Traditionally, PKA exists as a tetrameric holoenzyme consisting of a homodimer of regulatory subunits (RIα, RIβ, RIIα, or RIIβ; encoded by the PRKAR1A, PRKAR1B, PRKAR2A, PRKAR2B genes, respectively) bound to two catalytic subunits (Cα, Cβ, Cγ, or the related Cχ and Cy; encoded by PRKACA, PRKACB, PRKACG, PRKX, and PRKY, respectively) (Fig. 1, B and C) (Turnham and Scott, 2016; Taylor et al., 2021). Under physiologic conditions, PKA becomes active when the second messenger 3′,5′-cAMP binds to the cAMP binding domains (CBDs) of the regulatory subunits and unleashes activity of the catalytic subunits (Kim et al., 2006; Turnham and Scott, 2016) (Fig. 1C).
A. Fine-Tuning cAMP Levels
The level of cAMP in cells is tightly controlled by balancing production from adenylyl cyclase (AC) and degradation by phosphodiesterase (PDE) (Fig. 2A), of which there are 10 AC isoforms (Hanoune and Defer, 2001) and eight PDE families known to act on cAMP (an additional three PDEs are specific to cGMP) (Omori and Kotera, 2007). Upstream signals that feed into the cAMP-PKA pathway are largely provided by inputs from Gαs (stimulatory)-linked and Gαi (inhibitory)-linked heterotrimeric G protein–coupled receptors (GPCRs) on the cell surface (Fig. 2A). Gαs is encoded by GNAS, whereas Gαi is encoded by GNAI 1/2/3. GPCR activity can be modulated by a variety of extracellular ligands, such as hormones, ultimately controlling the activation of their intracellularly coupled G proteins. Heterotrimeric G proteins consist of α, β, and γ subunits, of which there are several isoforms of each, including four major Gα families (Gαs, Gαi, Gαq, Gα12/13). Upon activation, G proteins dissociate from the receptor and are capable of activating downstream effectors (Oldham and Hamm, 2008). The majority of AC isoforms reside at the membrane and are regulated by Gαs and Gαi (AC1-9). Additionally, some isoforms can be activated by Gβγ (AC 2/4/7), but conversely, for AC5 and AC6, activation of Gβγ and phosphorylation by PKA can initiate negative regulation of cyclase activity. Of note, some AC isoforms can be activated (AC1/3/8 through calmodulin) or inhibited (AC5/6) by physiologic levels of Ca2+ (Hanoune and Defer, 2001). Unlike the other isoforms, soluble AC (encoded by ADCY10) resides in the cytoplasm and inside the mitochondrial matrix, where it is responsive to changes in both calcium and bicarbonate (Tresguerres et al., 2011) (Fig. 2A). Additional details about adenylyl cyclase isoforms and their signaling activities have been previously reviewed (Hanoune and Defer, 2001; Schmid et al., 2014; Halls and Cooper, 2017; Sanchez-Collado et al., 2021).

(A) Signaling through the PKA pathway involves upstream activation of Gαs-coupled GPCRs, which in turn activate AC to produce cAMP. Activation of Gαi-coupled GPCRs negatively regulates AC and cAMP production. Soluble AC (sAC) also contributes to cAMP production with activation by Ca2+ and HCO3−. Levels of cAMP in the cell are controlled by production from various ACs as well as degradation by PDEs. The PKA holoenzyme is a tetrameric complex consisting of two R subunits and two C subunits. AKAPs coordinate regulatory subunits and substrates. Additional binding domains present on AKAPs facilitated the formation of protein complexes and targeting to discrete locations around the cell. Binding of cAMP to regulatory subunits causes dissociation of the holoenzyme, releasing catalytic subunits to phosphorylate substrates. (B) Expression of protein kinase A pathway components across normal tissues. Genes are grouped in families, and expression level is represented as the median of transcripts per million (TPM) (GTex Portal). The heatmap displays expression from 0 to 200 TPM in blue and above 200 TPM in teal, with darker shades representing higher expression values.
Much like AC isoforms, the PDEs also have tissue-specific expression patterns and nonredundant function (Fig. 2B; Supplemental Table 1). Adding to the complexity of cAMP dynamics, many variants exist for each PDE gene as a result of the use of alternate promoters and splicing effects. PDEs primarily differ in their amino terminus, which controls localization and regulation (Houslay and Adams, 2003; Bender and Beavo, 2006; Omori and Kotera, 2007). The majority of cAMP-hydrolyzing PDEs harbor PKA phosphorylation sites. Although the function of many sites remains unknown, in PDE3 and PDE4, PKA phosphorylation enhances catalytic activity, serving to provide negative feedback on cAMP levels. Interestingly, PDE3, whose cAMP-hydrolyzing activity can be competitively inhibited by cGMP, is regulated by phosphorylation from both PKA and the phosphoinositide 3-kinase (PI3K) pathway downstream of hormone and growth factor receptors (Bender and Beavo, 2006). In the PDE4 family, the long isoforms contain a PKA phosphorylation site, which can enhance PDE catalytic activity by 60%. PDE4B/C/D also have an extracellular-signal-related kinase (ERK) phosphorylation site that inhibits PDE activity. Activation of mitogen-activated protein kinase (MAPK) activity induces an initial increase in cAMP (through PDE inhibition), which by activating PKA will in turn stimulate PDEs, ultimately lowering cAMP levels again in a coordinated fashion. Conversely, short PDE4 isoforms, lacking the PKA phosphosite, are inhibited by ERK phosphorylation leading to increased cAMP, but this is complicated by differential upstream regulation of Raf-1 proto-oncogene (RAF1) and B-Raf proto-oncogene(BRAF) by PKA (see section 5. Gαs–Protein Kinase A Induced Therapeutic Resistance in Cancer) (Houslay and Adams, 2003; Bender and Beavo, 2006). Additionally, all PDE4 members can be recruited to β-arrestins to control GPCR/G protein–mediated signaling (Bender and Beavo, 2006). This fact may explain why there seems to a preference for PDE4 homozygous deletions in colorectal cancer, a tissue context that is responsive to GPCR-mediated prostaglandin signaling and pathway-dependent cell growth (see section 3. GNAS and Protein Kinase A Link Inflammation to Cancer Initiation). The function and roles of different PDE isoforms have been previously reviewed (Bender and Beavo, 2006; Omori and Kotera, 2007; DeNinno, 2012; Neves-Zaph, 2017; Blair and Baillie, 2019).
B. Regulatory Subunits
The PKA regulatory subunits each comprise an amino terminal dimerization/docking (D/D) domain that is joined by an intrinsically disordered linker segment to two consecutive CBDs at the carboxyl terminus (Fig. 1C). Of note, the four regulatory subunits are structurally similar but have diverse expression patterns and are functionally nonredundant. RIα and RIIα are ubiquitously expressed, whereas RIβ and RIIβ exhibit more tissue-specific expression (Kim et al., 2006) (Fig. 2B; Supplemental Table 1). The holoenzyme exists in an inactive state because the regulatory subunits’ inhibitory sequence (IS), embedded within the linker region, occupies the active site of the catalytic subunit, acting as a pseudosubstrate or substrate (Fig. 1C). The main difference between type I (RI-containing) and type II (RII-containing) holoenzymes is that the IS of RII subunits can be autophosphorylated, whereas RI subunits act as pseudosubstrates. This has important implications for how the holoenzyme assembles and inhibits activity. Consequently, formation of a high-affinity type I holoenzyme requires the binding of ATP and two divalent metal ions (i.e., Mg2+), whereas type II holoenzymes will form with high affinity independent of ATP binding (Herberg and Taylor, 1993; Herberg et al., 1999; Amieux and McKnight, 2002; Kim et al., 2006; Wu et al., 2007; Taylor et al., 2012; Knape et al., 2017; Lu et al., 2019; Walker et al., 2019).
C. Catalytic Subunits
Upon cAMP binding to the regulatory subunits, the catalytic subunits become free to phosphorylate their substrates (Fig. 2A). Cα1 and Cβ1 are ubiquitously expressed, whereas other C subunits and their splice variants display more limited, tissue-specific expression (Turnham and Scott, 2016; Søberg and Skålhegg, 2018; Taylor et al., 2021) (Fig. 2B; Supplemental Table 1). The catalytic subunit itself is composed of two lobes, a small N-lobe that contains the ATP binding site and a larger helix-rich C-lobe that is essential for substrate binding and coordinating the transfer of the phosphate from ATP to the substrate. The interface between the two lobes forms the active site cleft of the kinase (Knighton et al., 1991a) (Fig. 1C). Under physiologic conditions, the stable and fully active catalytic subunit is phosphorylated on its activation loop (Thr197) and C-terminal tail (S338) (Adams et al., 1995; Yonemoto et al., 1997). PKA facilitates the transfer of the gamma phosphate of ATP to serine or threonine residues preferentially in the context of the consensus Arg-Arg-x-Ser*/Thr*-hydrophobic motif, a phosphorylation motif that is quite similar to that of other AGC kinase family members (Kemp et al., 1977; Bramson et al., 1984).
D. Protein Kinase A Microdomains
Scaffolding molecules, known as A-kinase anchoring proteins (AKAPs), concurrently bind PKA regulatory subunits and protein substrates to form microdomains, or cAMP signaling islands, that facilitate substrate recognition, recruitment, and phosphorylation, thereby enhancing PKA substrate specificity (Langeberg and Scott, 2015) (Fig. 2A). Additional enzymes (kinases, phosphatases, GTPases), signal transducers (receptors, channels), and pathway regulators (PDEs) can also associate with AKAPs, contributing to their ability to modulate PKA signaling (Greenwald and Saucerman, 2011; Torres-Quesada et al., 2017). Together, these AKAP-coordinated complexes facilitate the convergence and crosstalk of discrete signaling subnetworks. For instance, glycogen synthase kinase 3β interacting protein (GSKIP) is capable of binding the PKA substrate glycogen synthase kinase 3β (GSK3β) to control β-catenin–dependent signaling, whereas AKAP11 binds GSK3β to drive β-catenin independent signaling (Dema et al., 2016). Moreover, AKAP complexes coordinate spatial specificity of the phosphorylation event and enable targeting of PKA activity to particular subcellular locations. Nearly 50 different AKAPs have been identified, but with differential expression patterns (Fig. 2B; Supplemental Table 1) and alternative spliceforms also adding to the diversity, many of their binding partners and physiologic roles are still not fully understood (Torres-Quesada et al., 2017). Detailed reviews of what is known about the role of AKAPs have been compiled previously (Wong and Scott, 2004; Skroblin et al., 2010; Welch et al., 2010; Bucko and Scott, 2020; Omar and Scott, 2020).
In addition to physically restricting substrate access, PKA signaling is also regulated spatially by controlling local cAMP pools. Historically, it was thought that these cAMP microdomains were generated by localized AC inputs and restrained by PDEs, impeding diffusion throughout the cell (Mika et al., 2012). Recent studies have challenged this concept, demonstrating that at physiologic concentrations, cAMP is largely in a bound state and only diffuses upon displacement from or saturation of binding sites (i.e., upstream receptor/AC stimulation). These binding sites buffer cAMP diffusion throughout the cell, enabling PDEs to directly control cAMP compartments in their vicinity (10–60 nm) (Bock et al., 2020). To this end, recent work has also shown that RIα drives liquid-liquid phase separation as a mechanism to actively sequester cAMP, further contributing to cellular cAMP buffering (Zhang et al., 2020). Further supporting this concept of localized PKA activation, recent evidence has demonstrated that at physiologic cAMP concentrations, the PKA holoenzyme (as assessed by AKAP79 and type II holoenzyme interactions) does not physically dissociate upon cAMP binding, but rather, the catalytic subunits remains associated with AKAP and capable of phosphorylating substrates within its immediate vicinity (15–25 nm) (Smith et al., 2017). Together, these findings highlight even greater specificity of PKA activation than previously recognized. Importantly, disruption of this organization has been shown to drive aberrant PKA activity (Nikolaev et al., 2010; Zhang et al., 2020).
E. Transcriptional Regulation
PKA is perhaps best known for its ability to phosphorylate and activate the cAMP responsive element binding protein (CREB) family of transcription factors, of which there are three members [CREB1, cAMP responsive element modulator (CREM), and activating transcription factor 1 (ATF-1), although CREM can act as a negative regulator]. The function of CREB was originally described by its ability to drive the development of long-term memory, a process known to require gene transcription. At the time, cAMP and PKA had been shown to enhanced neurotransmission between sensory and motor neurons, contributing to short-term memory (Brunelli et al., 1976; Castellucci et al., 1980; Kandel, 2012). Subsequent work revealed that persistent activation of PKA and CREB-mediated transcription facilitated the transition from short-term to long-term memory (Dash et al., 1990; Alberini et al., 1994; Kandel, 2012). It is now known that, upon activation, PKA translocates to the nucleus, where it phosphorylates CREB on serine 133 (Bacskai et al., 1993; Rosenberg et al., 2002; Altarejos and Montminy, 2011) (Fig. 3A). CREB phosphorylation recruits coactivators, CREB-binding protein (CBP) or p300, through direct binding of the KIX domain present in CBP/p300 (Parker et al., 1996). Finally, CREB and CBP/p300 bind to cAMP-response elements (CREs) in the genome to drive transcription of target genes (Montminy et al., 1986; Rosenberg et al., 2002; Altarejos and Montminy, 2011). CBP and p300 are histone acetyltransferases that enhance the ability of CREB to activate transcription by relaxing the chromatin structure at gene promoter regions and creating scaffolds for recruitment of RNA polymerase II complexes to the promoter (Kee et al., 1996; Altarejos and Montminy, 2011). Another class of coactivators, the cAMP-regulated transcriptional coactivators (CRTCs), are also critical to enhancing CREB-mediated transcription. Under basal conditions, CRTCs are phosphorylated by salt-inducible kinase 2 (SIK2) and AMP-activated protein kinase (AMPK) kinases and sequestered in the cytoplasm through phosphorylation-dependent interactions with 14-3-3 proteins (Altarejos and Montminy, 2011). CRTCs are dephosphorylated by phosphatases, including calcineurin, protein phosphatase 1 (PP1), and protein phosphatase 2A (PP2A), allowing them to translocate from the cytoplasm to the nucleus to facilitate CREB-mediated transcription (Fig. 3A) ( Rosenberg et al., 2002; Altarejos and Montminy, 2011; Sonntag et al., 2019). Of note, PP2A is emerging as a valuable therapeutic target in the treatment of PKA-driven cancers (see section V. Targeting the Gαs–Protein Kinase A Pathway Signalopathies).

(A) Protein kinase A drives CREB-mediated transcription. When hormone binds to Gαs-linked GPCRs on the cell surface, signaling through adenylyl cyclase stimulates cAMP production and PKA activation. Activation of Gαi-coupled GPCRs inhibits adenylyl cyclase and cAMP production. When active, C subunits translocate to the nucleus to phosphorylate CREB on serine 133. Phosphorylated CREB recruits coactivators like CBP to facilitate binding to CREs and transcription of target genes. Additional coactivators, like CRTCs, help to regulate CREB-mediated transcription. Phosphorylation of CRTCs by other kinases results in cytoplasmic sequestration, whereas dephosphorylation by phosphatase enables translocation to the nucleus. (B) cAMP binds and activates effectors beyond PKA. Binding of cAMP to CNG ion channels regulates channel opening and cation currents. HCN channels also bind cAMP to facilitate channel opening by membrane hyperpolarization. cAMP binds to EPAC to facilitate the exchange of GDP for GTP on the RAP family of small GTPases. POPDC proteins reside on the cell surface as dimers that bind cAMP.
Over 10,000 accessible CRE binding sites have been identified in humans, including some likely to represent alternative or bidirectional promoters. However, the majority reside within 200 base pairs of transcription start sites. Together, this accounts for regulation of over 4000 genes (Impey et al., 2004; Zhang et al., 2005). Genes vary in their dependence on coactivators and CREB occupancy, ensuring that transcriptional activation is finely tuned to specific PKA stimuli (Altarejos and Montminy, 2011). CREB target genes highlight most of the key physiologic processes we will discuss, including regulation of PKA pathway activity, cell cycle entry, mitochondrial homeostasis, and metabolism (Fig. 1A). Interestingly, many CREB target genes are themselves transcription factors (e.g., c-Jun, c-Fos), adding a temporal layer to the importance of PKA-driven transcription (Impey et al., 2004; Zhang et al., 2005). It is important to note, however, that PKA also regulates transcriptional programs independent of CREB. As we will discuss later, PKA phosphorylates components of other pathways (e.g., Wnt, sonic hedgehog, Hippo) to regulate their transcriptional output (see section 3. GNAS and Protein Kinase A Link Inflammation to Cancer Initiation and 4. GNAS–Protein Kinase A as Tumor Suppressors). Together, transcriptional effects and gene expression regulation permeate almost every role of PKA (physiologic or aberrant).
F. Metabolic Regulation
Another one of the major physiologic roles of PKA is in regulation of glucose and lipid metabolism. Excess glucose in the body can be stored as glycogen (glycogenesis) in the liver or skeletal muscles. Coordinated activities of PKA (in response to glucagon or β-adrenergic receptor stimulation) help to regulate the breakdown of glycogen and mobilization of glucose in times of low nutrient intake. For instance, PKA directly phosphorylates to inhibit glycogen synthase, one of the major enzymes responsible for glycogenesis, and at the same time phosphorylates to activate glycogen phosphorylase kinase, one of the major enzymes responsible for glycogen breakdown (Han et al., 2016; Yang and Yang, 2016). When glycogen stores become depleted, PKA also participates in gluconeogenesis to elevate glucose levels. PKA acts through direct phosphorylation and regulation of enzymes participating in gluconeogenesis as well as transcriptional activation (Yang and Yang, 2016). The transcriptional response of PKA is mediated by CREB, and as such, small interfering RNA (siRNA) knockdown of CREB in the liver decreases blood glucose levels and reduces expression of gluconeogenesis genes (Erion et al., 2009). Conversely, in a mouse model of CBP/CREB overactivity, gluconeogenesis is inappropriately activated during fed conditions, leading to glucose intolerance (Zhou et al., 2004). Genetic mouse models activating PKA Cα and RIα (dominant negative) also recapitulate these effects on glycogen and gluconeogenesis (Niswender et al., 2005; Willis et al., 2011; Yang and Yang, 2016).
Lipogenesis is another process by which glucose can be stored, in this case by conversion to fatty acids. Fatty acids are eventually stored as triglycerides in lipid droplets. When energy levels drop, fatty acids can be liberated by lipolysis. PKA is anchored to lipid droplets by an AKAP and known to activate lipolysis in adipose tissue through several mechanisms, most notably through phosphorylation of perilipin A (Rogne and Taskén, 2014; Yang and Yang, 2016). The so-called gatekeeper of lipolysis, perilipin covers the outer surface of lipid droplets, preventing the action of lipases (Rogne and Taskén, 2014). PKA phosphorylates perilipin to induce conformational changes that allow lipases to access the lipid droplet (Brasaemle et al., 2009). PKA can also phosphorylate and activate the lipases adipose triglyceride lipase and hormone-sensitive lipase, which participate in the multistep process of lipolysis, converting triglycerides to free fatty acids (Rogne and Taskén, 2014; Yang and Yang, 2016).
As the Gαs-PKA pathway is integral to many hormone-driven processes, it is not surprising that PKA is also heavily involved in steroidogenesis. Steroid hormones are small lipid signaling molecules derivative from cholesterol. PKA promotes cholesterol processing and steroid biosynthesis both directly through modulation of enzymes (cholesteryl ester hydrolase) and transcriptionally through phosphorylation and activation of transcription factors [CREB, steroidogenic factor 1 (SF-1), GATA binding protein 4 (GATA -4)] (Dyson et al., 2009; Manna et al., 2009). In addition to transcriptional regulation, PKA also regulates steroidogenic acute regulatory protein (StAR) post-translationally. StAR is important for transporting cholesterol into the mitochondria, where it is processed. PKA phosphorylation is strictly required for activation of StAR, an event that is facilitated in part by AKAP1 anchoring of PKA to the mitochondrial outer membrane (Dyson et al., 2009; Manna et al., 2009).
Given the direct regulation of both glucose and lipid by the Gαs-PKA pathway, many of the Gαs-PKA pathway signalopathies have hyperglycemic or obesity-related phenotypes. For instance, mutational activation of PKA (as in Cushing syndrome) can lead to hyperglycemia, and several pathway mutations are linked to development of diabetes mellitus (Sharma et al., 2015; Tengholm and Gylfe, 2017) (see section B. Endocrine and Metabolic Diseases). Although these mechanisms provide some explanation for the phenotypes in many Gαs-PKA pathway signalopathies, it is important to recognize that PKA’s role in metabolism is quite complicated, owing to the multilayer regulatory programs, including effects on enzyme activity, hormone secretion, and transcriptional responses.
G. Other cAMP Effectors
It is important to note that, although PKA is the major direct effector of cAMP, it is not the only one. When cAMP is free, it is capable of binding to and activating cyclic nucleotide–gated ion channels, exchange factors, and Popeye domain containing (POPDC) proteins (Fig. 3B). These additional cAMP-dependent signaling mechanisms are briefly described below.
Cyclic nucleotide–gated (CNG) channels are ion channels that participate primarily in the sensory processes of sight and smell, converting second messenger signals to voltage changes (Brown et al., 2006). CNG channels are nonselectively permeable to cations, but the action of Ca2+ predominates under physiologic conditions. Unlike other gated ion channels, CNG channels are not subject to desensitization; rather, they are regulated in their affinity for cyclic nucleotides. For instance, binding of Ca2+/calmodulin or post-translational modifications can alter the channels’ binding affinities. The various CNG channels also have differing innate affinities for cAMP versus cGMP, but in general, cAMP is the dominant signal in olfaction (Zagotta and Siegelbaum, 1996; Bradley et al., 2005). Sensory GPCRs function as signal detectors in both sight and smell processes. Olfactory GPCRs couple to Gαolf (encoded by GNAL), which functions like Gαs to stimulate AC and cAMP production, whereas rhodopsins (visual GPCRs) couple to transducin (Gαt) (encoded by GNAT1) to induce cGMP hydrolysis, explaining the importance of cAMP to olfaction (Julius and Nathans, 2012).
Another class of cyclic nucleotide–gated ion channels, known as hyperpolarization-activated, cyclic nucleotide–modulated (HCN) channels, function primarily at the sinoatrial (SA) node to maintain heartbeat. HCN channels are distinct from CNG channels in that they are regulated by membrane hyperpolarization in addition to binding of cyclic nucleotides (Brown et al., 2006; Biel, 2009). For HCN channels, the cyclic nucleotide binding domain serves an autoinhibitory function by making the channel more difficult to activate (through hyperpolarization) in the absence of cAMP (Wainger et al., 2001). In the SA node, stimulation of the sympathetic nervous system increases cAMP and facilitates channel opening in response to membrane hyperpolarization after an action potential. When activated, HCN channels allow the influx of cations, contributing to the slow depolarization during diastole and priming the SA node for initiation of another action potential. HCN channels can also play a role in other excitable tissues like neurons (Brown et al., 2006; Biel, 2009).
Although the roles of CNG and HCN channels are very specific for regulating currents, the roles of exchange protein directly activated by cAMP (EPACs) are much broader. As guanine nucleotide exchange factors, EPACs activate the small GTPases Ras-related protein 1 and 2 (RAP 1 and RAP2, respectively). There are two EPAC proteins, EPAC1 and EPAC2 (encoded by RAPGEF3 and RAPGEF4), which contain one and two CBDs, respectively. When cAMP binds to the CBD, a conformation change occurs to expose the critical residues that participate in the exchange of GDP for GTP to activate RAP1/2. EPAC1/2 are expressed in most tissues, and by modulating RAP activity, they play important roles in cell adhesion in many contexts. Much like PKA signaling, EPAC signaling is compartmentalized and controlled by local cAMP pools. EPACs use their domain structures, Dishevelled, Egl-10, and Pleckstrin (DEP) and Ras association (RA) domains, to target different cellular compartments and engage binding partners. Interestingly, PKA and EPAC participate in many of the same processes, with examples of both antagonistic and synergistic functions, and they have even been found in the same protein complexes. Of note, PKA is activated at much lower levels of cAMP than EPAC, providing another example of the dynamic responses to cAMP regulation (Gloerich and Bos, 2010).
The CBDs of PKA, CNG/HCN channels, and EPACs, are quite similar, but the POPDC proteins use a very different domain to bind cAMP, but still with a high affinity similar to that of PKA. POPDC proteins (encoded by POPDC1, POPDC2, and POPDC3) were named after Popeye the Sailor Man because they are highly expressed in striated muscle. POPDC proteins are heavily glycosylated and reside in the membrane, where they are involved in cell-cell contacts, vesicular transport, and epithelial morphology. They are expressed in many tissues but are primarily studied in the context of cardiac function and epithelial cell organization. Importantly, their dysfunction, downregulation, and mutation have been associated with arrhythmias, muscular dystrophy, and epithelial-to-mesenchymal transition effects in cancer (Schindler and Brand, 2016).
III. Mutational Landscape of the Gαs–Protein Kinase A Pathway Signalopathies
The Gαs-PKA pathway signalopathies represent a diverse group of diseases and disorders characterized by dysregulation of the Gαs-PKA pathway. As we will discuss in the next sections, the Gαs-PKA pathway signalopathies are defined by mutations, predominately in the Gαs subunit of GPCRs (encoded by GNAS) or the PKA holoenzyme (mainly PRKACA and PRKAR1A). Given the diversity of clinical phenotypes, many groups have aimed to understand the specific mechanisms of mutational activation (or inactivation). Here, we will highlight what is known about the structural and functional significance of disease-associated mutations and integrate available data from inherited (Landrum et al., 2020) and somatic mutation databases (Kim and Zhou, 2019; Tate et al., 2019) to identify broader mutational themes that contribute to the Gαs-PKA pathway signalopathies. Of note, in addition to drawing from publicly available databases, we also aim to highlight examples of mutational themes identified from the literature.
A. Mutations in GNAS
Mutations in GNAS are dominated by hotspot mutations at two residues, R201C/S/G/H/L and Q227L/K/R/H (Fig. 4A; Supplemental Tables 2 and 3). These residues are conserved across Gα subunits and reside within the switch I and switch II regions, respectively, which universally characterize GTPases, including small GTPases of the Ras superfamily. Switch I and switch II respond to changes in GTP and GDP binding by sensing the presence or absence of the gamma phosphate (Fig. 4, B and C). These residues are essential for GTPase activity, and thus, their mutation results in impaired GTPase function and constitutive activity (Sunahara et al., 1997; O'Hayre et al., 2013; Sprang, 2016). Recent work has also suggested that GNAS R201C may be capable of activating adenylyl cyclase and downstream signaling even in the presence of GDP, an event that is normally restricted to the GTP bound state (Hu and Shokat, 2018). Interestingly, R201 mutations are far more prevalent in human disease than Q227 (O'Hayre et al., 2013; Arang and Gutkind, 2020). This discrepancy is most striking in cancer, in which nearly 50% of all GNAS mutations are at R201, whereas only 2% are at Q227 (Fig. 4A; Supplemental Table 3). Little is known about why this preference occurs, but it could be linked to the biologic activity of the mutation, as is the case for another G protein, Gαq (encoded by GNAQ). For instance, uveal melanoma, the most common cancer of the eye, is almost exclusively caused by GNAQ mutations at residue Q209 (corresponding to GNAS Q227), whereas Sturge-Weber syndrome, characterized by angiomas or tumors of small blood vessels, is caused by GNAQ R183 mutations (corresponding to GNAS R201). GNAQ R183 mutants are responsive to signal termination by regulator of G protein signaling proteins, whereas Q209 mutants are not. This highlights that Q209 mutants are more active and consequently drive more extensive proliferation (O'Hayre et al., 2013; Shirley et al., 2013; Arang and Gutkind, 2020). Unlike Gαq, Gαs does not bind regulator of G protein signaling proteins as a mechanism to turn off signaling (Natochin and Artemyev, 1998a,b). Additionally, GNAS Q227 mutants have higher intrinsic activity than R201 mutants, contributing to greater proliferation and secretion (Landis et al., 1989; Ham et al., 1997). In the case of Gαs, fine-tuned regulation is critical, as too much or too little activity can be incompatible with life (Yu et al., 1998; Khan et al., 2018). Together, these findings suggest that Q227 mutations may not be tolerated in many contexts; thus, R201 mutations may be biologically selected.

GNAS mutational themes in disease. (A) Lollipop plots depict the location of GNAS mutation along the gene body in genetic diseases. Both activating and inactivating mutations are depicted. The height of the lollipop is representative of pathogenic mutation number (ClinVar database) (Landrum et al., 2020). Below the gene body, colored circles depict the location of cancer mutations (COSMIC database) ( Tate et al., 2019). The frequency of residue mutation (residue representing >1% of all GNAS mutations) is shown, with darker blue representing a larger proportion of GNAS mutations occurring at that residue. Hotspot mutations in the switch I and switch II domains are dominant in both genetic diseases and cancer. (B) Structure of the prototypical β2 adrenergic receptor (β2AR) coupled to the heterotrimeric Gαs G protein (protein data bank ID: 3SN6). Pathogenic mutations are shown in red spheres. Recurrent mutations are present in the nucleotide binding pocket. Other mutations are present at the receptor–G protein interface and in residues interacting with the Gβγ subunits. (C) Structure of Gαs binding to adenylyl cyclase (PDB: 1AZS) highlights the mutations clustered in the nucleotide binding pocket (switch I and switch II).
Similar to mutations, spliceforms of Gαs also seem to contribute to this tight regulation of activity, with differential splice preference in disease states, such as obesity, hypertension, and diabetes (Novotny and Svoboda, 1998). The long isoform (inclusion of exon 3) has a lower binding affinity for GDP, making it more easily exchanged for GTP and therefore more easily activated (Seifert et al., 1998). In fact, coupling of the long isoform to the glucagon receptor enhances glucagon binding affinity as much as 10-fold (Unson et al., 2000). Despite these findings, the direct disease-causing ability of either spliceform has yet to be established. Finally, although diseases may have preferential ways to activate Gαs, mutation of many different residues can disable Gαs activity, as missense mutations have been found in almost every exon of GNAS, with many of them leading to truncation mutations and haploinsufficiency (Weinstein et al., 2004) (Fig. 4A; Supplemental Table 2). Of note, there are also point mutations at the receptor–G protein interface (E392K and L388R) that are likely loss of function based on the patients’ clinical phenotype, suggesting that disruption of receptor–G protein contacts represents another mutational mechanism (Fig. 4B). Ultimately, these data highlight that achieving the proper balance of Gαs activity is critical, and thus, its dysregulation is closely tied to disease.
B. Mutations in Protein Kinase A Catalytic Subunits
Since PKA functions as a holoenzyme, the mutational themes in PKA are quite different than the hotspot mutations we observe in Gαs. Among the Gαs-PKA pathway signalopathies, Cushing syndrome caused by adrenocortical adenomas is the disease most commonly caused by mutations in PKA catalytic subunits and serves as an excellent example of activation themes exploited by mutations (see section 2. Cushing Syndrome and Adrenocortical Adenomas). Likely because of its ubiquitous expression and functional roles in many tissues (Fig. 2B; Supplemental Table 1), the majority of mutations occur in PRKACA or Cα. As mentioned previously, each catalytic subunit is composed of two lobes, the N-lobe, harboring the ATP binding site, and the C-lobe, responsible for substrate binding (Figs. 1C and 5A). The interface of these two lobes forms the active site, an interaction that is largely mediated by the binding of ATP and facilitates the opening and closing of this active site cleft along with substrate engagement. This interface also contacts the IS that is embedded in the intrinsically disordered linker region of each regulatory subunit (Figs. 1C and 5B). When the holoenzyme is inactive, the IS is locked into the active site of the C subunit, which prevents the binding of substrates (Johnson et al., 2001; Taylor et al., 2012). A sequence similar to the IS is also found in protein kinase inhibitor (PKI) isoforms, where it also acts as a pseudosubstrate. Briefly, PKIs are endogenous peptide inhibitors of PKA that are expressed in a variety of human tissues. When catalytic subunits are free, PKIs bind to and block PKA activation in response to cAMP, primarily operating in the cytoplasm or nucleus (Liu et al., 2020) (see section 2. Peptide Inhibitors of the Catalytic Subunit).
The most frequent mutation in PKA Cα is L206R, which lies near the interface of the N- and C-lobes, and contributes to the R:C interface as well as substrate recognition (Fig. 5, A and B; Supplemental Tables 2 and 3). Consequently, this mutation disrupts critical contacts and leads to PKA activation by multiple mechanisms (Walker et al., 2019). First, the L206R mutation disrupts interactions between Cα and the regulatory subunits, leading to constitutive activity even in the absence of cAMP (Calebiro et al., 2014; Röck et al., 2015). L206 (or 205 depending on numbering conventions), along with other residues, is part of a hydrophobic pocket that binds substrates as well as the regulatory subunits’ IS (Fig. 5B). Introduction of a more bulky, positively charged residue disrupts this hydrophobic interaction sterically and chemically (Moore et al., 2003; Taylor et al., 2012; Calebiro et al., 2014). As alluded to previously, the residues in the active site are critical for controlling the activity and regulation of Cα as well as for substrate recognition. They are important not only for intermolecular contacts but also for intramolecular or allosteric interactions. By measuring chemical shift perturbations through NMR, it is evident that wild-type and L206R Cα have dramatically different allosteric networks that alter the normal binding cooperativity between ATP and substrates. Ultimately, this results in an inability of L206R to achieve a fully closed state. Similarly, molecular dynamics simulations reveal that L206R has a much broader conformational range than wild-type Cα. Together, the disruption of normal allosteric interactions and protein conformations results in an altered substrate specificity (Walker et al., 2019, 2021). This altered substrate profile includes decreased activity toward canonical substrates and increase activity toward noncanonical substrates, particularly those with negatively charged (instead of hydrophobic) residues after the consensus phosphorylation motif (Arg-Arg-x-Ser*/Thr*-hydrophobic). Although the intrinsic activity of the Cα L206R does not seem to differ from the wild type, the altered substrate profile may contribute to aberrant signaling (Calebiro et al., 2014; Lubner et al., 2017; Luzi et al., 2018; Bathon et al., 2019; Walker et al., 2019, 2021).

Protein kinase A mutational themes. (A) Lollipop plots of PRKACA pathogenic mutations in genetic diseases (ClinVar database) (Landrum et al., 2020). The frequency of residue mutation in cancer (residues representing >1% of all PRKACA mutations) is depicted below (COSMIC database) (Tate et al., 2019). Darker blue represents that a greater proportion of PRKACA mutations occur at that residue. (B) Structure of Cα in complex with RIα (protein data bank ID: 5JR7). Pathogenic mutations are depicted as red spheres. PRKACA mutations lie at the interface of the catalytic and regulatory subunits, whereas PRKAR1A mutations are distributed throughout the protein. (C) As in (A), lollipop plots of genetic disease mutations in PRKACB and frequency of residue mutation in cancer below. (D) Structure of the R binding domain of AKAP10 in complex with RIα (PDB: 3IM4) [dotted line connects to the same region of RIα as shown in (B)]. Mutations of unknown significance (shown in yellow) reside within the D/D domain that mediates regulatory subunit dimerization and AKAP binding. (E) As in (A) and (C), lollipop plots and cancer residue frequency illustrate that no recurrent mutations occur in PRKAR1A.
Most Cushing syndrome mutations as well as cancer mutations in Cα (W197, L199_C200insW, C200_G201insV, S213R, E249Q) are located in the C-lobe near the active site cleft and contribute to peptide recognition. This region also includes the binding surface for the regulatory subunits (Fig. 5A; Supplemental Table 3). Because of their location in this critical region, it is thought that these mutations achieve PKA activation through mechanisms similar to L206R (Luzi et al., 2018; Walker et al., 2019; Walker et al., 2021). The E32V mutation is the only mutation that resides away from the active site, but similar to the other Cushing mutations, E32V also disrupts intramolecular allosteric interactions, leading to loss of binding cooperativity and aberrant activity (Ronchi et al., 2016; Walker et al., 2019, 2021). In fact, there is direct relationship between ATP-substrate binding cooperativity and intramolecular allosteric conformational changes, highlighting that this is a common feature disrupted by Cushing syndrome mutations (Walker et al., 2021). Recently, additional point mutations in both PRKACA and PRKACB have been described in several Gαs-PKA pathway signalopathies, including Cushing syndrome. Like many of the Cα mutations mentioned above, Cβ mutations S54L and H88R/N are located in a region critical for ATP and substrate binding near the active site. Interestingly, S54L and H88R/N both show increased sensitivity to cAMP as a result of reduced stability of the respective PKA holoenzymes and C:PKI interactions (Espiard et al., 2018; Palencia-Campos et al., 2020; Taylor et al., 2021) (Fig. 5C; Supplemental Tables 2 and 3). Similarly, other recently characterized mutations Cα G137R and Cβ G235R have reduced affinity for regulatory subunits and, consequently, increased PKA kinase activity at low cAMP levels. Although Cα G137R and Cβ G235R do not affect ATP binding, they do reside in the area that makes contacts with regulatory subunits as well as PKI (Palencia-Campos et al., 2020) (Fig. 5, A, B, and C; Supplemental Table 2). Together, these mutations highlight holoenzyme destabilization or defects in PKI signaling as alternative mechanisms to enhance PKA activity without altering intrinsic kinase activity (Espiard et al., 2018; Palencia-Campos et al., 2020).
C. Mutations in Protein Kinase A Regulatory Subunits
Destabilization of the PKA holoenzyme and disruption of regulatory-catalytic subunit contacts is the major mutational theme underlying the Gαs-PKA pathway signalopathies. Although we have already discussed the role of catalytic subunits in these interactions, mutation of regulatory subunits is actually the most frequently observed alteration (Fig. 5, B, D, and E; Supplemental Table 2). In fact, over 130 molecular defects in PRKAR1A, or RIα, have been associated with Gαs-PKA pathway signalopathies (PRKAR1A Mutation Database, https://prkar1a.nichd.nih.gov/). As is the case for PKA Cα, most mutations occur in Riα, likely due to its ubiquitous expression and functional importance in many tissues (Fig. 2B; Supplemental Table 1). These mutations span the length of the protein, ranging from missense mutations and premature stop codons to insertions and deletions, with deletions as large as 4 kb described (Kirschner et al., 2000a; Horvath et al., 2008, 2010). The loss-of-function nature explains why there are no recurrent mutations in PRKAR1A found in cancer (Fig. 5E; Supplemental Table 3). This pattern of gene mutations throughout the gene length is well established for known tumor suppressor genes in cancer (Vogelstein et al., 2013). Mutations may lead to altered function, alternative protein expression, and even absence of protein. Many of the premature stop codons or small insertions and deletions lead to nonsense mediated decay (NMD), representing 90% of PRKAR1A mutations (Greene et al., 2008; Bertherat et al., 2009; Horvath et al., 2010). NMD occurs at the mRNA level as a normal quality-control mechanism to prevent the translation of truncated proteins. Strong NMD mutations typically occur at least 50 base pairs upstream of the final exon-exon junction (Brogna and Wen, 2009). For these NMD mutations, the mutant protein is not expressed, leading to 50% reduction in RIα protein and consequently haploinsufficiency. PKA activity is ultimately enhanced because of disruption of the normal holoenzyme stoichiometry (Horvath et al., 2010). Alterations that occur in the last exon actually escape NMD and are translated (Veugelers et al., 2004). Interestingly, some of these mutations, specifically those coding for an elongated protein, are subject to proteasomal degradation and result in haploinsufficiency as well (Patronas et al., 2012).
Although most PRKAR1A mutations result in haploinsufficiency due to mRNA NMD or protein degradation, the mutations that successfully evade these quality-control mechanisms, forming alternative RIα protein, actually contribute to more severe disease (Meoli et al., 2008; Horvath et al., 2010). On a biochemical level, they are also incredibly informative of PKA holoenzyme dynamics. For instance, there are two regions of RIα that are critical to catalytic subunit binding, one of which is within the first CBD (CBD-A) (Fig. 5, B and E). Disruption of this interaction site by mutation results in increased PKA activity independent of cAMP levels, as the mutant RIα is unable to bind the catalytic subunit (Greene et al., 2008; Meoli et al., 2008). This is mirrored by large deletions that result in deletion of exon 3, which contains the IS, the other region critical to catalytic subunit binding (Greene et al., 2008; Horvath et al., 2008). There are also several mutations (D183Y, A213D, and G289W) that reside within the two cAMP binding domains (CBD-A and CBD-B) and have decreased binding affinity for cAMP but greater overall PKA activity. Other mutations, exhibiting similar decreased cAMP binding affinity, have been identified in critical regions such as the D/D domain (S9N), which alters protein conformation and disrupts the communication between the D/D domain, and the CBDs (Hamuro et al., 2004; Greene et al., 2008). These types of mutations may also disrupt AKAP scaffolding interactions, as the D/D domain mediates these contacts (Fig. 5, D and E). As mutations occur throughout the RIα protein, it is thought that many of the missense mutations located outside of functional domains may contribute to PKA activation through similar disruption of conformational communication (Hamuro et al., 2004; Veugelers et al., 2004; Greene et al., 2008).
Most of the RIα mutations we have discussed so far result in increased PKA activity. Conversely, there is a whole class of RIα mutations that suppress PKA activity, leading to different pathologies. Given the underlying importance of holoenzyme stability, it is not surprising that these mutations stabilize the holoenzyme, often rendering it less sensitive to dissociation by cAMP. To this point, we observe many of the acrodysostosis mutations (discussed further in section 7. Inactivating Parathyroid Hormone/Parathyroid Hormone–Related Peptide Signaling Disorder) cluster in the C terminus of the protein, the region where the two CBDs reside (Fig. 5E). Point mutations within the CBDs (CBD-A: Y175C, A213T and CDB-B: Q285R, G289E, A328V, R335L) as well as partial deletion of CBD-B (R368X, Q372X) make RIα resistant to cAMP, dampening PKA activity (Linglart et al., 2011; Rhayem et al., 2015; Bruystens et al., 2016). CBD-B is particularly important because binding of cAMP to CBD-B results in a conformation change that exposes CBD-A (Kim et al., 2007). Perhaps the most interesting finding is related to residues A213 and G289. As mentioned previously, their mutation decreases cAMP binding; however, depending on the residue, this can lead to completely different clinical presentations. A213T and G289E result in acrodysostosis and inhibit PKA activity, whereas A213D and G289W result in Carney complex disease and activate PKA activity. Interestingly, although all mutations display decreased cAMP binding, RIα G289W is rapidly degraded, resulting in PKA activation. RIα A213D on the other hand has a reduced degradation rate but appears to become inappropriately activated without holoenzyme dissociation and at extremely low cAMP levels. Of note, this increased activity is comparable with RIα WT at low cAMP levels but completely lost at high levels of cAMP (Rhayem et al., 2015).
D. Fusion Proteins: An Emerging Mutational Theme
As we discussed in the previous two sections, there are many mechanisms to disrupt normal PKA regulation and stability. The unexpected discovery of PKA fusion proteins in cancer has added yet another mechanism to the list. Honeyman et al., 2014 revealed that patients with fibrolamellar hepatocellular carcinoma (FL-HCC) express a chimeric RNA transcript that fuses the J domain of the molecular chaperone DnaJ homolog subfamily B member 1 (DNAJB1) in frame with PKA Cα (DNAJB1-PRKACA) because of an ∼400-kb deletion on chromosome 19 (Honeyman et al., 2014) (Fig. 6A). On a molecular level, the fusion protein retains kinase activity (Honeyman et al., 2014; Riggle et al., 2016) and normal contacts with PKI (Cheung et al., 2015) and Riα (Cao et al., 2019) and RIIβ (Lu et al., 2020) regulatory subunits. The fusion protein also retains interactions with AKAPs (including atypical AKAPs that associate with the amino terminal region of Cα) (Cheung et al., 2015; Riggle et al., 2016) and even interactions with heat shock protein 70 (HSP70) through the fused J domain (Turnham et al., 2019). Surprisingly, however, the fusion protein does disrupt normal RIα-mediated liquid-liquid phase separation and cAMP compartmentalization, potentially contributing to its oncogenic activity (Zhang et al., 2020). Furthermore, because the fusion protein is expressed from the DNAJB1 promoter, it results in relative overexpression compared with wild-type PKA Cα, which may be augmented by enhanced mRNA stability due to loss of 3′ untranslated region (UTR) regulation (Riggle et al., 2016). While the fusion protein maintains similar intrinsic kinase activity, the DNAJB1-PRKACA fusion protein may also achieve increased PKA activity due to increased responsiveness to cAMP, likely due to decreased holoenzyme stability and/or disruption of allosteric regulation (Cheung et al., 2015; Riggle et al., 2016; Lu et al., 2020). Importantly, the dynamic features of the PKI complex are also significantly altered (Olivieri et al., 2021). Recently, additional PKA fusion proteins involving ATP1B1 as the N-terminal fusion partner (ATP1B1-PRKACA and ATP1B1-PRKACB) have also been described to share a similar breakpoint as DNAJB1-PRKACA and exhibit increased catalytic subunit expression due to use of the ATP1B1 promoter (Nakamura et al., 2015; Singhi et al., 2020; Vyas et al., 2020) (Fig. 6A).

(A) Protein kinase A catalytic subunit fusion proteins identified in cancer. Colored lines on chromosome 1 (Chr 1) indicate the genomic position of PRKACB and ATP1B1 (green). Chromosome 19 (Chr 19) harbors DNAJB1 (red) and PRKACA (purple). Exon 1 of DNAJB1 or ATP1B1 is fused at the same position in PRKACA and PRKACB (exon 2–10). (B) RET/ptc2 fusion protein identified in papillary thyroid cancer fuses the N terminus of PRKAR1A on chromosome 17 (Chr 17, orange), including the D/D domain, with the tyrosine kinase domain of RET on chromosome 10 (Chr 10, pink). Two tyrosine residues are essential for mitogenic activity and participate in scaffolding interactions. (C) Prevalence of PKA pathway fusion proteins across cancer types (Fusion GDB) (Kim and Zhou, 2019). Among pathway genes, GNAS is the most common fusion partner.
Although the PKA catalytic subunit fusion proteins are certainly the most striking examples, they are not the only fusion proteins that exist within the PKA pathway. In papillary thyroid cancers (PTCs), fusions of PRKAR1A and the RET receptor tyrosine kinase have been described. Termed RET/ptc2, these chimeras fuse the N terminus of RIα with the tyrosine kinase domain of RET (Lanzi et al., 1992) (Fig. 6B; Supplemental Table 4). Interestingly, the D/D domain is the most important region of RIα required to mediate mitogenic activity when fused to the RET tyrosine kinase domain. Since RIα exists as a dimer, it is believed that the D/D domain facilitates fusion protein dimerization (a required step in normal receptor tyrosine kinase activation) and subsequent activation of RET, as the same proliferative effects can be observed with substitution of the epidermal growth factor receptor (EGFR) tyrosine kinase domain for that of RET (Durick et al., 1995, 1996). In fact, the contribution of dimerization domains from N-terminal fusions partners is well documented to drive constitutive dimerization and activation of other receptor tyrosine kinase fusion proteins (Nelson et al., 2017). Interestingly, there are two tyrosine residues conserved within the RET portion of the fusion protein that are also essential to mitogenic activity, likely as a result of their participation in scaffolding interactions when phosphorylated (Durick et al., 1995, 1996) (Fig. 6B). Although the effect of RET/ptc2 on PKA signaling is still unknown, given the importance of the Gαs-PKA pathway in thyroid pathophysiology, RET/ptc2 fusion could be a mechanism whereby activation of an oncogene (RET) and inactivation of a tumor suppressor (PRKAR1A) function together to drive transformation (Santoro and Carlomagno, 2013).
Given our mechanistic understanding of PKA fusion proteins, it is surprising that GNAS is actually the most common fusion partner, with fusion proteins present across many cancer types (Fig. 6C; Supplemental Table 4). Although little is known about the function of these fusion proteins, the prevalence in highly pathway-dependent cancers, such as those of the adrenal and thyroid glands, suggests that they could be functionally active in some way. Although GNAS lacks a truly recurrent fusion partner like DNAJB1-PRKACA, there are still some patterns that emerge. Interestingly, the majority of breakpoints cluster at similar genomic coordinates, fusing the 5′ coding sequence of GNAS with another gene. In addition to GNAS, other common pathway fusion partners include PDE4D and ADCY9 (Fig. 6C; Supplemental Table 4). Although these findings are certainly intriguing, much work is still required to understand whether these fusion proteins are expressed and functionally important. As we have seen with other mutations in the pathway, degradation (i.e., NMD of PRKAR1A mutants) could also be an important mutational mechanism used by fusion proteins.
E. Expanding the Mutational Themes
Although the Gαs-PKA pathway signalopathies are dominated by somatic and germline mutation of the key signaling nodes, GNAS, PRKACA, and PRKAR1A, additional mechanisms of pathway dysregulation continue to emerge, representing additional disease phenotypes (Fig. 7A; Supplemental Table 2). Mutations in several PDEs have be reported in Gαs-PKA pathway signalopathies characterized by both pathway activation and inhibition (see section IV. Human Gαs–Protein Kinase A Pathway Signalopathies). Further analysis is required to better understand the function and prevalence of these types of mutations in disease. As we highlight through this review, the role of specific GPCRs reaches across many Gαs-PKA pathway signalopathies. Recent studies have begun to understand the patterns of mutation in GPCRs, highlighting the importance of critical regions such as the DRY and NPxxY motifs in altering activity (Raimondi et al., 2019). This is a promising area of research from both a biologic and therapeutic perspective, helping to differentiate between passenger mutations and disease drivers. Additionally, mutations in the other subunits of the heterotrimeric G protein (i.e., Gβ1 or GNB1) , have been described as functionally significant (Brockmann et al., 2017; Zimmermannova et al., 2017). Unlike GPCRs and G proteins, the role of mutations in AKAP scaffolds remain largely unexplored. A prime example of the functional importance of AKAP9 mutations is in long-QT syndrome (Fig. 7A; Supplemental Table 2), in which patients suffer from irregular heartbeat due to issues with ionic currents in the heart. AKAP9 forms a critical complex with a potassium channel subunit, potassium voltage-gated channel subfamily Q member 1 (KCNQ1). Phosphorylation of KCNQ1 by PKA is required for repolarization after a cardiac action potential. The S1570L mutation in AKAP9 disrupts the KCNQ1 interaction, reduces phosphorylation, and most importantly renders the potassium channel functionally unresponsive to cAMP (Chen et al., 2007). Several reports have documented mutations in other pathway components, including PRKAR1B, a mutation thought to disrupt catalytic or AKAP binding, as well as gain-of-function mutations in ADCY5 (Fig. 7A; Supplemental Table 2). To date, these mutations have primarily been observed in neurologic and neurodegenerative diseases such as familial dyskinesia and Alzheimer disease (Chen et al., 2014; Wong et al., 2014; Marbach et al., 2021). Although we focus primarily on endocrine and neoplastic diseases in this review because of the strength of data linking genetics to disease mechanism, the role of PKA in neurologic diseases is certainly an emerging family of Gαs-PKA pathway signalopathies. For instance, PKA dysregulation may contribute to Alzheimer, Huntington, and Parkinson diseases, but these disease mechanisms and their therapeutic opportunities are still poorly understood (Dagda and Das Banerjee, 2015; Greggio et al., 2017).

(A) Network map of protein kinase A pathway mutations in genetic diseases. Outline of each node shows the functional significance of corresponding mutations, with benign variants or variants of unknown significance in green and pathogenic mutations or risk factors in red. Size of the node represents the number of mutations classified as pathogenic, likely pathogenic, or risk factor in ClinVar (Landrum et al., 2020). Pie charts within the node are colored by frequency of disease phenotypes associated with mutations in each node. Solids edges represent known FIs, with arrows indicating that some form of regulation exists between the nodes. Dashed edges represent FIs predicted by Reactome, and dotted edges indicate FIs predicted by STRING (score > 0.75). Disease phenotype abbreviations: BMIQ19, Body Mass Index Quantitative Trait Locus 19; LCCS8, lethal congenital contracture syndrome 8.
Overt mutation and genomic alteration are not the only mechanisms of pathway dysregulation. We have already discussed the potential role of aberrant splicing in GNAS, but many members of the Gαs-PKA pathway are subject to regulation by splicing, including tissue-specific isoforms of PKA catalytic subunits (Søberg et al., 2017) and signalosome-specific AKAP spliceforms (Wong and Scott, 2004). Furthermore, recent work has suggested that disease phenotypes may be associated with single nucleotide polymorphisms in specific GPCR isoforms (Marti-Solano et al., 2020). As we will discuss later, the role of autocrine and paracrine (oncocrine) pathway activation can also contribute to aberrant signaling. Overproduction of pathway ligands can certainly contribute to disease, as is the case for COX-2 overexpression–driven prostaglandin E2 (PGE2) production in colorectal cancer (see section 3. GNAS and Protein Kinase A Link Inflammation to Cancer Initiation). Finally, pathogenic mutation of PKA phosphosites is emerging as a mechanism of disease. For instance, Parkinson disease mutations in leucine rich repeat kinase 2 (encoded by LRRK2), highlight the specific mutation of PKA phosphosites known to regulate LRRK2 activity (Muda et al., 2014). It is important to synthesize the mutational themes and mechanisms of dysregulation that define the Gαs-PKA pathway signalopathies. This is a critical step necessary to connect the genomic and biochemical findings with clinical manifestations and ultimately catalyze the development of new, effective therapies.
IV. Human Gαs–Protein Kinase A Pathway Signalopathies
A. Infectious Diseases
1. Cholera
Perhaps one of the best examples of Gαs-PKA pathway–mediated pathophysiology is the severe diarrhea caused by infection with Vibrio cholera, or cholera. Cholera continues to be a global health concern, contributing to hundreds of thousands of deaths each year (Ali et al., 2015). Cholera toxin has a unique ability to ADP-ribosylate Gαs at arginine 201. The addition of an ADP-ribose group inhibits the GTPase activity of Gαs and renders it constitutively active in a manner similar to the disease-associated R201 mutations (discussed in section A. Mutations in GNAS) (Landis et al., 1989; Kaper et al., 1995). Overactivation of Gαs by cholera toxin leads to cAMP production and PKA activation in the intestinal epithelium (Fig. 8A). In crypt cells, PKA activity enhances secretion of Cl− into the intestinal lumen due to direct regulation of the cystic fibrosis transmembrane conductance regulator (CFTR) channel. Under normal physiologic conditions, the degree of phosphorylation of four PKA phosphosites controls the degree of CFTR channel opening. Thus, overactive PKA in response to cholera leads to maximal channel opening. In villous cells, PKA also functions to decrease Cl− absorption by inhibiting Na+/Cl− cotransporters and Na+/H+ exchangers (Goodman and Percy, 2005). As a result of osmotic imbalance, water rapidly moves out of cells into the intestinal lumen, overwhelming reabsorption mechanisms and producing severe, watery diarrhea and dehydration that can prove deadly if left untreated (Fig. 8A). Interestingly, patients with cystic fibrosis are resistant to the effects of cholera toxin as a result of mutations in the CFTR channel. Notably, the majority of patients harbor the F508del mutation in the regulatory region of CFTR. This mutation causes PKA phosphorylation defects that alter trafficking through the endoplasmic reticulum and Golgi to the cell surface as well as disrupt the conformational cues induced by PKA phosphorylation that are critical to channel opening (Kaper et al., 1995; Goodman and Percy, 2005; Bharati and Ganguly, 2011; Chin et al., 2017).

Gαs-PKA pathway signalopathy pathophysiology. (A) Pathophysiology of cholera. Cholera is an intestinal parasite that enters the digestive tract when consumed via contaminated water. In the intestinal epithelium, cholera toxin ADP-ribosylates and activates Gαs, leading to overactivation of PKA. PKA directly phosphorylates the CFTR to facilitate channel opening. Efflux of chloride ions disrupts normal ionic gradients, and water passes into the intestinal lumen to compensate. Consequently, the clinical manifestations of cholera include watery diarrhea and dehydration. (B) Cushing syndrome pathophysiology. ACTH is secreted by the pituitary gland in the brain and travels through the bloodstream to the adrenal gland located on top of the kidney. ACTH binds to the melanocortin receptor (MC2R) on the surface of adrenocortical cells to activate PKA and stimulate cortisol secretion. In Cushing syndrome, loss-of-function mutation in RIα (or gain-of-function mutation in Cα) leads to persistent PKA activation and excess cortisol secretion. Clinical manifestations of the disease exacerbate the effects of cortisol and include hypertension, hyperglycemia, and obesity. (C) Fibrous dysplasia pathophysiology. Fibrous dysplasia is a postzygotic disease caused by activating mutation in GNAS. Persistent activation of PKA in mesenchymal stem cells impairs proper differentiation to adipocyte, chondrocyte, and osteogenic lineages. In particular, accumulation of osteogenic precursors shifts the balance of osteoblasts and osteoclasts to favor bone resorption by osteoclasts. Resulting clinical manifestation of the disease includes brittle bone and frequent fracture or deformity.
B. Endocrine and Metabolic Diseases
1. Carney Complex
Carney complex is a rare disease that is characterized by multiple neoplasms of both endocrine (commonly adrenal, pituitary, or thyroid glands and gonadal tissues) and nonendocrine tissues (commonly heart, skin, or eye). First described in 1985, only about 750 individuals have been diagnosed worldwide (Correa et al., 2015). Interestingly, 70% of the cases are familial, following autosomal dominant inheritance patterns, with the majority of patients having inactivating mutations in PRKAR1A (Kirschner et al., 2000b; Bertherat et al., 2009). Additionally, 35% of sporadic cases are also caused by these same mutations (Kirschner et al., 2000b). In fact, Carney complex was the first disease to be associated with mutations in the PKA holoenzyme (Kamilaris et al., 2019). As mentioned previously, the vast majority of mutations are not actually expressed due to NMD, creating PRKAR1A haploinsufficiency, ultimately resulting in catalytic subunit hyperactivity (Bertherat et al., 2009). Aligned with this concept, a patient with Carney complex with copy number gains in PRKACB has also been documented (Forlino et al., 2014).
Carney complex is a heterogeneous disease with typical onset around age 20, but some patients have even been diagnosed as children (Correa et al., 2015). Interestingly, patients with PRKAR1A mutations tend to present at a younger age with specific phenotypes (Bertherat et al., 2009). Most patients present with Cushing syndrome (see section 2. Cushing Syndrome and Adrenocortical Adenomas) and endocrine phenotypes. One of the most common physical characteristics is the presence of pigmented skin lesions, like café-au-lait spots, caused by the hyperproliferation of melanocytes (also seen in McCune-Albright syndrome; see section 4. Fibrous Dysplasia and McCune-Albright Syndrome). Another common characteristic is cardiac myxoma, a neoplasm of the heart. Cardiac myxoma represents a major cause of mortality in Carney complex because of its rapid growth and recurrence, resulting in obstruction of blood flow in the heart (see section 3. Cardiac Myxoma) (Wang et al., 2018b). Finally, the most common endocrine phenotype is primary pigmented nodular adrenocortical disease, affecting up to 60% of patients with Carney complex. As the name suggests, it manifests as pigmented nodules on the adrenal gland (Bertherat et al., 2009). This results in adrenocorticotropic hormone (ACTH)-independent Cushing syndrome, which is discussed in the next section. Interestingly, primary pigmented nodular adrenocortical disease can occur outside of Carney complex and not only is caused by mutations in PRKAR1A but can also be caused PDE8B or PDE11A mutations (Bertherat et al., 2009; Kamilaris et al., 2019) (Fig. 7A; Supplemental Table 2). This highlights that overactive PKA is a driver of this disease, regardless of how it is achieved.
Similarly, the physical manifestations of the disease are in line with the importance of PKA signaling to the cell types affected by Carney complex. In these tissues, normal programs such as growth and development and energy metabolism are driven by the hormone-GPCR-Gαs-PKA signal transduction axis (see section F. Metabolic Regulation for more information on energy metabolism). Acting through cAMP second messengers, PKA mediates systemic responses to hormones of the pituitary, adrenal gland, thyroid, parathyroid, and hypothalamus as well as more local responses in tissue such as the pancreas, kidney, liver, and gonads (Tilley and Fry, 2015). Of note, individual mutations in their cognate GPCRs can also cause endocrine phenotypes related to Carney complex or other Gαs-PKA pathway signalopathies (Lania et al., 2006). However, when dysregulation of this signaling occurs through loss of RIα function, it typically results in neoplastic growth and tumorigenesis across these tissues. In fact, as evidence to the importance of PKA in global growth and development, Prkaca knockout mice weigh 65% less than control littermates and exhibit a significant growth delay (Skålhegg et al., 2002).
2. Cushing Syndrome and Adrenocortical Adenomas
Cushing syndrome is a rare disease that affects around two individuals per million per year across the world (Steffensen et al., 2010). It can present with very broad symptoms, including hypertension, hyperglycemia, obesity, skin changes, mood disorders, and other hormonal changes. Although these symptoms can have multiple etiologies, Cushing syndrome is specifically characterized by exposure to excess cortisol (Sharma et al., 2015). Cortisol is a hormone that helps control the stress response by regulating blood pressure and blood sugar as well as dampening the immune response. The release of cortisol is regulated by ACTH, which is secreted by the pituitary glands at the base of the brain. Once in the bloodstream, ACTH travels to the adrenal gland, located on top of the kidneys, where it binds the melanocortin receptor (MC2R). MC2R is a Gαs-linked GPCR located on the surface of the adrenocortical cells, which when stimulated activates PKA to trigger cortisol secretion (Fig. 8B).
Cushing syndrome has many etiologies, including overuse of glucocorticoid medication, ACTH-secreting pituitary tumors (termed Cushing disease), or cortisol-secreting adrenocortical adenomas (Sharma et al., 2015). Although rare, Cushing syndrome can also have genetic causes that converge on overactivation of the PKA pathway. One of the most common genetic causes of Cushing syndrome is the PRKACA L206R mutation. As mentioned previously, L206R disrupts regulatory subunit contacts, leading to constitutive PKA activity. This mutation, along with loss-of-function mutations in PRKAR1A, underlie ACTH-independent Cushing syndrome (Fig. 8B). Similarly, germline PRKACA copy number gains (Beuschlein et al., 2014; Lodish et al., 2015) and somatic PRKACB S54L mutations can also cause cortisol-producing adrenocortical adenomas/hyperplasias and Cushing syndrome (Espiard et al., 2018). Somatic mutations in GNAS and PDE8D have also been identified (Espiard et al., 2018). In general, patients with PKA gene mutations have earlier onset of disease with more comorbidities. There is some evidence, at least for germline PRKACA amplifications, that this is a dose-dependent effect, with patients harboring PRKACA triplication having the most severe symptoms and earliest onset (Lodish et al., 2015). Interestingly, patients with GNAS and PRKACA mutations have smaller tumor sizes, which is a sign that the tumor is capable of efficient cortisol production and secretion (Goh et al., 2014). This finding is also in line with the role of cAMP in controlling regulated exocytosis, which contributes to hormone secretion in endocrine cells. For instance, in the pituitary, cAMP increases the size of secretory granules (Seino and Shibasaki, 2005), and in the adrenal gland, basal PKA signaling is required to maintain the vesicle pools that are primed and ready to be exocytosed (Nagy et al., 2004). In general, increase in intracellular Ca2+ is the main driver of exocytosis, but cAMP can also modulate the response at several different levels through mechanisms involving both PKA and EPAC.
Although Cushing syndrome is the most prominent diagnosis, primary macronodular adrenal hyperplasia is a related disorder that reflects a spectrum of disease ranging from subclinical hypercortisolism all the way to overt Cushing syndrome. Of note, it can also be part of the manifestations of McCune-Albright syndrome (see section 4. Fibrous Dysplasia and McCune-Albright Syndrome) (De Venanzi et al., 2014). It is characterized by large functional nodules on the adrenal gland that alter cortisol secretion. Although rare, primary macronodular adrenal hyperplasia can be caused by activating mutations in MC2R (encoding MC2R) (Hiroi et al., 1998; Swords et al., 2004) or GNAS (Fragoso et al., 2003; Hsiao et al., 2009).
3. Cardiac Myxoma
Cardiac myxomas (CMs) can occur in the context of Carney complex, and this accounts for about 7% of all CM cases (Milunsky et al., 1998). The vast majority of the patients with Carney complex have loss-of-function mutations in PRKAR1A (70%) (Bertherat et al., 2009; Wang et al., 2018b). For these patients, CMs typically present earlier in life (with frequent reoccurrence) and can affect any chamber of the heart with multiple lesions. Conversely, isolated sporadic CMs typically occur as a single lesion in middle-aged women (mean age 51 years) and preferentially in the left atria (Carney, 1985; Reynen, 1995; Stratakis et al., 2001). Interestingly, it is estimated that anywhere from 31% (Maleszewski et al., 2014) to 64% (He et al., 2017) of isolated sporadic CMs are also caused by loss-of-function mutations in PRKAR1A. Although the vast majority of CMs are sporadic, there are also a few reports of familial CMs not associated with Carney complex. Typically, these familial mutations follow autosomal dominant inheritance. For instance, in one family, both the father (44 years of age) and daughter (20 years of age) developed CM as a result of the V164D frameshift deletion (c.491_492delTG) in PRKAR1A. The woman’s uncle and brother did not harbor the mutation and had no signs of CM to date (Ma et al., 2019). CMs are the most common primary tumor in the heart, and although they are benign, they can cause significant morbidity and mortality because of their location (Reynen, 1995). The mechanism of tumorigenesis for CM is not fully understood, but it is thought that mesenchymal stem cells (MSCs) from the endocardium and epicardium are the cell of origin (Di Vito et al., 2015). Effects on this MSC population may also account for GNAS mutations found in intramuscular and cellular myxomas (>90% GNAS mutants) (Sunitsch et al., 2018). Of note, MSCs are also the cell of origin for fibrous dysplasia, which is discussed in the next section.
4. Fibrous Dysplasia and McCune-Albright Syndrome
Fibrous dysplasia (FD) is a rare skeletal disorder that is characterized by painful and brittle bones that are prone to fracture and deformity. The clinical presentations can be very heterogeneous, affecting one bone (monostotic) or multiple bones (polyostotic) with variable severity. FD can also present with additional manifestation of café-au-lait spots or endocrine hyperfunction, which is termed McCune-Albright syndrome (MAS) (Feller et al., 2009; Riminucci et al., 2010). Additionally, if FD presents with intramuscular myxomas, tumors of musculoskeletal soft tissue, it is termed Mazabraud syndrome. FD/MAS is caused by postzygotic somatic activating mutations in GNAS (GNAS R201C/H) (Fig. 8C); thus, the disease is not inherited. The heterogeneity of FD/MAS results from somatic mosaicism, wherein some cells inherit the defect, whereas others do not. The tissues involved in FD/MAS arise from all three embryonic germ layers (ectoderm, endoderm, mesoderm), suggesting that in most cases the mutation may be acquired prior to gastrulation, before cell lineage decisions are made (Riminucci et al., 2006; Feller et al., 2009).
Recent studies by our groups and others have demonstrated that expression of GNAS activating mutations in mesenchymal/skeletal stem cells is necessary and sufficient to drive FD development in mouse models (Zhao et al., 2018). Interestingly, germline expression of the FD mutation is embryonic lethal (Khan et al., 2018), but when expression is induced during embryogenesis or postnatally, FD lesions develop rapidly (Zhao et al., 2018). The severity of the disease, however, is not linked to stage of development in which the mutation is acquired but, rather, the degree to which mutated cells contribute to critical functions within the tissues (Riminucci et al., 2006; Feller et al., 2009). For instance, patients with a higher ratio of mutated cells to normal cells in the osteogenic progenitor pool will develop more severe FD, whereas patients with a higher ratio of normal cells to mutant cells will display milder phenotypes. In fact, isolation of bone marrow stroma progenitors from patients with FD revealed that the stroma is a mosaic of mutant and normal cells. Mosaic stromal marrow engrafts into immunocompromised mice, whereas purified mutant marrow fails to engraft (Bianco et al., 1998). Therefore, it has been proposed that there is a “critical mass” of mutated cells that are necessary to drive symptomatic disease (Riminucci et al., 2006; Feller et al., 2009).
Under normal physiologic conditions, bone is constantly being remodeled, which is a balance between bone production by osteoblasts and bone resorption by osteoclasts. Overactivation of Gαs signaling through PKA induces proliferation of osteogenic precursors but impairs proper differentiation of osteoblasts and mineralization while enhancing osteoclast differentiation (Riminucci et al., 1997; Zhao et al., 2018) (Fig. 8C). Ultimately, this shifts the balance toward bone resorption, which is a histologic marker of FD in patients.
5. Acromegaly, Gigantism, and Pituitary Tumors
Acromegaly and gigantism are rare diseases characterized by overproduction of growth hormone (GH). GH is normally secreted by the pituitary gland into the bloodstream, where it travels to the liver to stimulate insulin-like growth factor-1 production and growth of bones and body tissues. Gigantism occurs early in childhood before growth plate fusion, resulting in dramatic vertical growth, whereas acromegaly occurs in adulthood and is characterized by growth and swelling of many body tissues, including hands, feet, nose, lips, jaw, and brow (Hannah-Shmouni et al., 2016). In most cases, acromegaly and gigantism are caused by somatotropinoma or GH-secreting pituitary tumors. The majority of GH-secreting pituitary tumors occur sporadically, but there are a few examples of familial cases. The most common sporadic alteration in acromegaly is GNAS activating mutations (40%–60%) (Freda et al., 2007; Hage et al., 2018). Typically, these patients have smaller tumors but very high GH secretion, highlighting again the physiologic role of the cAMP in secretion. Of note, no mutations have been identified in the PRKACA or PRKACB (Larkin et al., 2014), and GNAS mutations specifically enrich in GH-secreting pituitary tumors over other subtypes of pituitary tumors (Bi et al., 2017). In about 10% of gigantism, patients have very-early-onset disease (before the age of 4), known as X-linked acrogigantism (XLAG). In addition to overproduction of GH, patients with XLAG also overproduce the hormone prolactin. XLAG is caused by duplications in GPR101, an orphan GPCR on the X chromosome. XLAG predominates in females, but some males also acquire sporadic mutations (Iacovazzo and Korbonits, 2016; Gadelha et al., 2017). Additionally, there have been two independent families that display GPR101 duplications. GPR101 is predicted to couple to Gαs and has been show to stimulate cAMP production in vitro; however, there is some evidence it could couple to Gαi as well (Bates et al., 2006; Martin et al., 2015; Iacovazzo and Korbonits, 2016).
Acromegaly and gigantism are also associated with Carney complex and McCune-Albright syndrome, but in these cases, it is generally caused by hyperplasia of the somatotrophs, GH-secreting cells in the pituitary, instead of overt tumors. In Carney complex, most patients have PRKAR1A loss-of-function mutations, leading to PKA activation and GH and prolactin excess, but only about 10% of patients actually present with acromegaly. For McCune-Albright syndrome, a smaller percentage of patients have pituitary involvement, but of those, 36% develop gigantism, whereas the other 64% develop acromegaly (Boikos and Stratakis, 2007; Gadelha et al., 2017).
6. Hyperthyroidism
Hyperthyroidism is a disease in which the thyroid gland is overactive, producing too much of the hormones that control metabolism, triiodothyronine and tetraiodothyronine. This leads to increased appetite and unintentional weight loss, rapid and irregular heartbeat, restlessness, and potentially goiter (enlargement of the thyroid gland) (De Leo et al., 2016). Hyperthyroidism can have many causes, but as previously mentioned, it can be a component of Carney complex and McCune-Albright syndrome. Whether patients present as part of a broader syndrome, these nonautoimmune hyperthyroidisms can be caused by activating mutations in the thyroid-stimulating hormone receptor (TSHR, encoded by TSHR) or GNAS. As a GPCR, TSHR couples to Gαs to control secretion of triiodothyronine and tetraiodothyronine, but activating mutations in this pathway can cause thyroid adenomas that autonomously secrete hormones (Hébrant et al., 2011; Lacka and Maciejewski, 2015). Of these thyroid adenomas, 5%–10% are caused by GNAS mutations, and 70%–80% are caused by TSHR mutations (Palos-Paz et al., 2008; Nishihara et al., 2009). A recent report suggested that for hot thyroid nodules (nodules that preferential take up radioactive iodine, generally with excess thyroid-stimulating hormone secretion), GNAS and TSHR are the only driver mutations, with a clear preference for TSHR mutations (Stephenson et al., 2020). Over 30 different mutations in TSHR have been documented. Some mutations have been identified in adenomas as well as sporadic and familial cases, whereas others have preference for specific subsets (Hébrant et al., 2011). The reason for this preference is a balance between mutation expression and strength of activation. Strong clonal mutations are likely to cause adenomas and sporadic hyperthyroidism, whereas weaker germline mutations expressed in all cells are likely to cause familial cases. Although there is no defined syndrome, it is probable that particularly strong germline TSHR mutations are embryonic lethal since thyroid hormones are critical to fetal development (Lacka and Maciejewski, 2015).
7. Inactivating Parathyroid Hormone/Parathyroid Hormone–Related Peptide Signaling Disorder
Unlike the other diseases discussed so far, inactivating parathyroid hormone/parathyroid hormone–related peptide signaling disorder (iPPSD), represents a heterogeneous group of disorders that is characterized by inactivating defects in the Gαs-PKA signaling pathway. Clinical features of this disease are diverse and overlapping among subtypes. Common features include skeletal deformities (brachydactyly, short stature), obesity, cognitive impairment, and hormone insensitivity, leading to improper mineral metabolism and delayed reproductive development, among other manifestations (Mantovani and Elli, 2018, 2019). The current iPPSD nomenclature encompasses diseases such as Blomstrand chondrodysplasia/Eiken syndrome, pseudohypoparathyroidism, acrodysostosis, Albright hereditary osteodystrophy, and progressive osseous heteroplasia, but the specific distinctions are beyond the scope of this review (Mantovani and Elli, 2019). Here, we will focus on the molecular underpinnings of the iPPSD subtypes.
The clinical features of iPPSD highlight the physiologic roles of PTH signaling in a wide variety of developmental and homeostatic mechanisms. PTH is secreted from the parathyroid glands located in the neck to regulate calcium and phosphate homeostasis by signaling through the parathyroid hormone receptor (PTHR). PTHR is a Gαs-coupled GPCR that is expressed at particularly high levels in the bone and kidney. Not surprisingly, inactivating mutations in PTHR (PTH1R) cause iPPSD1 with predominately skeletal defects. Gαs itself is also subject to heterozygous loss-of-function mutations or, more commonly, genomic imprinting that reduces Gαs mRNA and protein levels by around 50% (iPPSD2/3) (Turan and Bastepe, 2015; Mantovani and Elli, 2019). Clinical phenotypes, particularly heterotopic ossification, are recapitulated in mice with Gnas knockout in mesenchymal progenitor cells (Regard et al., 2013). GNAS is also subject to tissue-specific maternal imprinting or loss of paternally imprinted methylation patterns in particular regions on the GNAS locus. Patients with loss of function in Gαs display variable resistance to hormones, including PTH, thyroid-stimulating hormone, gonadotropin, and GHRH, which determine their clinical manifestations (Mantovani and Elli, 2018, 2019). For instance, all patients of these subtypes display bone and adipose phenotypes due to biallelic expression of Gαs in these tissues, whereas individuals with maternally inherited loss of function will present with additional cognitive and endocrine phenotype due to paternal imprinting of Gαs in these tissues (Mantovani et al., 2004; Long et al., 2007; Mouallem et al., 2008; Turan and Bastepe, 2015). In line with the importance of the Gαs-PKA signaling pathway, mutations in RIα, PDE4D, and PDE3A characterize the remainder of the molecularly defined iPPSD subtypes (iPPSD4/5/6) (Mantovani and Elli, 2019). Of particular note, mutations in PDE3A further highlight the importance of cAMP in driving the pathophysiology of iPPSD. As mentioned previously, PDE3 family members can hydrolyze both cAMP and cGMP. Interestingly, mutations in PDE3A have been shown to enhance the cAMP-hydrolyzing activity without altering enzymatic activity toward cGMP, ultimately resulting in reduced cellular cAMP levels (Maass et al., 2015; Ercu et al., 2020).
C. Neoplasms and Carcinomas
Thus far, we have highlighted the role of PKA signaling in neoplasms of the adrenal, pituitary, thyroid, gonads, and even heart due to both germline or somatic mutations in the pathway, all members of the broad and overlapping endocrine and metabolic Gαs-PKA pathway signalopathies. Many of these neoplasms are monogenetic and inherently accompanied by endocrine hyperactivity, a process in which it is evident overactive Gαs-PKA signaling is the driver of pathophysiology. In the context of cancer, however, disease is rarely the result of a single mutation but, rather, a complex polygenetic network subject to the biology of diverse tissues and other modulatory inputs like inflammation and immune evasion. With the precision medicine revolution and rapid advances in cancer genomics, we can finally begin to appreciate a broader role of Gαs-PKA in cancer as both an oncogenic driver and tumor suppressor. By leveraging our knowledge of mutational themes and Gαs-PKA–mediated pathophysiology, we can begin to understand many cancers as emerging Gαs-PKA pathway signalopathies.
1. GNAS–Protein Kinase A as Oncogenes: Beyond Endocrine Tumors
A real shock to the field came with the discovery of a PKA fusion protein that drives a rare form of liver cancer (<1% of cases), known as FL-HCC (Honeyman et al., 2014). Affecting children and young adults with no underlying pathology, FL-HCC could not be more different from the majority of liver cancers, which affect adults with liver damage commonly due to viral infection or alcoholism. As mentioned previously, patients with FL-HCC were found to express an in-frame fusion of DNAJB1 with PKA Cα (DNAJB1-PRKACA) that resulted in increased PKA activity due to relative overexpression of the catalytic subunit (Riggle et al., 2016), but importantly, overexpression of PRKACA does not completely recapitulate the oncogenicity of the fusion protein (Kastenhuber et al., 2017) (see section D. Fusion Proteins: An Emerging Mutational Theme) (Fig. 6A). To date, across multiple studies, DNAJB1-PRKACA has been identified in nearly 80% of patients with FL-HCC (Cornella et al., 2015). Of note, several patients with FL-HCC lacking the DNAJB1-PRKACA fusion protein, but with a history of Carney complex and other tumors, exhibited a complete loss of RIα protein instead (Graham et al., 2018). Recent studies have pointed to an even broader role of PKA fusion proteins, including additional fusions with PRKACB and ATP1B1, suggesting that they may also be driver oncogenes in extrahepatic cholangiocarcinoma, intraductal oncocytic papillary neoplasms, and intraductal papillary mucinous neoplasms (IPMNs) of the pancreas and bile duct (Nakamura et al., 2015; Singhi et al., 2020; Vyas et al., 2020) (Fig. 6A).
Although DNAJB1-PRKACA in FL-HCC clearly establishes PKA as an oncogenic driver, broader analysis of cancer genomes by our group revealed that GNAS is the most highly mutated G protein, harboring mutations in over 4% of all sequenced tumors to date, with the majority representing hotspot mutations (O'Hayre et al., 2013; Wu et al., 2019; Arang and Gutkind, 2020). Surprisingly, we and others have noted that among GNAS mutated cancers, there is a clear enrichment of gastrointestinal cancers, including colorectal adenocarcinoma (4%–10%), stomach adenocarcinoma (6%–10%), and pancreatic adenocarcinoma (5%–12%), a finding which extends to GPCRs and other G protein subunits (O'Hayre et al., 2013; Innamorati et al., 2018; Wu et al., 2019; Arang and Gutkind, 2020). GNAS and PKA also seem to be particularly important to neuroendocrine cancers of the pancreas, prostate, liver, and lung (Deeble et al., 2007; Boora et al., 2015; Kastenhuber et al., 2017; Innamorati et al., 2018; Coles et al., 2020). Expanding on these observations, we find that GNAS mutation frequency is even more significant in less-studied cancers, such as those of the bone (40%) and the peritoneum (53%) (Fig. 9A; Supplemental Table 5). Although GNAS mutation is recognized for its importance in cancer and is routinely included in clinical sequencing panels, such as FoundationOne (https://www.foundationmedicine.com/), analysis of the broader pathway reveals that mutations occur at every node. There are particularly good examples of each, such as ADCY2 mutations in liver (20%), PDE4D mutations in prostate (25%), and SPHK1-interactor and AKAP domain-containing protein (SPHKAP) in skin (26%) (Fig. 9A; Supplemental Table 5). Given that there are many genes representing each node of the pathway, when we consider the mutation frequency of each gene family, it becomes clear that some gene families are preferentially mutated in certain tissues; for instance, GNAS mutations predominate in hormone-sensitive tissues (Fig. 9B; Supplemental Table 5). Somewhat strikingly, we find that adenylyl cyclase mutations constitute the bulk of the mutations across many tissue types. Intriguingly, AKAPs are mainly mutated in the stomach and pancreas, whereas PKA catalytic subunits have a consistent low level of mutation across most tissues (Fig. 9B; Supplemental Table 5). Of important note, most patient samples harbor only one or two pathway mutations (57%), with the majority of those (41%) being single pathway mutations (Supplemental Table 5). As we discussed previously, there is limited knowledge on the functional importance of mutations within these other nodes of the pathway (see section E. Expanding the Mutational Themes), but given the emergence of genomic medicine and the success of targeted therapies, the role of the Gαs-PKA pathway in cancer certainly warrants further study. For the remainder of this review, we will highlight examples of the clinical and biologic function of the Gαs-PKA pathway in cancer.

Protein kinase A pathway mutations in cancer. (A) Frequency of specific pathway gene mutation across several tumor and cancer types. Heatmap is colored by mutation frequency (0% to >50%), with darker purple representing higher mutational frequency. All gene mutations from whole genome sequencing data sets are included (COSMIC database) (Tate et al., 2019). (B) Frequency of pathway mutation grouped by gene family across tumor and cancer types. A sample is considered to have a pathway mutation if it harbors at least one mutation in a family gene member.
2. Mucin Production Drives Clinical Phenotypes
One of the most striking and clinically relevant features of GNAS mutant cancers is their high level of mucin production across several tissue types (lung, stomach, bile duct, pancreas, appendix, colorectum, and gonads) (Innamorati et al., 2018). Mucins are large glycoproteins, either secreted or membrane-bound, with important physiologic and homeostatic roles. In the intestine, mucin provides the first line of defense against microbes and is critical to preserving epithelial barrier integrity. Mucins also have important structural roles to help physically maintain the microvilli architecture that is so important to intestinal function (Pelaseyed and Hansson, 2020). Consequently, the dysregulation of mucin can have profound impacts on disease. For instance, mucin 2 (encoded by Muc2) knockout mice have defects in goblet cell differentiation. This results in increased epithelial cell proliferation and migration coupled with decreased apoptosis and lack of acidic mucin production. Ultimately, these Muc2 knockout mice spontaneously develop tumors in the small and large intestine that progress to invasive carcinoma (Velcich et al., 2002). Interestingly, the Gαs-PKA pathway is known to directly regulate MUC2 expression through the G protein–coupled E-type prostanoid receptor 4 in the intestine. PKA-mediated activation of CREB triggers binding to the CRE in the MUC2 promoter and transcriptional upregulation (Nishikawa et al., 2013; Dilly et al., 2017). In pancreatic ductal cells, GNAS mutation is known to dramatically increase the expression of another mucin, MUC5AC. MUC5AC is one of the predominant mucins overexpressed in IPMNs of the pancreas, which commonly harbor GNAS hotspot mutations (discussed below) (Ideno et al., 2013; Komatsu et al., 2014). Transcriptional upregulation of mucin production is also augmented by the role of cAMP and PKA in vesicular transport. PKA is involved in constitutive transport of vesicles through the trans-Golgi network to the cell surface (Muñiz et al., 1996). Specifically, AKAPs anchor PKA to the cytoplasmic surface of the endoplasmic reticulum (AKAP1) and Golgi (AKAP1/9), where it can be activated in response to extracellular stimulation (Rios et al., 1992; Huang et al., 1999; Ma and Taylor, 2008; Mavillard et al., 2010).
At a molecular level, mucin overexpression in cancer has been implicated in dysregulation of cell polarity and disruption of proper cell-cell contacts. Further, mucin can facilitate aberrant oncogenic signaling, such as β-catenin activation, and receptor tyrosine kinase oligomerization and activation (Kaur et al., 2013; Pelaseyed and Hansson, 2020; Pothuraju et al., 2020). Mucin is also thought to play an important role in modulating the tumor microenvironment, serving as a bridge to nutrient-rich stroma through neoangiogenesis as well as by providing immunosuppressive mechanisms to evade immune surveillance. In addition to biologic effects on the tumor microenvironment, mucin can also serve as a physical barrier, sequestering local growth factors and protecting neoplastic cells from cytotoxic agents (Hollingsworth and Swanson, 2004; Kaur et al., 2013). Consequently, mucinous adenocarcinoma (in which >50% of the tumor mass is mucin) and tumors with a mucinous component (<50% of tumor mass is mucin) are implicated, with poor prognosis and chemoresistance across many tissue types (Schiavone et al., 2011; Lee et al., 2013; Kajiyama et al., 2014; Asare et al., 2016; Xie et al., 2018). Of note, pseudomyxoma peritonei (PMP) is one of the most devastating examples of mucin dictating clinical outcomes, for which the 5-year survival rate of high-grade disease is only 23% (Nummela et al., 2015). PMP is an extremely rare subtype of mucinous adenocarcinoma (typically originating from the appendix) in which the peritoneal cavity is colonized by mucin-secreting neoplastic cells. The excess mucin (>90% of tumor volume, dominated by MUC2 and a lesser extent MUC5AC) (O'Connell et al., 2002) overtakes the peritoneum, obstructing normal intestinal function and ultimately killing the patient. GNAS hotspot mutations are found in 63% of all PMPs, including both low- and high-grade disease (56% and 70%, respectively). Currently, the only therapeutic options for these patients are reductive surgery and intraperitoneal chemotherapy, which have significant treatment-associated morbidity. Thus, targeting the Gαs-PKA pathway as a means to limit mucin production has been proposed for patients with PMP (Nummela et al., 2015). Interestingly, in recurrent PMP, patients with GNAS mutations have poorer outcomes after chemotherapy, but it is uncertain whether this is because of the biology of GNAS mutants or whether GNAS is a biomarker of therapeutic resistance (discussed in section 5. Gαs–Protein Kinase A Induced Therapeutic Resistance in Cancer) (Pietrantonio et al., 2016).
When considering the prevalence of GNAS mutations in PMP, among other cancer subtypes, another trend that becomes rapidly apparent is a co-occurrence with mutations in the KRAS proto-oncogene (encoded by KRAS) Interestingly, 63%–72% of GNAS mutant PMPs also harbor KRAS mutations (Nummela et al., 2015; Ang et al., 2018). Furthermore, in mucinous neoplasms of the appendix, 69% of patients with GNAS mutations actually harbor GNAS and KRAS comutations. Nearly all of these patients had low-grade histology (Alakus et al., 2014). Another study corroborated this, finding that 50% of patients with low-grade appendiceal mucinous neoplasm were positive for both GNAS and KRAS mutations (Nishikawa et al., 2013). Interestingly, 38%–43% of patients with IPMNs of the pancreas, which are analogous low-grade lesions of the pancreas, harbor both GNAS and KRAS mutations (Molin et al., 2013; Amato et al., 2014). Furthermore, 58% of villous adenocarcinomas of the colorectum, which are characterized by noninvasive tissue architecture (similar to low-grade appendiceal mucinous neoplasm and IPMN) and profound mucin production are also GNAS and KRAS comutants (Yamada et al., 2012). Together these co-occurrence patterns highlight that GNAS and KRAS mutation give rise to unique biology in neoplastic diseases that cannot be achieved be either gene alone.
3. GNAS and Protein Kinase A Link Inflammation to Cancer Initiation
Consistent with clinical evidence that GNAS mutations are predominantly found in benign, noninvasive lesions, mouse models reveal that GNAS mutation alone is insufficient to induce epithelial tumorigenesis (Wilson et al., 2010; Patra et al., 2018). Our team showed that, in the context of KRAS mutations in the pancreas, GNAS drives lesions toward the cystic lineage; together, these comutants form well differentiated, mucinous cysts that resemble IPMNs, instead of noncystic pancreatic intraepithelial neoplasias. Somewhat counterintuitively, GNAS R201C expression does not accelerate KRAS-driven progression to pancreatic adenocarcinoma (PDAC). Instead, inactivation of tumor suppressors, like p53 (TP53), cyclin dependent kinase kinhibitor 2a (CDKN2A), or SMAD family member 4 (SMAD4), are needed to facilitate efficient progression to PDAC (Ideno et al., 2018; Patra et al., 2018). Interestingly, in the context of PDAC, GNAS R201C expression through activation of PKA actually attenuates aggressiveness and invasiveness due to epithelial differentiation (Pattabiraman et al., 2016; Ideno et al., 2018). This is supported by clinical evidence that patients with GNAS mutant have a better overall survival in appendix cancer (Ang et al., 2018). However, in small cell lung cancer, a neuroendocrine disease, GNAS and PKA activity is critical to cancer stem cell maintenance and increases rate of initiation and progression (Coles et al., 2020). This suggests that GNAS and PKA can play disparate roles within the various stages from neoplastic initiation to carcinogenic progression. Analysis of colorectal tissues on this spectrum from adenoma to carcinoma revealed that the frequency of GNAS mutation drops with progression. For instance, adenomas had the highest frequency of mutation, followed by carcinomas with residual benign adenoma, carcinomas with adenoma, and regions of invasion, and finally, no mutants were detected in pure carcinomas (Zauber et al., 2016). This suggests that in epithelial tissues, GNAS is most important in early initiation events. Indeed, several studies have highlighted that GNAS mutation can accelerate tumorigenesis (Wilson et al., 2010; Ideno et al., 2018; Patra et al., 2018; Coles et al., 2020). Given that tumors are heterogeneous, GNAS may confer a selective advantage initially, in which context additional mutational insults, like KRAS and subsequently TP53, can drive malignant growth ultimately independent of GNAS mutation. To this end, sequencing of normal human colon crypts unsurprisingly shows that KRAS and TP53 mutations are rare, suggesting that they are more important in intermediate and late events. However, reanalysis of available data highlights 55% of normal crypts harbored GNAS mutations (five of nine subjects), supporting the notion that GNAS may be important in neoplastic initiation and tumorigenesis (Lee-Six et al., 2019).
This idea of GNAS mutations participating in neoplastic initiation tracks well given the established Vogelgram of colorectal cancer (CRC) mutation accumulation. In the original model, KRAS mutations participated in intermediate events, facilitating the progression of adenomas, whereas TP53 loss served as the final barrier to carcinogenesis (Fearon and Vogelstein, 1990). As we have gained more understanding of the molecular events involved in carcinogenesis, COX-2–mediated inflammation has been defined as one of the earliest events in initiation (Markowitz and Bertagnolli, 2009). COX-2 is not expressed under normal conditions but is rapidly upregulated in response to stress and inflammatory stimuli. Naturally, COX-2 has become a prominent biomarker in colorectal cancer and many others, including lung (Hida et al., 1998), pancreas (Tucker et al., 1999), breast (Ristimäki et al., 2002), liver (Shiota et al., 1999), esophagus (Zimmermann et al., 1999), cervix (Ryu et al., 2000), and skin cancer (Buckman et al., 1998). COX -2 is the inducible form of the COX enzymes, which converts arachidonic acid to lipid signaling molecules, including prostaglandins and thromboxanes. These inflammatory mediators are ligands for a number of GPCRs in the prostanoid family (Hata and Breyer, 2004). Most notably, PGE2 is the ligand for two Gαs-coupled GPCRs, E-type prostanoid receptors 2 and 4 (encoded by the PTGER2 and PTGER4 genes, respectively). PGE2 has been shown to increase proliferation in colon cancer cells and mediate activation of β-catenin (through Gαs) and other mitogenic signaling molecules, like phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt) (through Gβγ effects) (Castellone et al., 2005).
Frequent and early genomic alteration in the adenomatous polyposis coli (APC) gene are often concurrent with COX-2 overexpression in early initiation events of CRC (Fearon and Vogelstein, 1990; Markowitz and Bertagnolli, 2009), thus highlighting the interplay between their regulated pathways. APC acts a major tumor suppressor in CRC, inhibiting the Wnt–β-catenin signaling route (Kolligs et al., 2002). The Wnt pathway is a major determinant of cell fate decisions, helping to promote stem cell maintenance and tissue renewal from embryogenesis to adulthood. Consequently, these normal programs are frequently co-opted by disease. Wnt signaling controls β-catenin, a coactivator that drives transcription through binding of nuclear transcription factors (i.e., T-cell factor or TCF). When the pathway is inactive, β-catenin is sequestered in the cytoplasm by a protein complex termed the destruction complex and ultimately targeted for degradation (Fig. 10A). This destruction complex consists of key molecules like GSK3, casein kinase 1α (CK1α), Axin, and APC. CK1α and GSK3 provide the phosphorylation signals that target β-catenin for ubiquitination and degradation. Canonically, the pathway becomes activated by extracellular Wnts or changes in adherens junctions (Angers and Moon, 2009; Valenta et al., 2012). However, the Gαs-PKA pathway can modulate β-catenin activity at several levels (Fig. 10A). When activated by receptors, Gαs has been shown bind to Axin, leading to the stabilization and activation of β-catenin (Castellone et al., 2005). Many components of the destruction complex are also phosphorylated by PKA. The predominant mechanisms highlight the ability of PKA to phosphorylate and inhibit GSK3, releasing β-catenin to enter the nucleus (Fang et al., 2000). This, coupled with direct PKA phosphorylation of β-catenin to inhibit ubiquitination and degradation, helps drive β-catenin–mediated transcription (Hino et al., 2005). These mechanisms have important biologic consequences, including stem cell maintenance and tissue regeneration and repair (Goessling et al., 2009; Wang et al., 2016). Crosstalk with the Gαs-PKA pathway is also particularly important for the endocrine Gαs-PKA pathway signalopathies (Walczak and Hammer, 2015). For instance, β-catenin expression is very strong in adrenal tumors and Carney complex caused by genetic defects in the Gαs-PKA pathway (Almeida et al., 2012). This contributes to dysregulated Wnt signaling and loss of cell cycle control (Almeida et al., 2010). In CRC, activation of the Wnt–β-catenin pathway by Gαs-PKA may represent a key event in CRC initiation and progression, whether it is achieved by mutations in GNAS or perhaps more often by PGE2 and COX-2–initiated, Gαs-linked GPCR signaling (Castellone et al., 2005; Wu et al., 2019).

Aberrant protein kinase A pathway activity leads to dysregulation of signaling and transcriptional programs. (A) Wnt and PKA activity drive β-catenin–mediated gene transcription. Canonically, Wnt binds to Frizzled receptors and coreceptors like LPL receptor related protein 6 (LRP 6) on the surface of the cell to inhibit the activity of the destruction complex. Destruction complex members include APC, GSK3, CK1α, and Axin. Inhibition of this complex releases β-catenin to drive target gene transcription through the transcription factor TCF. Production of PGE2 through COX-2 leads to activation of Gαs-coupled GPCRs, E-type prostanoid receptor 2 (EP2), and E-type prostanoid receptor 4 (EP4). Activation of Gαs leads to direct phosphorylation and inhibition of GSK3 as well as stabilizing phosphorylation of β-catenin. These effects coupled with the direct binding of Gαs to Axin lead to accumulation of β-catenin and activation of target gene transcription. (B) PKA inhibits Hippo pathway and YAP/TEAD-mediated transcription. The Hippo pathway is regulated by a kinase cascade whereby the upstream kinase MST phosphorylates and activates LATS kinase. Phosphorylation of YAP by LATS inactivates YAP through cytoplasmic sequestration and degradation. PKA-mediated phosphorylation of LATS, among other mechanisms, also inhibits YAP activity and consequently blocks target gene transcription through TEAD. (C) PKA regulates hedgehog (HH) signaling in the cilium to inhibit GLI transcriptional activity. When HH ligand is present, it binds to and inhibits the receptor Patched (PTCH1), allowing the Gαi-like GPCR SMO to traffic to the ciliary membrane. SMO inhibits cAMP production and PKA activity, allowing GLI-mediated transcription to proceed. When HH ligand is absent, PTCH1 constitutively inhibits SMO and allows the Gαs-coupled GPCR GPR161 to traffic to the ciliary membrane. When present at the membrane, GPR161 stimulates cAMP production and PKA activity. PKA in turn phosphorylates and inhibits GLI, eventually leading to its degradation.
Aligned with this perspective, PGE2 dramatically increases intestinal tumor burden in CRC mouse models, and the inhibition of PGE2 production with COX-2 inhibitors, such as by nonsteroidal anti-inflammatory drugs (NSAIDs), reduces tumor burden (Hansen-Petrik et al., 2002; Kawamori et al., 2003; Wang and DuBois, 2010). In humans, retrospective studies have revealed a reduced incidence of colorectal cancer with prolonged NSAID use, and NSAIDs can directly reduce polyp size and number in patients with familial CRC. Unfortunately, the clinical response to NSAIDs is incomplete, and long-term use can have limiting toxicities (Giardiello et al., 1993; Brown and DuBois, 2005). Of available NSAIDs, aspirin has been used successfully long-term in cardiovascular disease. In these patient populations, aspirin has also been shown to reduce CRC incidence and mortality. Interestingly, the benefit of aspirin in chemoprevention was most pronounced after 10 years (Chan et al., 2005; Chubak et al., 2015; Drew et al., 2016). One mechanism by which aspirin is thought to reduce mortality is by preventing metastasis, particularly in the progression of local adenoma to metastatic disease (Rothwell et al., 2012). Given the consistent efficacy of aspirin and other NSAIDs in chemoprevention, numerous clinical trials have tested their efficacy in other settings. Notably, NSAIDs have shown efficacy in some adjuvant settings but failed when operating as single-agent chemotherapeutics (Brown and DuBois, 2005; Wang and DuBois, 2010). The clinical efficacy of NSAIDs as chemopreventive agents, but failure as chemotherapeutics, highlights the true complexity of prostaglandin signaling. It is likely that PGE2 and others participate in autocrine and paracrine signaling loops that involve both tumor, stroma, and immune components. To this end, Gαs-PKA activation, downstream of the proton-sensing GPCR GPR68, has been shown to drive the secretion of interleukin 6 (IL- 6) from cancer-associated fibroblasts and subsequent proliferation of PDAC in trans (Wiley et al., 2018). Further, Gαs-linked GPCRs, like prostanoid (Zelenay et al., 2015; Böttcher et al., 2018; Pelly et al., 2021) and adenosine receptors (Visser et al., 2000; Novitskiy et al., 2008; Young et al., 2014; Young et al., 2018), contribute to tumor immune evasion and drive immune suppression by dampening T-cell responses, as well as interfering with immune cell migration and maturation. For example, these mechanisms can include direct PKA-mediated phosphorylation of C-terminal Src kinase (CSK) and other components involved in T-cell receptor signaling and activation, as well as PGE2-mediated suppression of chemokine production and dendritic cell recruitment (Wehbi and Taskén, 2016; Böttcher et al., 2018). Recent evidence also points to the specific role of PKA Cβ2 (an immune-specific spliceform) in regulating immune responses in inflammatory disease (Moen et al., 2017). Together, this highlights that the Gαs-PKA pathway can participate in tumor initiation and progression through autocrine and paracrine (oncocrine) mechanisms (Wu et al., 2019). Even in the absence of overt mutations, these oncocrine signals can have important effects throughout the tumor microenvironment, including contributions to a cancer immune evasion and therapeutic resistance (see section 5. Gαs–Protein Kinase A Induced Therapeutic Resistance in Cancer).
4. GNAS–Protein Kinase A as Tumor Suppressors
Thus far, our discussions of the Gαs-PKA pathway in cancer have focused on the role of GNAS and DNAJB1-PRKACA as oncogenes. Paradoxically, however, there are several examples in which the Gαs-PKA pathway functions as a tumor suppressor. A study by our group unexpectedly found that genetic ablation of Gnas or inhibition of PKA in the epidermis was sufficient to drive basal cell carcinoma, with dramatic expansion of the stem cell compartment residing in the hair follicle. Conversely, overactivation of the pathway with the GNAS R201C mutation drove the same stem cell population to terminal differentiation and exhaustion. Mechanistically, stem cell expansion in the hair follicle is controlled by PKA-mediated repression of yes-associated protein (YAP) and glioma-associated oncogene (GLI) transcriptional activity, with no effect on other stem cell programs like Wnt (Iglesias-Bartolome et al., 2015). Of note, PKA has been shown to repress YAP activity in pancreatic cancer (in which PKA functions as an oncogene) but still induce a differentiation phenotype (Ideno et al., 2018). Much like GNAS and PKA, YAP has also been shown to behave as either an oncogene or a tumor suppressor depending on the cellular context.
The Hippo pathway controls growth, differentiation, and cell death, balancing these processes to ensure proper organ development and size. In mammals, YAP and tafazzin (TAZ) are the main effectors that regulate transcriptional output through binding to transcription factors like TEA domain transcription factor (TEAD) in the nucleus. YAP/TAZ are regulated by phosphorylation from upstream kinases large tumor suppressor kinase 1 and 2 (LATS 1/2), whereby phosphorylation induces YAP/TAZ cytoplasmic sequestration and subsequent degradation (Fig. 10B). LATS1/2 in turn can be regulated by many upstream signals, including GPCRs. Gαs-coupled GPCRs activate LATS1/2 to repress YAP/TAZ (Yu et al., 2012). PKA directly phosphorylates LATS1/2 to enhance its kinase activity, and mutation of the PKA phosphosites abrogates PKA regulation of LATS1/2 while other regulatory mechanisms remain intact (Kim et al., 2013) (Fig. 10B). Physiologically, this is important because PKA is known to induce adipogenesis and neurogenesis through suppression of YAP (Kim et al., 2013; Yu et al., 2013). In general, YAP phosphorylation and inactivation is critical for cell cycle exit and terminal differentiation, and it is thought that PKA contributes to this regulation (Lee et al., 2008; Kim et al., 2013). This can explain in part why many neoplasms and cancers characterized by Gαs-PKA pathway activation are of well differentiated histology and typically less proliferative or low-grade (as discussed previously, see section 3. GNAS and Protein Kinase A Link Inflammation to Cancer Initiation).
In line with the additional effects of Gαs on GLI in basal cell carcinoma, low GNAS expression is also a feature of the sonic hedgehog (SHH) subtype of medulloblastoma (SHH-MB). Medulloblastoma is the most common pediatric brain cancer, with SHH-MB representing 30% of patients (Kijima and Kanemura, 2016). Within this subtype, activation of the SHH pathway (through multiple mechanisms) is thought to drive tumor initiation. Interestingly, patients with SHH-MB with low GNAS expression have significantly worse prognosis compared with patients with high GNAS expression (50% 5-month survival versus 100% 5-month survival). Similar to the hair follicle model, knockout of Gnas in neural progenitor cells induced expansion of this stem cell population in neonatal mice that progressively developed into a tumor resembling medulloblastoma by adulthood. The tumors were marked by upregulation of GLI and SHH signaling with no effect on the Wnt pathway, a pattern that matches the signature of patients with SHH-MB (He et al., 2014). Around 6% of patients with SHH-MB actually have GNAS mutations, including frameshift and nonsense inactivating mutations (He et al., 2014; Huh et al., 2014; Kool et al., 2014). Perhaps more surprisingly through, around 80% of SHH-MBs overexpress C-X-C motif chemokine receptor 4 (CXC R4), which is a Gαi-coupled GPCR (Sengupta et al., 2012). These patients are typically younger (∼50% were infants) with desmoplastic histology (He et al., 2014). Although CXCR4 is not often mutated, CXCR4 and its ligand, C-X-C motif chemokine ligand 12 (CXCL 12), are markers of poor prognosis and earlier onset in other brain tumors, like gliomas (Calatozzolo et al., 2006; Bian et al., 2007). For these patients, cAMP elevating agents, such as PDE inhibitors, have been proposed as potential therapeutic options (Rao et al., 2016).
The importance of Gαs in the SHH-MB subtype of pediatric brain cancer reflects the fundamental importance of Gαs-PKA in brain development. As a testament to its importance, Gnas homozygous knockout mice are embryonic lethal (Yu et al., 1998). Similarly, only 27% of Prkaca homozygous knockout mice survive past weaning (Skålhegg et al., 2002). As mentioned previously, both Cα1 and Cβ1 are ubiquitously expressed and capable of some degree of compensation. Therefore, it is not surprising that Cα and Cβ1 double knockout mice are embryonic lethal. Restoration of one allele in either gene (Cα or Cβ1) confers survival, but mice die from severe neural tube defects. Histologically, these mice have an expansion of cell types that are dependent on hedgehog (HH) signaling (Huang et al., 2002).
In a more pathway-specific fashion, PKA is known to regulate HH signaling, both SHH and Indian hedgehog, within the context of cilia. Interestingly, the ciliary structure is essential to proper signaling and development controlled by the HH pathway, a feature that is not shared by other developmental programs. The GLI family of transcription factors are the main effectors that respond to upstream stimulus from HH ligands. In the absence of pathway stimulation, GLI is sequestered and eventually degraded (Carballo et al., 2018). AKAPs position PKA at the base of the cilium, where the catalytic subunit phosphorylates GLI to facilitate GLI’s proteolytic processing and degradation, ultimately preventing transcriptional activation (Fig. 10C). Recent work has demonstrated that the Gαs-coupled GPCR, GPR161, contains an AKAP domain enabling it to directly recruit PKA to cilia (Bachmann et al., 2016). Of note, GPR161 is regulated by trafficking and only capable of signaling when it is present on the ciliary membrane (Bangs and Anderson, 2017). Activation of GPR161, among other Gαs-coupled GPCRs, is important to trigger production of cAMP and subsequent PKA activation. Generally, PKA activity is quite high when HH ligand is absent (Tschaikner et al., 2020). However, when HH is present, the Gαi-like-coupled GPCR Smoothened (SMO) traffics to the cilium to trigger a reduction in cAMP levels and inhibition of PKA activity (Ogden et al., 2008), allowing full-length GLI to activate transcription. This trafficking is regulated by the binding of HH to its receptor patched homolog 1 (PTCH1) at the membrane, thereby relieving the inhibition on SMO (Bangs and Anderson, 2017). Recent evidence has also demonstrated that SMO can directly inhibit PKA through binding to the free catalytic subunits at the membrane (Arveseth et al., 2021) (Fig. 10C). Numerous other GPCRs, such as CXCR4 (Gαi-coupled) and PAC1 (Gαs-coupled), can also contribute to the modulation of ciliary cAMP levels and PKA activity, although some of these roles are complex and cell type–dependent (Niewiadomski et al., 2013; Mukhopadhyay and Rohatgi, 2014; Schou et al., 2015; Mykytyn and Askwith, 2017; Amarante et al., 2018; Tschaikner et al., 2020). Ultimately, the degree of GLI transcriptional output is dependent on the level of PKA activity as a balance of these various inputs (Tschaikner et al., 2020). Consequently, the overexpression of PKA Cα is sufficient to inhibit SHH-stimulated proliferation and induce differentiation (Barzi et al., 2010). Recently, several mutations in Cα and Cβ, which display increased sensitivity to cAMP, show reduced HH pathway activation (Palencia-Campos et al., 2020). Conversely, deletion of Gαs in the mouse augments SHH signaling with developmental defects that mirror PKA deletion, or deletion of other negative regulators of the SHH pathway (Regard et al., 2013). SHH signaling is particularly important in guiding development of the nervous system and limb patterns, whereas Indian hedgehog is important in skeletal development (Bangs and Anderson, 2017). This explains why patients with loss-of-function mutation in the Gαs-PKA pathway can develop SHH-MB or severe skeletal deformities as part of iPPSD (discussed previously, see section 7. Inactivating Parathyroid Hormone/Parathyroid Hormone–Related Peptide Signaling Disorder). Furthermore, recent reports have described mutations in PRKACA that cause skeletal ciliopathies (Palencia-Campos et al., 2020; Hammarsjö et al., 2021).
5. Gαs–Protein Kinase A Induced Therapeutic Resistance in Cancer
Our discussions have already highlighted some features of the Gαs-PKA pathway that contribute to therapeutic resistance, including supporting an immune suppressive tumor microenvironment, and clinical evidence of poor outcomes and chemoresistance due to mucinous disease. Here, we will focus on additional evidence of the therapeutic resistance potential of the Gαs-PKA pathway in cancer.
Building on the evidence of GNAS and KRAS functioning as codrivers of carcinogenesis, several unbiased studies have identified Gαs and PKA as key drivers of resistance to MAPK pathway inhibition. In metastatic melanoma, about half of all patients have BRAF mutations and are primarily treated with BRAF inhibitors. Although most patients have clinical responses, approximately 20% of patients with BRAF mutation have intrinsic resistance to BRAF inhibitors (Sanchez et al., 2018). Unfortunately, many initial responders later develop acquired resistance from genetic (60%) or epigenetic and transcriptomic (40%) changes, primarily through reactivation of MAPK signaling outputs (Kakadia et al., 2018). Several studies have aimed at understanding these mechanisms of resistance and reactivation. Gain-of-function open reading frame and CRISPR activation screens in BRAF V600E melanomas have been used to identify programs that confer resistance to multiple BRAF and MAPK inhibitors. Surprisingly, GPCRs were consistently among the top hits, many of them being Gαs-coupled (Johannessen et al., 2013; Konermann et al., 2015). Downstream, ADCY9 and PKA Cα also confer resistance to MAPK inhibitors, with PKA Cα having a higher score than even RAF1. Further analysis revealed that PKA via CREB was able to activate transcriptional programs that MAPK normally activates (Johannessen et al., 2013). In melanocytes, there is a fine balance between MAPK control of proliferation and cAMP control of differentiation (Dumaz et al., 2006). This balance is achieved in part because PKA can phosphorylate and inhibit RAF1, while BRAF continues signaling downstream to ERK (Cook and McCormick, 1993; Dhillon et al., 2002). Interestingly , when RAS is mutated, RAF1 predominantly signals to ERK, a program that BRAF controls when it is mutated. This type of compensatory crosstalk is the basis for PKA-mediated resistance to MAPK pathway inhibition. Of note, this crosstalk is not present in all cell types, e.g., fibroblasts (Dumaz et al., 2006).
As we discussed previously, inflammatory signaling through COX2-PGE2-Gαs contributes to the pathogenesis of many cancers. Recently, this pathway has also been implicated as a mechanism of resistance to combination BRAF and MAPK pathway inhibition in BRAF V600E colorectal cancer. Using a high-throughput kinase activity screen, the SRC proto-oncogene(SRC) was identified as having increased activity after inhibitor treatment. SRC in particular was shown to initiate a proinflammatory autocrine loop mediated by PGE2 and Gαs that was sensitive to COX2 inhibition. Dramatically, the addition of a COX2 inhibitor to two or three drug combinations targeting the MAPK pathway led to greater rates of tumor regression in patient-derived xenograft resistance models (Ruiz-Saenz et al., submitted manuscript). The mechanisms of resistance through COX2-PGE2-Gαs and PKA include survival of cancer stems cells as well as immune suppression (Tong et al., 2018). In BRAF V600E mutant melanoma, for instance, COX-2 was shown to drive tumor immune escape, a response that underlines the preclinical synergy of COX-2 inhibitors in combination with immune checkpoint blockade (Zelenay et al., 2015). Similarly, the ability of PKA to drive tumor immune evasion has also limited the efficacy of other immune-based therapies such as chimeric antigen receptor T cells (CAR-Ts) (Newick et al., 2016). This suppression of CAR-Ts and T cells in general is mediated by PKA/AKAP associations that negatively regulate T-cell function (Ruppelt et al., 2007). Interestingly, disruption of this PKA/AKAP interaction can improve CAR-T efficacy and enhance tumor killing (Newick et al., 2016). Building on the understanding of these immune suppressive mechanisms (see section 3. GNAS and Protein Kinase A Link Inflammation to Cancer Initiation), NSAIDs as well as prostanoid and adenosine receptor antagonists are being investigated as agents to combat tumor immune evasion and enhance the clinical efficacy of immune therapies (Leone et al., 2015; Hamada et al., 2017; Take et al., 2020). Finally, the Gαs-PKA pathway has effects on migration and metastasis. This role is somewhat controversial, as PKA has been shown to drive epithelial differentiation, instead of the epithelial-to-mesenchymal transition phenotypes generally recognized as metastatic (Pattabiraman et al., 2016). However, PKA is also known to play a role in cytoskeletal changes through direct AKAP interactions that are required for many of the hallmarks of cell migration (Howe, 2004). Importantly, it seems that these effects are context-dependent, since Gαs and PKA serve as a central regulatory hub integrating many signaling pathways and biologic functions.
PKA can also contribute to therapeutic resistance by co-opting other normal mechanisms, including energy adaptation. The mitochondria are the main producers of energy in the cell; thus, maintaining mitochondrial homeostasis is critically important to cell health. Mitochondrial homeostasis represents a dynamic balance between fusion (joining) and fission (division) events. PKA is particularly well studied in its ability to inhibit mitochondrial fission through phosphorylation of dynamin-related protein 1 (DRP 1), a dynamin-like GTPase. DRP1 functions to bring mitochondrial membranes close to each other to facilitate fission events. PKA phosphorylation at serine 637 inhibits DRP1 GTPase activity and recruitment to the mitochondria (Chang and Blackstone, 2007). By inhibiting fission, fusion is allowed to proceed, resulting in elongated mitochondria and increased respiration. Increased cAMP and PKA activity has also been linked to decreased mitophagy and ultimately control of mitochondrial recycling; however, it remains unclear whether this is primarily due to increased fusion or additional effects of cAMP and PKA. Together, the actions of the cAMP-PKA pathway on the mitochondria provide a prosurvival signal (Di Benedetto et al., 2018; Ould Amer and Hebert-Chatelain, 2018). Under physiologic conditions of low nutrients, cells elongate mitochondria to compensate. Interestingly, this physiologic adaptation can be exploited by cancer cells, which, although somewhat counterintuitive, rely heavily on glycolysis for energy (Vander Heiden et al., 2009). For instance, KRAS transformed cells die in low-glucose conditions, but activation of cAMP/PKA rescues their survival under these conditions. PKA-mediated activation of mitochondrial respiration ramps up oxidative phosphorylation and ATP levels (Acin-Perez et al., 2009; Palorini et al., 2013; Ould Amer and Hebert-Chatelain, 2018). Coupled with reduction in reactive oxidative species and increased autophagy, cAMP and PKA metabolically rewire cells to promote survival (Palorini et al., 2013; Palorini et al., 2016; Ould Amer and Hebert-Chatelain, 2018). Under physiologic conditions of low nutrients, PKA also liberates energy from glycogen and lipid stores through direct phosphorylation, as well as transcriptional regulation, of the enzymes involved in these processes (Rogne and Taskén, 2014; Yang and Yang, 2016) (see section F. Metabolic Regulation). However, it remains unclear to what extent cancer cells exploit these energy sources. Together, energy adaptation mechanisms and prosurvival signals provide some insight as to why GNAS and PKA serve as biomarkers of therapeutic resistance in many cancer types and further why GNAS and KRAS often comutate in cancer.
Finally, the role of Gαs and PKA in resistance can be seen clinically in breast cancer, a tissue type in which GNAS mutations are rarely found. One study profiled circulating free DNA before and after treatment with targeted therapy in metastatic, human epidermal growth factor receptor 2–positive (HER2+) breast cancer. Surprisingly, they found that GNAS mutations were only present in patients that were resistant to targeted therapy (Ye et al., 2017). Similarly, PRKACA transcripts were elevated in HER2+ patients that were resistant to trastuzumab (HER2 inhibitor) (Moody et al., 2015). In vitro models of resistance have also demonstrated that knockdown of PRKAR2A, to activate PKA, confers partial resistance to trastuzimab (Gu et al., 2009). Unlike in melanoma, this resistance could not be explained by MAPK pathway reactivation but, rather, by restoration of antiapoptotic signaling (Moody et al., 2015). In another subtype of breast cancer, estrogen receptor–expressing, patients receive antiestrogen therapies such as tamoxifen. Tamoxifen binds to estrogen receptor α to induce a conformation that prevents its activation and signaling. Interestingly, PKA has been found to phosphorylate the estrogen receptor α, an interaction coordinated by AKAP13. This phosphorylation prevents the inhibitory conformational change induced by tamoxifen and renders tamoxifen ineffective (Michalides et al., 2004; Bentin Toaldo et al., 2015). GNAS amplifications have been identified in 20% of HER2+ breast cancers and 13% of hormone receptor–positive breast cancers (Kan et al., 2010). Although further studies are required, it is tempting to suggest that GNAS amplification may serve as a biomarker, predicting resistance to therapy in breast cancer. Here, we have highlighted several known mechanism of therapeutic resistance, but there are certainly additional mechanisms yet to be described. Together, these findings highlight again the diversity and complexity of Gαs and PKA signaling and their roles in the diversity of the Gαs-PKA pathway signalopathies.
V. Targeting the Gαs–Protein Kinase A Pathway Signalopathies
Given the breadth of the Gαs-PKA pathway signalopathies, it is tempting to imagine how valuable a magic bullet PKA drug, potentially a life-changing resource for families with germline Gαs-PKA pathway signalopathies, like Carney complex, could be. Although throughout this review we have often distilled diseases down to mutational themes, all circling back to simple activation or inactivation of the Gαs-PKA pathway, we have also taken care to highlight the complexity that underlies all of these signaling events. We must acknowledge the role of local microdomains and specific isoforms that allow PKA to mediate disparate yet parallel functions and of course recognize the diverse inputs that modulate their activity. This complexity may seem like a liability at first glance. However, as we continue to understand the specifics of each signaling defect more deeply, it may provide a unique opportunity to carve out a therapeutic window. Current standard of care for the Gαs-PKA pathway signalopathies, particularly those characterized by developmental defects or neoplasia, involve surgical and palliative treatments (Sharma et al., 2015; Javaid et al., 2019). These treatments do not address the true cause of the disease but instead highlight the value of targeted approaches.
A. Targeting G Protein–Coupled Receptors and G Protein–Coupled Receptor Ligands
When considering how to target the Gαs-PKA pathway signalopathies, the natural first step lies at the cell surface with receptors. GPCRs are the target of approximately one-third of all clinically approved small-molecule drugs (Santos et al., 2017). Nearly every family of GPCR has been targeted by either an approved drug or one in clinical development, including both small molecules and peptides. GPCR drugs have proven to be tremendously effective in diseases such as heart failure and asthma, in which drugs targeting β-adrenergic receptors, among others, can improve heart function and cause airways dilation, respectively (Wang et al., 2018a; Wendell et al., 2020). As the Gαs-PKA pathway signalopathies largely focus on genetic diseases with endocrine and neoplastic phenotypes, here we will focus on the therapeutic potential of GPCRs in these settings, with the ability to modulate both Gαs- and Gαi-coupled receptors with agonists and antagonists, depending on the role of the pathway in the disease. This strategy has already proven effective in several Gαs-PKA pathway signalopathies. For example, somatostatin receptor analogs have been used to treat acromegaly for years, and a new analog, pasireotide, was recently approved for Cushing syndrome (Freda, 2002; McKeage, 2013). Somatostatin is the endogenous peptide ligand for the Gαi-coupled somatostatin family of GPCRs (SSTRs), but its use is limited clinically because of its extremely short half-life. Several peptide analogs have been developed to improve the half-life and with variable selectivity for somatostatin receptor subtypes. In acromegaly, 50%–60% of all patients benefit from somatostatin analogs, showing reduced GH and insulin-like growth factor-1 secretion as well as tumor shrinkage; however, surgery is often still the first line of therapy (Freda, 2002). In Cushing syndrome, pasireotide specifically targets SSTR5, which is highly expressed on ACTH-secreting pituitary tumors. Activation of SSTR5 reduces ACTH secretion and subsequently cortisol secretion. However, SSTR5 is also expressed on pancreatic β-cells, in which pasireotide inhibits insulin secretion and can exacerbate hyperglycemia, even contributing to the development of diabetes mellitus as a side effect in some patients (McKeage, 2013; Colao et al., 2014). To counteract these adverse events, patients are often administered GLP -1 agonists, targeting the Gαs-coupled glucagon-like peptide-1 receptor (GLP1R) (Colao et al., 2014). GLP-1 agonists are commonly used to treat type II diabetes and obesity apart from Cushing syndrome because of their ability to increase insulin secretion and control appetite (Miller et al., 2014). Although GPCRs have proven to be great targets, no clinical drugs are available to target Gαs or Gαi directly (Campbell and Smrcka, 2018).
Other therapeutic approaches related to GPCRs are aimed at limiting ligand production, as is the case for many of the drugs used to treat Cushing syndrome and hyperthyroidism, which broadly inhibit steroidogenesis or hormone synthesis to limit hormone production (Sharma et al., 2015; De Leo et al., 2016). In the case of adrenal or pituitary adenomas that automatously secrete hormone, surgical removal of the tumor is a common approach (Sharma et al., 2015). For hereditary hyperthyroidism, patients typically receive radioactive iodine or surgery to remove the thyroid, but antithyroid drugs may also be used to interfere with thyroid hormone production, as some patients present at a young age (Hébrant et al., 2011; De Leo et al., 2016). Similarly, we have also discussed the use of COX-2 inhibitors as a means to limit prostaglandin production in colorectal cancer (see section 3. GNAS and Protein Kinase A Link Inflammation to Cancer Initiation). As evident from clinical studies, the side effects of this type of approach can largely limit the efficacy (Brown and DuBois, 2005). Furthermore, some patients, particularly those with genetic mutation of the PKA holoenzyme, are inherently resistant to these types of upstream modulation.
B. Targeting the Protein Kinase A Holoenzyme Directly
Although most kinases are manipulated by selective protein kinase inhibitors that target the active site cleft, there are a variety of ways to interfere with the PKA holoenzyme. In addition to small-molecule inhibitors, such as H89, that mimic ATP (Hidaka et al., 1984), high-affinity inhibitory peptides have been derived from the endogenous PKI (Cheng et al., 1985). In addition, analogs of cAMP differentially target type I versus type II regulatory subunits (Schwede et al., 2000), and isoform-selective peptides can disrupt holoenzyme targeting (Wang et al., 2014, 2015; Bendzunas et al., 2018). Although many of these strategies hold promise, currently there are no clinical-grade drugs that target PKA specifically.
1. ATP Analog Inhibitors of the Catalytic Subunit
The most commonly used small-molecule inhibitors are the high-affinity, ATP-competitive isoquinolinesulfonyl protein kinase inhibitors, such as H89, H7, and H8 (Hidaka et al., 1984; Chijiwa et al., 1990; Engh et al., 1996; Lochner and Moolman, 2006); natural product derivative KT-5720 (Kase et al., 1987); or staurosporine (Meggio et al., 1995). Although these are very effective inhibitors, they have low specificity and inhibit several other kinases in the AGC family of protein kinases and hence should not be considered specific inhibitors (Lochner and Moolman, 2006; Murray, 2008). Of course, these inhibitors also do not discriminate between the PKA isoforms, thus limiting their clinical translatability.
2. Peptide Inhibitors of the Catalytic Subunit
To overcome the concerns of specificity, derivatives of the substrate-competitive, heat-stable PKI (encoded by PKIA, PKIB, and PKIG) can be used. PKI(5-24) has low-nanomolar inhibition constants and is absolutely specific for PKA (Cheng et al., 1985). PKI(5-24) can be modified by myristylation, which allows for membrane permeation (Eichholtz et al., 1993); however, it can also be expressed recombinantly in cells to overcome delivery issues. A hydrocarbon-stapled version of a PKI-derived sequence provides another excellent tool as a membrane-permeable, highly selective inhibitor of the catalytic subunits acting with low-subnanomolar affinity (Manschwetus et al., 2019).
3. Bisubstrate Inhibitors of the Catalytic Subunit
A combination of the two cosubstrate inhibitors, ATP and peptide, would be the logical consequence, and indeed, such bisubstrate analog inhibitors termed ARC-type inhibitors have been developed by linking an adenosine analog (either an adenosine derivative or ATP inhibitor) and an arginine-rich peptide (Lavogina et al., 2010). A series of ARC-type inhibitors have been designed with low-nanomolar or even picomolar affinities and efficacy against PKA Cα and Cβ (Ricouart et al., 1991; Enkvist et al., 2006; Enkvist et al., 2007; Lavogina et al., 2010; Nonga et al., 2020). Recent work has demonstrated that ARC inhibitors can also be engineered to have greater selectivity for mutant Cβ over wild-type Cβ (Nonga et al., 2020). Although ARC inhibitors have primarily been used as tool compounds, including fluorescently conjugated ARCs, recent advances have drastically improved their pharmacokinetic properties, making them poised for future application in a therapeutic context (Lavogina et al., 2010).
4. Targeting the Regulatory Subunits with cAMP Analogs
In contrast to the ATP analog inhibitors that target the catalytic subunit, cAMP analogs have been engineered with specificity for the two classes of regulatory subunits (RI and RII). Both activators and inhibitors have been developed (Christensen et al., 2003). Achieving PKA regulatory subunit specificity has been a special challenge, as other proteins such as cGMP-dependent protein kinases, EPACs, CNG channels, PDEs, and cyclases all have cyclic nucleotide binding domains (Berman et al., 2005; Holz et al., 2008) (see section G. Other cAMP Effectors). By modifying the oxygens of the cyclic phosphate, chemists generated cAMP agonists (Sp analogs) and antagonists (Rp analogs). Global inhibition can be achieved with the Rp analogs, which bind to but do not promote dissociation of the holoenzyme (Rothermel and Parker Botelho, 1988; Christensen et al., 2003). By comparing the activity of type I inhibitors, like Rp-8-Br-cAMPS, with the activity of nonselective inhibitors, like Rp-cAMPS, it is possible to discriminate between the activities of the two holoenzymes (Gjertsen et al., 1995; Christensen et al., 2003; Farquhar et al., 2008). Similarly, the combination of different agonists can achieve some level of isoform-specific activation, but this still remains a challenge in the field (Robinson-Steiner and Corbin, 1983). However, leveraging regulatory subunit agonists and antagonists has facilitated the high-quality purification of PKA holoenzymes as well as free regulatory subunits (Bertinetti et al., 2009; Hanke et al., 2011). Unfortunately, many of these cAMP analogs suffer from poor membrane permeability, limiting their efficacy if delivered extracellularly. To overcome this, membrane-permeable versions of the cAMP analogs have been developed as prodrugs. When cleaved by cytosolic esterases, the analog is free to act inside the cell (Chepurny et al., 2013; Schwede et al., 2015). Care must be taken, however, because the effective concentration of the released nucleotide inside the cell may vary, and extremely high levels of cAMP may perturb other cyclic nucleotide signaling.
5. Inhibitors of A-Kinase Anchoring Protein Binding
PKA specificity is also highly dependent on targeting to specific sites in the cell. Targeting is typically mediated by binding to AKAPs that contain a high-affinity helical binding motif that interacts with the D/D domains of the regulatory subunits. Naturally, peptides have been developed to disrupt this interaction, nonselectively perturbing the interactions with both type I and type II interactions (Carr et al., 1992; Herberg et al., 2000). Over time, this led to development of peptides specific to type I or type II, although these peptides still suffered from limited cell permeability (Calejo and Taskén, 2015). Now, isoform-specific, cell-permeant stapled peptides have been engineered that can selectively disrupt the targeting of type I and type II holoenzymes (Wang et al., 2014, 2015; Kennedy and Scott, 2015; Bendzunas et al., 2018). Unfortunately, these peptides still lack clinical utility because of their unfavorable pharmacokinetics and relative inability to distinguish among specific AKAP interactions (Calejo and Taskén, 2015). Reagents have also been developed to disrupt other AKAP binders, such as PDEs and phosphatases, but as AKAPs have multiple binding partners, it has been difficult to translate this disruption to direct modulation of cellular consequences (Bucko and Scott, 2020; Omar and Scott, 2020). To begin to answer these difficult questions of microdomain dynamics, a promising new tool has been developed using AKAP targeting sequences as a means to localize drug delivery to specific PKA microdomains, such as those present at the centrosome. Although this approach, called local kinase inhibition, is still in its infancy, conceptually it holds a lot of promise in understanding AKAP interactions more directly and ultimately enhancing the specificity of PKA modulation (Bucko et al., 2019). Finally, small-molecule AKAP disrupters represent another promising approach with potential for clinical translation. Protein-protein interactions have been notoriously difficult to target with small molecules, but the advances in high-throughput screening have made this approach more feasible (Calejo and Taskén, 2015). Several groups have applied these approaches recently to identify disrupters of AKAP interactions (Gold et al., 2013; Schächterle et al., 2015). Although there are real challenges, huge potential lies in the ability to apply these small-molecule disrupters to specific AKAP complexes in diseases settings; for instance, disruption of the PKA/AKAP interactions that mediate immune suppression in T cells in cancer (see section 5. Gαs–Protein Kinase A Induced Therapeutic Resistance in Cancer).
6. Emerging Approaches
As we discussed, many of the Gαs-PKA pathway signalopathies are driven by specific hotspot point mutations, like GNAS R201C or PRKACA L206R, so generating mutation-specific drugs could be a viable therapeutic option. Recently, this strategy has shown clinical promise, most notably by targeting the mutant cysteine of KRAS G12C with drug electrophiles (Ostrem et al., 2013). It has been proposed that this same method could also be applied to target GNAS R201C mutants in cancer (Visscher et al., 2016). Although PKA is not amenable to targeting with drug electrophiles, the PKA Cα L206R mutation does have reduced affinity for its endogenous inhibitor PKI compared with wild-type PKA Cα, whereas the small-molecule inhibitor H89 still retains its efficacy. This opens the possibility of exploiting this differential binding to selectively target PKA Cα mutants. However, significant challenges remain, as H89 retains its efficacy because it is an ATP-competitive inhibitor. As alluded to previously, this class of drug is susceptible to multiple off-target effects on other kinases, making it a liability in the clinical setting (Luzi et al., 2018). In an effort to identify drugs that do not act as ATP-competitive inhibitors of PKA, high-throughput screening platforms based on fluorescence polarization have been developed and proven capable of identifying allosteric agonists and antagonists (Saldanha et al., 2006; Brown et al., 2013). Some promise has also been shown for antisense oligonucleotides targeting RIα in combination with chemotherapy in cancer (Goel et al., 2006; Almeida et al., 2012). The mechanism is not completely understood, but the compensatory increase in RIIβ protein could be important in restoring the balance of type I and type II holoenzyme signaling (Nesterova et al., 2000). Similarly, although many of the PKA Cα mutations have been linked to altered substrate profiles and decreased preference toward canonical substrates. It is plausible that restoring activity toward key substrates may also serve as an additional therapeutic avenue (Lubner et al., 2017).
C. Degraders of Pathway Components
Another promising approach to targeting the Gαs-PKA pathway directly is in targeting the stability of pathway components. This strategy has garnered huge interest in the past few years with the development of small-molecule inhibitors termed proteolysis targeting chimeras (PROTACs). PROTACs consist of an element targeting the protein of interest as well as an element targeting an E3 ubiquitin ligase that are linked together, facilitating target degradation through endogenous ubiquitin-proteasome system (UPS) machinery (Gao et al., 2020). This technology has been hailed for its exquisite specificity and ability to target “undruggable” proteins because it can take advantage of any binding site on the protein and does not require that the binding interferes with catalytic activity (Mullard, 2021). PROTACs hold particular promise for targeting the Gαs-PKA pathway because several components of the pathway are already known to be regulated by the UPS, including GPCRs, G proteins, PKA, PDEs, and AKAPs (Rinaldi et al., 2015). For instance, under physiologic conditions, the UPS contributes to desensitization of GPCRs at the plasma membrane after stimulation (Rinaldi et al., 2015; Skieterska et al., 2017). Furthermore, levels of Gαs and PKA catalytic subunits are also regulated by ubiquitination and degradation in response to pathway stimulation (Naviglio et al., 2004; Nagai et al., 2010; Rinaldi et al., 2019). In contrast to desensitization mechanisms that control receptor and G protein in response to stimulus, the UPS can also provide feedforward regulation of pathway activity, as is the case for regulation of PKA regulatory subunits. Specifically, regulatory subunits associate with Praja2, a RING E3-ubiquitin ligase that also functions as an AKAP. When PKA becomes activated, the catalytic subunit dissociates from the regulatory subunits and phosphorylates Praja2, stimulating the ubiquitination and degradation of the regulatory subunits, thereby potentiating PKA activity (Lignitto et al., 2011). Interestingly, PKA is also capable of regulating the stability of other proteins through the UPS (VerPlank et al., 2019). For instance, cAMP signaling has been shown to downregulate levels of p300 and sirtuin 6 (SIRT 6) through their ubiquitin-dependent proteasomal degradation (Jeong et al., 2013; Kim and Juhnn, 2015).
To date, PDE4 and CBP/p300 represent the only Gαs-PKA pathway components with small-molecule degraders designed against them (Ohoka et al., 2017; Vannam et al., 2021). As PROTACs and targeted degrader technology advances, components of the Gαs-PKA pathway certainly represent promising targets. With the first PROTACs now demonstrating positive clinical responses and favorable safety profiles (Mullard, 2020), there is also tremendous potential to translate these compounds into clinical drugs for use in the Gαs-PKA pathway signalopathies.
D. Targeting Protein Kinase A Indirectly
Given the significant hurdles in targeting PKA directly, another therapeutic strategy is to modulate cAMP levels. The tool compound forskolin, an activator of adenylyl cyclase (Seamon et al., 1981), is commonly used, and adenylyl cyclase inhibitors are less common (Bitterman et al., 2013). PDE-targeting drugs have been much more tractable clinically. Inhibitors targeting cAMP-hydrolyzing PDEs are approved for the treatment of cardiovascular, airway, and inflammatory diseases (PDE3 and PDE4 inhibitors), but to our knowledge have not been used to treat any Gαs-PKA pathway signalopathies. Unfortunately, these drugs are largely limited by side effects (Boswell-Smith et al., 2006). Currently, there are several compounds in development aimed to minimize side effects by targeting specific PDE4 isoforms as well as PDE7 and PDE8 (Martinez and Gil, 2014). For the Gαs-PKA pathway signalopathies, the application of cAMP-specific PDE inhibitors is particularly promising for the treatment of SHH-MB (Rao et al., 2016). Although many of the Gαs-PKA pathway signalopathies exploit activation of the Gαs-PKA pathway, PDE activators may also have therapeutic benefit. Recently, a novel positive allosteric modulator of PDE4 showed promise in models of autosomal dominant polycystic kidney disease, a disease driven by chronically elevated cAMP (Omar et al., 2019). Other mechanisms of targeting PKA indirectly include activation of phosphatases. Results in vivo have suggested that inhibition of PKA via activation of the phosphatase PP2A may be a valuable therapeutic approach in small cell lung cancer (Coles et al., 2020). However, given the relative nonselectively of phosphatases like PP2A, further work is necessary to establish the translational potential of this type of therapeutic approach.
E. Synthetic Lethality Approaches
Finally, given the complexity of the Gαs-PKA pathway, particularly in polygenetic diseases like cancer, finding specific, context-dependent vulnerabilities could be extremely valuable. Synthetic lethality stems from the idea that, in cancer, if you target one gene program either genetically or with a drug, you may shift the reliance of that cancer to another program. By specifically leveraging the vulnerabilities of the cancer cell over normal cells, targeting a secondary program will ultimately prove lethal to the cancer while sparing the normal tissue (Kaelin, 2005). The most notable example of this is the use of poly(ADP-ribose) polymerase inhibitors in breast cancer type 1/2 susceptibility protein (BRCA1/2) mutant cancer that are DNA damage–deficient (Ashworth and Lord, 2018). Synthetic lethalities are largely identified by large chemical or genetic screens (Kaelin, 2005). To this end, recent work by our group has demonstrated that this approach is feasible to identify synthetic lethal vulnerabilities in a Gαq-driven cancer, uveal melanoma, and is now the subject of ongoing clinical trials (Feng et al., 2019; Paradis et al., 2021) (ClinicalTrials.gov, NCT04720417). Furthermore, recent work has also shown that cancer cell growth driven by the DNAJ-PRKACA fusion protein in liver cells can be selectively targeted by HSP70 inhibitors because of a scaffolding interaction unique to the fusion protein (Turnham et al., 2019). As we discussed throughout this review, alteration of the Gαs-PKA pathway is accompanied by unique phenotypes. Ultimately, these unique cell states could be leveraged to exploit single and multimodal synthetic lethal therapies for the treatment of the Gαs-PKA pathway signalopathies.
VI. Conclusion
For the first time, we have defined the Gαs-PKA pathway signalopathies as a family of germline, postzygotic, and somatic diseases driven by dysregulation of the Gαs-PKA pathway. The Gαs-PKA pathway signalopathies cover a diverse range of pathophysiology, and this diversity mirrors the physiologic roles of Gαs-PKA pathway signaling, contributing to fundamental processes from gene transcription and intracellular trafficking to cellular differentiation and organismal development. On a cellular level, owing to isoform specificity and scaffolding interactions, PKA is localized to distinct microdomains. This feature enables PKA to integrate signals from multiple inputs and participate at multiple levels within the same physiologic process. Similarly, PKA is also uniquely poised to mediate the same molecular action across multiple areas of physiology (i.e., regulation of ion channels). Consequently, the Gαs-PKA pathway signalopathies can be characterized by diseases that exploit either pathway activation or inactivation. We find that the major themes of activation include aberrant upstream inputs (GPCR and Gαs activation) as well as disruption of PKA holoenzyme stability (loss of RIα or loss of R:C contacts), with recent evidence also suggesting the role of an altered PKA substrate profile. Conversely, there are many ways to inactivate the pathway, affecting almost every signaling node without consistent hotspot mutations.
From a clinical perspective, these mutational themes are primarily represented in monogenetic, endocrine, bone, and metabolic disorders, largely altering hormone function and developmental events. With this review, we now highlight how the same mutational themes, depending on the tissue and cell context, enable the Gαs-PKA pathway to act as both an oncogenic driver and a tumor suppressor in cancer. Dysregulated signaling through the Gαs-PKA pathway is accompanied by unique phenotypes in cancer, including enhanced mucin production, which makes GNAS, in particular, a promising biomarker. However, as genomics has informed us about the ability of GNAS to cooperate with KRAS in cancer initiation, it has also failed to appreciate the complex connections within the tumor microenvironment. These complex interactions ultimately contribute to the ability of the Gαs-PKA pathway to drive therapeutic resistance.
Naturally, PKA has been the target of significant drug development efforts, but unfortunately, kinase cross-reactivity and complex biology have proven to be substantial hurdles. Conceptually, the tetrameric holoenzyme structure provides a unique landscape for bispecific compounds to flourish. An idea that could even extend to targeting of specific microdomains using AKAP motifs. Promising new approaches are aimed at targeting the pathway with degraders as well as leveraging context specificity to target synthetic lethal interactions. With these new perspectives on the capabilities of the Gαs-PKA pathway and its promise as a therapeutic target, there is a tremendous opportunity to explore new connections among the Gαs-PKA pathway signalopathies, linking seemingly disparate fields through a common signaling mechanism. More importantly, by synthesizing the field, we hope to provide a blueprint for therapeutic advances in treating the human Gαs-PKA pathway signalopathies.
Acknowledgments
The authors would like to acknowledge Dr. Alexandr Kornev at the University of California, San Diego, for his help preparing PKA structure graphics. The authors would also like to thank Dr. Daniela Bertinetti at the University of Kassel and Dr. Bjørn Steen Skålhegg at the University of Oslo for their helpful discussions related to cAMP analogs and mouse models of PKA, respectively. Finally, the authors would like to acknowledge the use of BioRender.com in creating many of our graphics.
Authorship Contributions
Performed data analysis: D.J.R., F.R., N.A.
Wrote or contributed to the writing of the manuscript: D.J.R., F.W.H., S.S.T., J.S.G.
Footnotes
D.J.R. and this work were supported by the National Institutes of Health National Institute of General Medical Sciences Cellular and Molecular Pharmacology Training Grant [T32-GM007752], the National Science Foundation Graduate Research Fellowship Program, and the National Cancer Institute Cancer Systems Biology Consortium [U54-CA209891]. F.R. was supported by the Italian Ministry of University and Research through the Department of excellence “Faculty of Sciences” of Scuola Normale Superiore. The research leading to these results also received funding from the Italian Association for Cancer Research (AIRC) under My First AIRC Grant (MFAG) 2020 - ID. 24317 project – P.I. Raimondi Francesco. F.W.H. was supported by the Federal Ministry of Education and Research (BMBF) [16GW0270 TargetRD] and the Kassel graduate school “clocks.”
↵
 This article has supplemental material available at pharmrev.aspetjournals.org.
This article has supplemental material available at pharmrev.aspetjournals.org.

Abbreviations
- AC
- adenylyl cyclase
- ACTH
- adrenocorticotropic hormone
- AKAP
- A-kinase anchoring protein
- AKAP
- domain-containing protein
- Akt
- protein kinase B
- AMPK
- AMP-activated protein kinase
- APC
- adenomatous polyposis coli
- AT F-1
- activating transcription factor 1
- BRAF
- B-Raf protooncogene
- BRCA1/2
- breast cancer type 1/2 susceptibility protein
- C
- catalytic
- CAR-T
- chimeric antigen receptor T cell
- CBD
- cAMP binding domain
- CBP
- CREB-binding protein
- CDKN2A
- cyclin dependent kinase kinhibitor 2a
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CK1α
- casein kinase 1α
- CM
- cardiac myxoma
- CNG
- cyclic nucleotide–gated
- COX
- cyclooxygenase
- CRC
- colorectal cancer
- CRE
- cAMP response element
- CREB
- cAMP responsive element binding protein
- CREM
- cAMP responsive element modulator
- CRTC
- cAMP-regulated transcriptional coactivator
- CSK
- C-terminal Src kinase
- CXCL12
- C-X-C motif chemokine ligand 12
- CXCR4
- C-X-C motif chemokine receptor 4
- D/D
- dimerization/docking
- DEP
- Dishevelled, Egl-10 and Pleckstrin
- DNAJB1
- DnaJ homolog subfamily B member 1
- DRP1
- dynamin-related protein 1
- EGFR
- epidermal growth factor receptor
- EPAC
- exchange protein directly activated by cAMP
- ERK
- extracellular-signal-related kinase
- FD
- fibrous dysplasia
- FI
- functional interaction
- FL-HCC
- fibrolamellar hepatocellular carcinoma
- GATA-4
- GATA binding protein 4
- GH
- growth hormone
- GLI
- glioma-associated oncogene
- GLP1R
- glucagon-like peptide-1 receptor
- GPCR
- G protein–coupled receptor
- GSK3
- glycogen synthase kinase 3
- GSKIP
- glycogen synthase kinase 3β interacting protein
- HCN
- hyperpolarization-activated, cyclic nucleotide–modulated
- HER2
- human epidermal growth factor receptor 2
- HH
- hedgehog
- HSP70
- heat shock protein 70
- IL-6
- interleukin 6
- IPMN
- intraductal papillary mucinous neoplasm
- iPPSD
- inactivating PTH/parathyroid hormone–related peptide signaling disorder
- IS
- inhibitory sequence
- KCNQ1
- potassium voltage-gated channel subfamily Q member 1
- KRAS
- KRAS proto-oncogene
- LAT S1/2
- large tumor suppressor kinase 1
- LRP6
- LDL related protein 6
- LRRK2
- leucine rich repeat kinase 2
- MAPK
- mitogen-activated protein kinase
- MAS
- McCune-Albright syndrome
- MC2R
- melanocortin receptor
- MSC
- mesenchymal stem cell
- Muc2
- mucin 2
- NMD
- nonsense mediated decay
- NSAID
- nonsteroidal anti-inflammatory drug
- PDAC
- pancreatic adenocarcinoma
- PDE
- phosphodiesterase
- PGE2
- prostaglandin E2
- P I3K
- phosphoinositide 3-kinase
- PKA
- protein kinase A
- PKI
- protein kinase inhibitor
- PMP
- pseudomyxoma peritonei
- POPDC
- Popeye domain containing
- P P1
- protein phosphatase 1
- PP2A
- protein phosphatase 2A
- PROTAC
- proteolysis targeting chimera
- PTC
- papillary thyroid cancers
- PTCH1
- patched homolog 1
- PTH
- parathyroid hormone
- PTHR
- parathyroid hormone receptor
- R
- regulatory
- RA
- Ras association
- RAF1
- Raf-1 proto-oncogene
- RAP1
- Ras-related protein 1
- RAP2
- Ras-related protein 2
- SA
- sinoatrial
- SF-1
- steroidogenic factor 1
- SHH
- sonic hedgehog
- SHH-MB
- sonic hedgehog medulloblastoma
- SIK2
- salt inducible kinase 2
- siRNA
- small interfering RNA
- SIRT6
- sirtuin 6
- SMAD
- SMAD family member 4
- SMO
- Smoothened
- SP HKAP
- SP HK1-interactor and SRC, SRC protooncogene
- SSTR
- somatostatin family of GPCRs
- StAR
- steroidogenic acute regulatory protein
- TAZ
- tafazzin
- T CF
- T-cell factor
- T EAD
- T EA domain transcription factor
- T GF-(b)
- transforming growth factor (b)
- T P53
- p53
- TSHR
- thyroid-stimulating hormone receptor
- UPS
- ubiquitin-proteasome system
- UT R
- untranslated region
- XLAG
- X-linked acrogigantism
- YAP
- yesassociated protein.
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
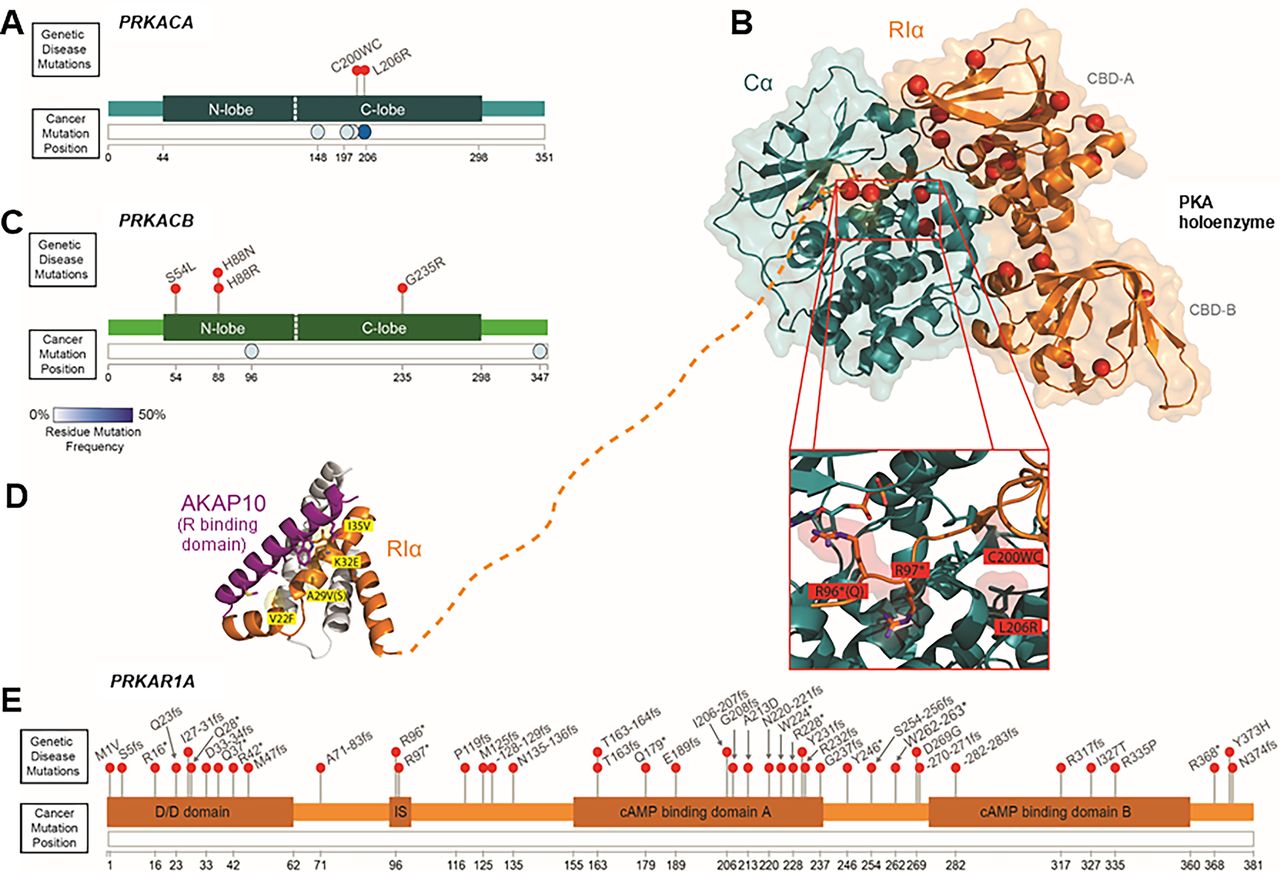{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. Gαs–Protein Kinase A Pathway Basics
- III. Mutational Landscape of the Gαs–Protein Kinase A Pathway Signalopathies
- IV. Human Gαs–Protein Kinase A Pathway Signalopathies
- V. Targeting the Gαs–Protein Kinase A Pathway Signalopathies
- VI. Conclusion
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI