


Abstract
Injury to or disease of the nervous system can invoke chronic and sometimes intractable neuropathic pain. Many parallel, interdependent, and time-dependent processes, including neuroimmune interactions at the peripheral, supraspinal, and spinal levels, contribute to the etiology of this “disease of pain.” Recent work emphasizes the roles of colony-stimulating factor 1, ATP, and brain-derived neurotrophic factor. Excitatory processes are enhanced, and inhibitory processes are attenuated in the spinal dorsal horn and throughout the somatosensory system. This leads to central sensitization and aberrant processing such that tactile and innocuous thermal information is perceived as pain (allodynia). Processes involved in the onset of neuropathic pain differ from those involved in its long-term maintenance. Opioids display limited effectiveness, and less than 35% of patients derive meaningful benefit from other therapeutic approaches. We thus review promising therapeutic targets that have emerged over the last 20 years, including Na+, K+, Ca2+, hyperpolarization-activated cyclic nucleotide–gated channels, transient receptor potential channel type V1 channels, and adenosine A3 receptors. Despite this progress, the gabapentinoids retain their status as first-line treatments, yet their mechanism of action is poorly understood. We outline recent progress in understanding the etiology of neuropathic pain and show how this has provided insights into the cellular actions of pregabalin and gabapentin. Interactions of gabapentinoids with the α2δ-1 subunit of voltage-gated Ca2+ channels produce multiple and neuron type-specific actions in spinal cord and higher centers. We suggest that drugs that affect multiple processes, rather than a single specific target, show the greatest promise for future therapeutic development.
I. Introduction
A. Clinical Presentation and Pharmacotherapy of Neuropathic Pain
Pain is the most common reason for people to seek medical attention (Salter, 2014). Despite this, nociceptive pain is a vital physiologic process that signals actual or potential tissue damage (Iadarola and Caudle, 1997). By so doing, it protects the individual from injury and secures the survival of the species. By contrast, direct injury to neural tissue can produce nerve or neuropathic pain that lasts for months or years after any injury has healed (Costigan et al., 2009b; Moulin et al., 2014). Exact prevalence of neuropathic pain within the global population is unknown, but most studies put estimates at between 1.5% and 8%, equating to between 100 million and 560 million people worldwide (Gilron et al., 2006; Torrance et al., 2006, 2013; Salter, 2014; Bouhassira and Attal, 2016).
Neuropathic pain is defined formally as “pain caused by a lesion or disease of the somatosensory system” (Jensen et al., 2011). It can be caused by traumatic nerve, spinal cord, or brain injury (including stroke) or can be associated with diabetic, human immunodeficiency virus/AIDS, and postherpetic neuropathies, or with multiple sclerosis (Treede et al., 2008) or cancer, and/or the toxic effects of chemotherapeutic agents (Xiao et al., 2007; Schmidt et al., 2010).
Clinical diagnosis of neuropathic pain depends on evaluation of the following criteria: 1) pain with a distinct neuroanatomical distribution, 2) a medical history that suggests a lesion or disease of the nervous system, 3) a confirmatory test to demonstrate neuroanatomical distribution, and 4) a confirmatory test to demonstrate a lesion or disease of the nervous system (Treede et al., 2008). Definite neuropathic pain is categorized as fulfilling all of these criteria, whereas probable neuropathic pain is categorized as fulfilling criteria 1 and 2 without evidence from 3 to 4. There has been an increasing awareness of neuropathic pain in the scientific literature over the past 40 years, suggesting that it is more commonplace than previously thought. The prevalence of the phrase neuropathic pain in books as measured using Google “Ngram” Viewer has increased more than fourfold between 1992 and 2008.
The various manifestations of neuropathic pain are notoriously resistant to the actions of nonsteroidal anti-inflammatory drugs and opioids (Yekkirala et al., 2017). In stark contrast to the profound effectiveness of opioids in nociceptive pain, there is no similar panacea for the treatment of neuropathic pain. Any pain that is opioid resistant is likely neuropathic. A recent meta-analysis of clinical trial data (Finnerup et al., 2015) supported the use of tricyclic antidepressants, serotonin–noradrenaline uptake inhibitors such as duloxetine and the gabapentinoids, pregabalin (PGB), and gabapentin (GBP) as first-line treatments. Tramadol and controlled-release opioids are recommended as second-line treatments and cannabinoids as third-line treatments. Fourth-line treatments include methadone, lamotrigine, lacosamide, tapentadol, and botulinum toxin (Moulin et al., 2014). Other drugs in current clinical use include carbemazepine, lidocaine patch, capsaicin patch, and ziconotide (Table 1).
Drugs currently used in the clinical management of neuropathic pain
Barriers to the development of effective new treatments include the diversity of pathophysiological situations, different pain etiologies, genetic predispositions (Mogil, 2012a; Zorina-Lichtenwalter et al., 2016; Sexton et al., 2017), and different ethnicities (Hastie et al., 2012) encountered in the clinic. This is compounded by epigenetic modifications in humans (Stone and Szyf, 2013) and by the limited ability of rodent models to predict clinical efficacy (Mogil, 2017; Patel et al., 2017; Sexton et al., 2017; Yekkirala et al., 2017). Despite the realization that pain processing in males differs from that in females (Mogil, 2012b; Mifflin and Kerr, 2013; Sorge et al., 2015; Dodds et al., 2016; Melchior et al., 2016; Dickie et al., 2017; Sorge and Totsch, 2017), many preclinical studies have been done on male rodents to avoid possible complications imposed by the estrous cycle. It is also established that neonatal injury can affect manifestation of pain in adults (Beggs et al., 2012a). Lastly, ongoing and/or paroxysmal stimulus-independent pain is a major issue with patients, but this is difficult to assess in animal models (Calvo et al., 2012).
Despite these issues, recent advances in understanding the etiology of neuropathic pain have paved the way for development of new therapeutic approaches. At present, however, none of these rationally developed compounds have attained the status of monoamine uptake inhibitors and gabapentinoids as first-line treatments. Several reviews of the pharmacotherapy of neuropathic pain have appeared in the last few years; some are broadly based (Kremer et al., 2016; Yekkirala et al., 2017), whereas others discuss the use of specific agents such as cannabinoids (Luongo et al., 2017), amitriptyline (Moore et al., 2015), Ca2+ and/or Na+ channel blockers (Waxman and Zamponi, 2014; Zamponi et al., 2015; Patel et al., 2017), and agents that potentiate GABAergic inhibition (Zeilhofer et al., 2012a).
Because gabapentinoids were originally developed as anticonvulsant agents (Offord and Isom, 2015), much is still to be learned about the mechanism of their antiallodynic effect (Bannister et al., 2017b). This means that the drugs that are used most frequently are the least well understood. In the following sections, we will overview recent advances in understanding the etiology of pain and how it has led to rational drug development and to a further, yet incomplete, understanding of gabapentinoid action.
B. Organization of the Spinal Dorsal Horn
Although a full description of the organization of the dorsal horn is beyond the scope of the present review, the following paragraphs provide background to recent advances in the understanding of spinal mechanisms that initiate neuropathic pain. We will show how these insights have led to the identification of therapeutic targets and to improved understanding of gabapentinoid action. For additional information on dorsal horn organization and nociceptive processing, the reader is referred to recent reviews (Braz et al., 2014; Peirs et al., 2015; Cordero-Erausquin et al., 2016; Peirs and Seal, 2016; Abraira et al., 2017; Todd, 2017) and original papers (Lu and Perl, 2003, 2005; Santos et al., 2007; Yasaka et al., 2010).
Tissue damage or perturbation capable of causing pain is detected by free nerve endings in peripheral and visceral structures and relayed by first-order sensory neurons (primary afferents) to second-order sensory neurons in the dorsal horn of the spinal cord. Glutamate and neuropeptides such as substance P and calcitonin gene-related peptide mediate neurotransmission between primary afferent and second-order sensory neurons. Glutamate interacts with Ca2+-permeable and Ca2+-impermeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and with N-methyl D-aspartate (NMDA) receptors, but not with kainate receptors (Tong and MacDermott, 2006). The cell bodies of first-order sensory neurons reside in the dorsal root ganglia (DRG).
Although all primary afferent neurons are widely believed to be glutamatergic (West et al., 2015), it has been suggested that GABA-mediated inhibitory interactions may play a role in nociceptive processing at the level of the DRG (Du et al., 2017). If these findings are verified, they will provoke considerable debate and reassessment of current ideas of sensory and nociceptive processing. It is possible that primary afferent neurons release GABA from their cell bodies in DRG, yet release glutamate from their terminals in the spinal cord.
The gray matter of the spinal cord is divided into the 10 laminae of Rexed (Fig. 1A) (Rexed, 1952). Nociceptive information is received in lamina I, lamina II (substantia gelatinosa; Fig. 1, A and B), and, to a lesser extent, lamina V (Todd, 2010; Zeilhofer et al., 2012b; West et al., 2015; Peirs and Seal, 2016). Although lamina I contains interneurons for modulation and projection neurons for transmission of nociceptive information, lamina II contains mainly interneurons that project to lamina I. In spite of its importance, there is still very much that is unknown about dorsal horn circuitry and how it relates to the pathophysiology of chronic pain (Sandkuhler, 2009; Todd, 2010; Prescott et al., 2014; Peirs and Seal, 2016). Aspects of dorsal horn physiology and organization that are salient to pain etiology, drug development, and gabapentinoid action are described below.

(A) Rexed laminae of the spinal cord. Laminae I and II form the marginal zone and substantia gelatinosa, respectively, and together these make up the superficial dorsal horn. Within the dorsal horn, Aβ tactile and hair afferents end mainly in laminae III–VI with some extension into lamina IIi with distribution dependent on function. Aδ hair-follicle afferents extend across the lamina II/III border. Aδ nociceptors end mainly in lamina I, occasionally branching to laminae V and X. Peptidergic, nociceptive C fiber afferents synapse mainly in lamina I and IIo. Nonpeptidergic C fibers occupy the inner part of lamina II. (B) Acutely isolated spinal cord slice from a 30-day-old rat. The substantia gelatinosa is clearly visible as a translucent band under infrared differential interference contrast optics. (C) Diagram to illustrate the principal synaptic connections in the superficial dorsal horn. From Peirs and Seal (2016). Reprinted with permission from American Association for the Advancement of Science.
1. Lamina I/Marginal Zone
This contains both interneurons and projection neurons and receives inputs from Aδ- and C-peptidergic primary afferents fibers (Torsney, 2011; Cordero-Erausquin et al., 2016) (Fig. 1C). Aδ fibers convey the immediate first pain evoked by tissue injury. Peptidergic afferent fibers, which contain substance P and calcitonin gene-related peptide, are involved in transmitting the second pain that results from injury-induced inflammation. Unlike non-nociceptive C fibers, they do not bind the plant isolectin B4 (IB4) (Stucky and Lewin, 1999; Fang et al., 2006) and instead express transient receptor potential (TRP) channel type V1 (TRPV1) channels, tropomyosin receptor kinase A neutrophin receptors, and voltage-gated sodium channel type 1.9 (Nav1.9) tetrodotoxin-resistant Na+ channels (Fang et al., 2002). Lamina I also receives input from excitatory vertical cells in lamina II. Output neurons to higher centers express neurokinin NK1 receptors (Yu et al., 2005) that are activated by substance P (Fig. 1C).
2. Lamina II/Substantia Gelatinosa
Unlike lamina I, lamina II does not contain neurons that project directly to higher centers (Fig. 1C). It is divided into three sublaminae.
a. Laminar IIo (outer)
Laminar IIo (outer) receives glutamatergic nociceptive input from lamina I as well as polysynaptic input from deeper laminae. It contains glutamatergic stalked or vertical cells (Fig. 1C), which project back to lamina I and typically display a delayed firing pattern in response to a depolarizing current command (Yasaka et al., 2010). Their dendrites penetrate deeper regions of lamina II and lamina III (Todd, 2010; Peirs and Seal, 2016). Another population of excitatory neurons known as central cells expresses short (<400 μm) rostrocaudal projections (Todd, 2010, 2017) (Fig. 1C).
Inhibitory interneurons in lamina IIo typically display a tonic discharge pattern and islet cell morphology and project rostrocaudally for 400 μm or more (Grudt and Perl, 2002; Todd, 2010). They receive some of their excitatory synaptic input from non-nociceptive, rapidly conducting Aβ fibers (Daniele and MacDermott, 2009), and this may provide a cellular basis for the attenuation of pain by activation of innocuous sensory pathways (Zeilhofer et al., 2012b). It is likely that islet cell terminals release both glycine and GABA and that both neurotransmitters may be packaged in the same vesicles (Keller et al., 2001).
b. Lamina IIid (inner dorsal)
Neurons in lamina IIid receive direct input from C nonpeptidergic afferents (Lu and Perl, 2003; Cordero-Erausquin et al., 2016) (Fig. 1C). These afferents generally do not convey nociceptive information and bind the plant isolectin IB4. They express P2X3 receptors for ATP and the RET–tyrosine kinase–GFRα receptor complex for glial cell line–derived neurotrophic factor (Molliver et al., 1997).
c. Lamina IIiv (inner ventral)
Lamina IIiv (inner ventral) contains excitatory interneurons that express calretinin or protein kinase Cγ (Peirs and Seal, 2016). Immunohistochemical staining for this layer of protein kinase Cγ–expressing neurons has been used to define the boundary between lamina II and III (Hughes et al., 2003; Stebbing et al., 2016; Boyle et al., 2017).
3. Lamina III
This receives peripheral input from Aβ/Aδ myelinated or lightly myelinated primary afferents (Fig. 1C). Inputs project to parvalbumin-containing inhibitory GABA/glycinergic neurons and to excitatory interneurons. NR1-expressing projection neurons also reside in lamina III (Todd, 2010).
4. Laminae IV, V, and VI
These laminae contain wide dynamic range (projection) neurons that receive direct Aβ fiber input from primary afferents as well as polysynaptic inputs that convey nociceptive information from superficial laminae (Peirs and Seal, 2016). Their rate of discharge is thus correlated to the type of input, with painful sensation eliciting high frequency discharge and innocuous stimuli generating more modest discharge (Dalal et al., 1999). These laminae are involved in the complex processing of tactile information such as object size, shape, texture as well as vibration, and direction of stimulus movement. This encoded information is relayed to higher centers (Abraira et al., 2017).
II. Mechanisms of Neuropathic Pain and Potential Therapeutic Targets
Neuropathic pain is a maladaptive response of the nervous system to damage. The signs and symptoms include allodynia (pain in response to an innocuous stimulus), hyperalgesia (increased pain response to a noxious stimulus), spontaneous pain (electric-shock–like or shooting pain), and, occasionally, causalgia or incessant burning pain (Costigan et al., 2009b). Some patients experience anesthesia dolorosa or loss of sensation, but persistence of pain from the site of injury (Wall et al., 1979). Neuropathic pain is also seen in complex regional pain syndromes I and II as a result of neurogenic inflammation and/or pathologic cross talk between sensory and sympathetic nerves (Abdulla and Smith, 1997; McLachlan and Hu, 1998; Yen et al., 2006; Calvo et al., 2012; Magnussen et al., 2015). For these reasons, neuropathic pain is often referred to as the “disease of pain.” Neuropathic pain is marked by changes in normal sensory signaling at the level of the periphery, spinal cord, and brain (thalamus and cortex) that occur over the course of weeks or months. These lead to alterations in genomic expression and differences in cortical structures (Costigan et al., 2009b; Sandkuhler, 2009; Alvarado et al., 2013; Tajerian et al., 2013, 2014; Luo et al., 2014). Thus, the pathophysiological changes responsible for the onset of neuropathic pain are distinct from those responsible for its chronic presentation. This distinction between the onset phase and the maintenance phase of neuropathic pain becomes particularly relevant when attempting to relate findings in animal models with the presentation of pain in the clinic. Many animal studies relate to pain onset and do not address the long-term persistence of pain, which is more relevant to the clinical situation.
This review concentrates on neuropathic pain as generated by peripheral nerve injury, as this is the focus of most experimental studies (Kim et al., 1997; Decosterd and Woolf, 2000; Calvo et al., 2012; Stemkowski and Smith, 2013). It has been suggested, however, that injury to the spinal cord per se may engage peripheral mechanisms to generate chronic pain (Yang et al., 2014b). In the clinic, peripheral nerve injury can reflect traumatic injury but is more frequently a consequence of diabetic-, postherpetic-, or human immunodeficiency virus–related neuropathies or complex regional pain syndromes I or II. In all situations, release of inflammatory mediators at the site of injury triggers alterations in the properties of primary afferent neurons (Watkins and Maier, 2002; Scholz and Woolf, 2007). Their excitability is increased, and this leads to the appearance of ectoptic, stimulus-independent activity (Wall and Devor, 1983). This altered activity results from changes in the properties and/or expression of various types of voltage-gated Na+, K+, and Ca2+ channels (Waxman et al., 1999; Abdulla and Smith, 2001b, 2002; Cummins et al., 2001; Stemkowski and Smith, 2012b; Bourinet et al., 2014; Waxman and Zamponi, 2014; Daou et al., 2016). There are also changes in the function of Na+-K+ ATPases (Venteo et al., 2016), intracellular Ca2+ handling (Hogan et al., 2014; D’Arco et al., 2015; Yilmaz and Gold, 2016; Yilmaz et al., 2017), TRP channels (Basso and Altier, 2017), and hyperpolarization-activated cyclic nucleotide–gated (HCN) channels (Hogan and Poroli, 2008; Emery et al., 2011; Noh et al., 2014; Young et al., 2014).
It has been known for 40 years that injury to sensory axons can bring about changes in the organization of the spinal cord sensory map (Devor and Wall, 1978), and that sensory disturbances relating to neuropathic pain can be attributed to changes in excitability of the injured neuron and abnormal ongoing and evoked discharge from ectopic neural pacemaker sites (Govrin-Lippmann and Devor, 1978; Wall and Devor, 1983). These ideas were confirmed by showing that ectopic discharge from the neuroma induced by nerve injury or from dorsal root ganglia could be silenced by lidocaine (Devor et al., 1992). The idea of central sensitization was first put forward by Clifford Woolf in the early 1980s to describe the cascade of events that are attributed to the maladaptive changes in plasticity of sensory processing that occur in neuropathic pain (Woolf, 1983; Woolf and Thompson, 1991; Woolf and Mannion, 1999; Latremoliere and Woolf, 2009).
A. Excitation–Inhibition Balance in Neuropathic Pain
Following peripheral nerve injury or neuropathy, central sensitization within the spinal cord results from changes in synaptic transmission; excitatory synaptic processes are enhanced, and inhibitory processes are attenuated (Kuner, 2010; Prescott et al., 2014; West et al., 2015). The intrinsic properties of dorsal horn neurons such as rheobase, threshold, excitability, and/or input resistance are little changed by peripheral nerve injury (Balasubramanyan et al., 2006). This finding is consistent with the likelihood that ongoing ectopic activity in peripheral nerves is required to drive and maintain central sensitization (Devor, 2006; Pitcher and Henry, 2008; Gold and Gebhart, 2010; Vaso et al., 2014; Daou et al., 2016; Sexton et al., 2017).
Numerous parallel and interdependent pathophysiological processes in the periphery, spinal cord, and higher centers contribute to the onset of neuropathic pain. This very much complicates the search for effective therapeutic targets. Although gabapentinoids may only bind to one specific target, the α2δ-1 subunit of voltage-gated Ca2+ channels (Gee et al., 1996; Field et al., 2006; Offord and Isom, 2015), this interaction has multiple consequences for synaptic transmission, neuron-selective actions, and network excitability at the spinal level (Biggs et al., 2014; Alles and Smith, 2016; Alles et al., 2017) and within higher brain centers (Suzuki et al., 2005; Eroglu et al., 2009; Suto et al., 2014; Crosby et al., 2015; Patel and Dickenson, 2016a; Bannister et al., 2017b); this multiplicity of effect may explain their therapeutic effectiveness. The appreciation of this concept has led to the evidence-based development of drugs that will affect multiple targets (Gadotti et al., 2015; Zamponi et al., 2015).
Details of pathophysiological processes that contribute to the development of neuropathic pain are outlined in the following sections. In addition, and where relevant, we identify experimental drugs that target these changes. Some of these substances have not yet been tested in the clinic, whereas others have yielded disappointing results in phase I or phase II trials. Table 2 lists substances that remain as viable entities for use as future therapeutic development.
Examples of drugs in development that remain viable candidates for future therapeutic interventions
B. The Colony-Stimulating Factor 1, Microglia, Purinergic Ionotropic 2X4, ATP, Brain-Derived Neurotrophic Factor, GABA-Chloride Cascade
1. Brain-Derived Neurotrophic Factor and a Shift in Neuronal Chloride Gradient
Coull et al. (2003) showed that a decrease in transmembrane chloride gradient occurred in rat dorsal horn lamina I neurons following peripheral nerve injury as a result of a reduction in expression of the potassium-chloride exporter, potassium-chloride exporter 2 (KCC2). The resulting accumulation of intracellular Cl− can cause normally inhibitory GABAergic, anionic, outward synaptic currents to become inward excitatory currents (Coull et al., 2003; Prescott et al., 2006).
It was corroborated that this change was due to a reduction in KCC2 expression as a knockdown of spinal KCC2 in noninjured rats reduced pain thresholds and resulted in neuropathic pain behaviors (Coull et al., 2003, 2005). Hence, an injury that causes neuropathic pain results in a marked change in dorsal horn function such that the net excitability of nociceptive lamina I neurons is increased. It was also shown more recently that alterations in KCC2 expression in deep dorsal horn neurons are confined to nociceptive neurons that project via the spinothalamic tract, whereas wide dynamic range neurons that are activated by a variety of sensory modalities (Dalal et al., 1999) were unaffected (Lavertu et al., 2014).
The Salter and de Koninck (Coull et al., 2005) groups have also shown that brain-derived neurotrophic factor (BDNF) release from activated spinal microglia is responsible for the aforementioned depolarizing shift in anion gradient observed in lamina I neurons in neuropathic pain. In this study, administration of activated microglia or application of BDNF produced the shift in anion gradient seen after nerve injury. Also, blocking signaling between BDNF and its cognate receptor [tropomyosin receptor kinase B (TrkB)] reversed pain behaviors (allodynia) and the shift in anion gradient. Lastly, allodynia and the shift in anion gradient were shown to be prevented by blocking release of BDNF from microglia by treatment with interfering RNA against BDNF.
It was shown recently that BDNF potentiates excitatory NMDA receptor-mediated currents through activation of TrkB and phosphorylation of the GluN2B subunit by the Src-family kinase Fyn (Hildebrand et al., 2016). Interestingly, this potentiation appears to require the coincident BDNF-mediated Cl− disinhibition. The exact molecular mechanism of this interaction remains to be elucidated as it does not appear to reflect increased NMDA receptor availability as a result of GABA-induced depolarization (Hildebrand et al., 2016).
2. Role of ATP
It is also known that, following peripheral nerve injury, there is an increase in the expression of the ATP-gated ionotropic purinoceptor, 2X4 (P2X4R), and in the metabotropic purinoceptor, 2Y12 (P2Y12R), in microglia. This increase in expression parallels the increase in pain hypersensitivity (Tozaki-Saitoh et al., 2008; Trang et al., 2011, 2012). The mechanism by which P2X4R upregulation leads to BDNF release from microglia is attributed to influx of Ca2+ through P2X4Rs, activation of p38 mitogen-activated kinase, and a consequent increase in synthesis and SNARE-mediated exocytosis of BDNF (Trang et al., 2009). Interestingly, this mechanism is specific to male mice as microglia are not required for mechanical sensitivity to pain in female mice in which adaptive immune cells are involved (Sorge et al., 2015; Sorge and Totsch, 2017). Sex differences in pain processing are currently a very active area of investigation with obvious clinical implications (Mogil, 2012b; Mifflin and Kerr, 2013; Dodds et al., 2016; Mifflin et al., 2017).
3. Signaling between Injured Peripheral Nerve and Spinal Microglia
The transformation of resting microglia into a phenotype that expresses P2X4R has been ascribed to the release of chemical mediators from injured primary afferent fibers. These include the chemokines, fractalkine (Milligan et al., 2004; Grace et al., 2014), chemokine (C-C motif) ligand (CCL)2, and CCL21 (Biber et al., 2011; Toyomitsu et al., 2012). Recent work, however, discounts these mediators and instead favors colony-stimulating factor 1 (CSF-1) as the primary effector of microglial transformation in neuropathic pain (Guan et al., 2016; Okubo et al., 2016). It has been suggested that the injury-induced release of inflammatory mediators such as interleukin (IL)-1β from satellite glial cells in DRG promotes induction of Csf1 in the cell bodies of primary afferent neurons (Lim et al., 2017). As will be discussed below, fractalkine and CCL2 are involved in other aspects of the etiology of neuropathic pain. CSF-1 is released from primary afferents and acts on microglia to induce various genes, including that for the ATP receptor, P2X4R, as well as microglial proliferation and self-renewal (Guan et al., 2016). The membrane adaptor protein DAP12 is required for nerve injury–induced upregulation of P2X4R, but not for microglial proliferation. The relationship between CSF-1, P2X4R, ATP, and BDNF is illustrated schematically in Fig. 2A. It remains to be determined whether the release of CSF-1 from primary afferents is vesicular and depends on neuronal activity. This is important to know as it may help to explain how injury-induced ectopic activity in primary afferents leads to central sensitization.

(A) Diagram to illustrate some of the processes leading to central sensitization. Classic inflammatory mediators released at the site of injury alter the properties of primary afferent fibers such that they become hyperexcitable and/or spontaneously active. CSF-1 released from primary afferents changes the phenotype of microglia that they start to express new receptors, including purinergic ionotropic 2X4. ATP released from dorsal horn neurons interacts with P2X4Rs on microglia to promote release of BDNF, which interacts with neurons to increase dorsal horn excitability. (B and C) Diagrams to show how peripheral nerve injury increases ATP release from dorsal horn neurons by upregulating the vesicular nucleotide transporter VNUT.
BDNF is also released from sensory neurons in an activity-dependent fashion (Balkowiec and Katz, 2000), but it remains to be determined whether this process contributes to the onset of neuropathic pain or whether it is associated with more physiologic and reversible central sensitization as is seen in some types of inflammatory pain.
4. Source of ATP in Central Sensitization
After peripheral nerve injury, increased spinal levels of ATP reflect release from the dorsal horn neurons themselves and not from primary afferents, astrocytes, or microglia. This increased release is brought about by upregulation of the vesicular nucleoside transporter (VNUT; Fig. 2, B and C) (Masuda et al., 2016). Neurotransmitters are transported from the cytoplasm into synaptic vesicles via specific transporter proteins: vesicular GABA transporter in inhibitory neurons and various isoforms of the vesicular glutamate transporter in excitatory neurons. Synaptic vesicles in most, if not all, neurons contain ATP that is released along with the primary neurotransmitter. VNUT serves to transport cytosolic ATP into synaptic vesicles. When it is upregulated after nerve injury, vesicles contain a high level of ATP, which is released and stimulates P2X4R on microglia. As mentioned above, this promotes release of BDNF (Trang et al., 2009). The mechanism whereby peripheral nerve injury produces a trans-synaptic upregulation of VNUT in dorsal horn neurons remains to be elucidated.
C. Increased Excitatory Drive to Excitatory Neurons and Decreased Excitatory Drive to Inhibitory Neurons
Chronic constriction injury (CCI) or axotomy of the sciatic nerve produces an electrophysiological footprint of neuropathic pain in the substantia gelatinosa (Biggs et al., 2010; Smith, 2014) (Fig. 3A). In this footprint, neurons have been classified according to their firing pattern as tonic, delay, irregular, phasic, or transient (Balasubramanyan et al., 2006), and the effects of CCI noted as increases or decreases in the amplitude and frequency of spontaneous excitatory postsynaptic current (sEPSC) or miniature excitatory postsynaptic current (mEPSC). Given the established relationship between neuronal firing pattern and its neurotransmitter phenotype (Yasaka et al., 2010; Punnakkal et al., 2014), the footprint reflects an increased excitatory drive to putative excitatory delay-firing and transient-firing neurons (Fig. 4A) and a decreased excitatory drive to putative inhibitory tonic-firing neurons (Fig. 4B) (Bailey and Ribeiro-da-Silva, 2006; Balasubramanyan et al., 2006; Chen et al., 2009; Lu et al., 2009). Both of these changes would be expected to produce an overall increase in dorsal horn excitability. Decreased excitatory drive to inhibitory neurons involves both pre- and postsynaptic changes, including a decreased contribution of Ca2+-permeable AMPA receptor to sEPSCs (Chen et al., 2016). Increased excitatory drive to excitatory neurons also involves presynaptic effects and a possible increased contribution of Ca2+-permeable AMPA receptor to postsynaptic events (Chen et al., 2013).

(A) Diagram to illustrate the electrophysiological footprint of neuropathic pain seen in substantia gelatinosa following peripheral nerve injury (Biggs et al., 2010). Five cell types characterized by their firing pattern, tonic, delay, irregular, phasic, or transient (Balasubramanyan et al., 2006), are listed at the top of the scheme, and the frequencies and amplitudes of sEPSCs and mEPSCs are listed at the right of the scheme. Upward arrows in green-colored squares indicate a nerve injury–induced increase in each type of synaptic event in each neuron type. Downward arrows in red-colored squares indicate a nerve injury–induced decrease. nd = not determined; horizontal double-headed arrow indicates no change. (B) Similar presentation to show cell-type–specific effects of gabapentin in substantia gelatinosa of nerve-injured animals. (C) Superimposition of patterns from (A) and (B). Yellow shading represents phenomena that were affected in one way by nerve injury and the opposite direction by gabapentin.
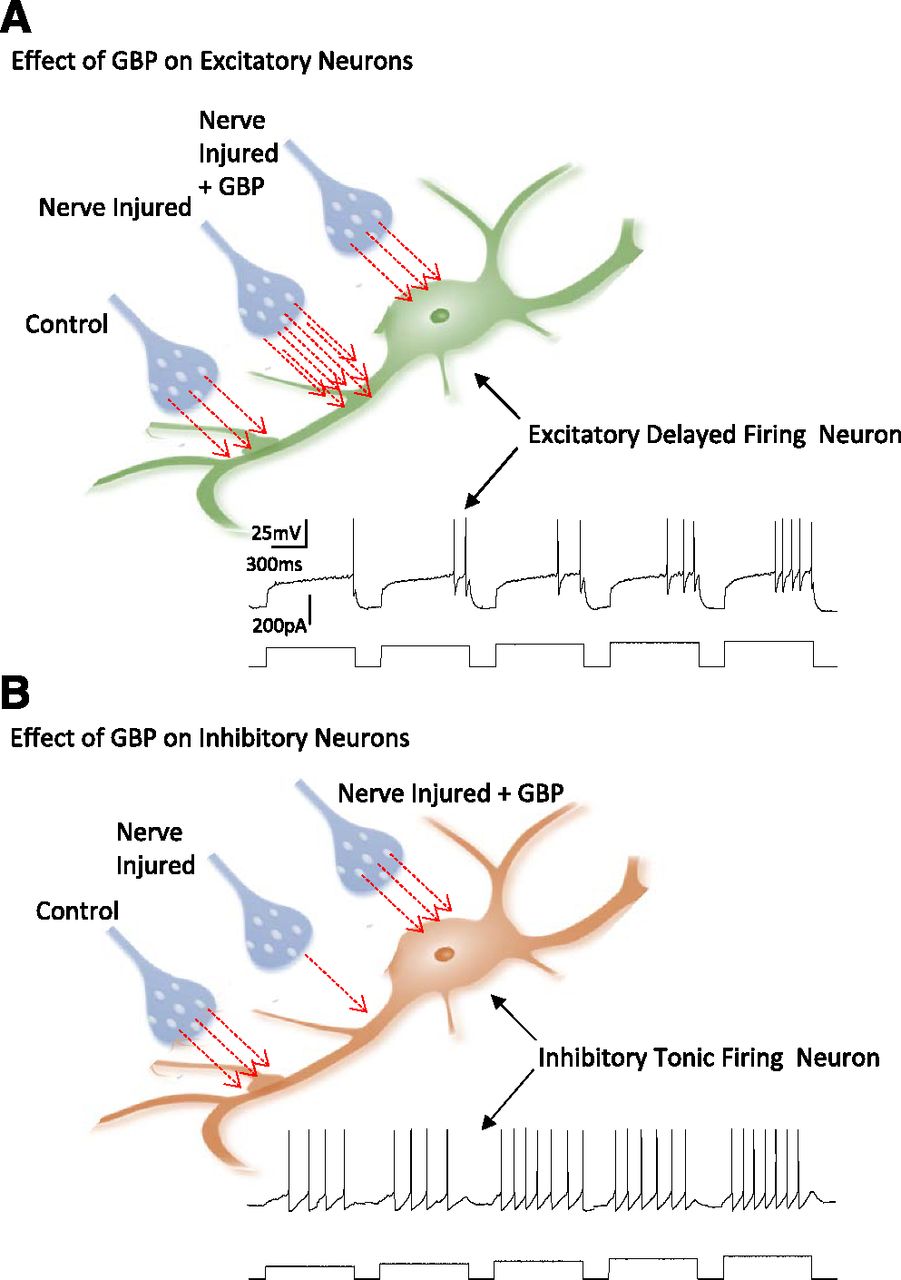
(A) Diagram to illustrate increase in excitatory synaptic transmission onto excitatory dorsal horn neurons following CCI and reversal of these changes following GBP treatment. The inset shows the characteristic delay-firing pattern of excitatory neurons. Red arrows illustrate relative quantity of glutamate released onto control excitatory neurons, onto neurons in animals subject to CCI, and onto CCI neurons following treatment with GBP. The inset shows the characteristic delay-firing pattern of excitatory neurons. (B) Diagram to illustrate decrease in excitatory synaptic transmission onto inhibitory dorsal horn neurons following CCI and restoration of glutamate release following GBP treatment. The inset shows the characteristic tonic-firing pattern of inhibitory neurons. Red arrows illustrate relative quantity of glutamate released onto control-inhibitory neurons, onto neurons in animals subject to CCI, and onto CCI neurons following treatment with GBP.
1. Role of Brain-Derived Neurotrophic Factor
Treatment of substantia gelatinosa neurons in organotypic culture with BDNF for 5 to 6 days produces an electrophysiological footprint (Lu et al., 2006; Biggs et al., 2012) that is very similar to that produced by CCI in vivo (Biggs et al., 2010; Smith, 2014). Both BDNF and CCI increase sEPSC and mEPSC amplitude and frequency in putative excitatory delay-firing neurons, and a more detailed analysis shows that both manipulations also unveil a new population of large mEPSC events, which are absent in neurons from sham-operated rats and control organotypic cultures (Biggs et al., 2010; Smith, 2014). CCI-induced attenuation of excitatory synaptic drive to inhibitory neurons is associated with functional loss of Ca2+-permeable AMPA receptors in the postsynaptic membrane (Chen et al., 2016). BDNF has also been reported to activate presynaptic NMDA receptors via a TrkB-dependent mechanism (Chen et al., 2014). Somewhat counterintuitively, this has been proposed to decrease neurotransmitter release (Bardoni et al., 2004), which may contribute to attenuation of synaptic events in inhibitory neurons of the substantia gelatinosa.
As would be expected, spinal cord slices obtained from animals subject to CCI and those in organotypic cultures exposed to BDNF for 5 to 6 days displayed an overall increase in excitability. This was observed as an increase in the amplitude of Ca2+ responses evoked by 35 mM K+ (Lu et al., 2007; Alles et al., 2017). In addition, activated microglia-conditioned medium increased overall organotypic slice excitability, and preincubation of slices with a TrkBd5 (recombinant receptor, which sequesters BDNF) inhibited this effect (Lu et al., 2009). This provides further evidence for the role of microglia-derived BDNF as a major driver of central sensitization (Coull et al., 2005; Biggs et al., 2010; Trang et al., 2011; Smith, 2014).
2. Brain-Derived Neurotrophic Factor as a Drug Target?
Given the seminal role of BDNF in the onset of neuropathic pain and findings that suggest its involvement in pain maintenance (Wang et al., 2009), one may posit that BDNF antagonists would be an effective treatment (Sah et al., 2003). Because BDNF is involved in so many other essential processes, such as neuronal growth, survival, and synaptogenesis (Reichardt, 2006), and in processes related to memory (Malcangio and Lessmann, 2003; Montalbano et al., 2013), systemic blockade of BDNF function in pain patients would doubtless cause undesirable side effects, including cognitive impairment and the onset of depression (Autry and Monteggia, 2012; Smith, 2014).
3. Use of Botulinum Toxin
Botulinum toxin represents another means by which to control neurotransmitter release. Although it is currently a fourth-line treatment (Finnerup et al., 2015), there is increasing interest in its use in the management of neuropathic pain (Oh and Chung, 2015; Baron and Binder, 2016; Park and Park, 2017; Yaksh et al., 2017). Botulinum A cleaves the synaptic protein SNAP-25 and thereby decreases the Ca2+ responsiveness of the secretory machinery and attenuation of neurotransmitter release (Trudeau et al., 1998). Because the toxin blocks the release of both inhibitory and excitatory transmitters, its mechanism of action has been debated (Baron and Binder, 2016). Actions may include attenuation of neurogenic inflammation in the periphery. The suggestion that the substantia gelatinosa is “primarily an excitatory network” (Santos et al., 2007) in which only 25% of the neurons are inhibitory (Boyle et al., 2017) may account for the actions of botulinum toxin at the spinal level.
D. Altered Sensory Processing and Generation of Allodynia
Following nerve injury, attenuation of GABAergic and/or glycinergic transmission leads to aberrant processing of sensory information within the dorsal horn (Baba et al., 2003; Torsney and MacDermott, 2006; Prescott et al., 2014). In addition to changes in chloride gradient (Coull et al., 2003, 2005) and decreased excitatory drive to inhibitory neurons (Balasubramanyan et al., 2006; Lu et al., 2009; Leitner et al., 2013), this may involve a loss of GABAergic terminals (Lorenzo et al., 2014) or reduced glycine release (Imlach et al., 2016). Tactile and innocuous information is carried by Aβ fibers, which synapse primarily onto dorsal horn neurons in laminae III and IV (Abraira et al., 2017), whereas noxious information carried by C and Aδ fibers is transmitted to the superficial laminae I and II (Peirs and Seal, 2016) (Fig. 1C). GABAergic and glycinergic inhibition normally separates these two modalities by suppressing the activity of pre-existing excitatory synaptic circuits (Lu et al., 2013). However, when this inhibition is compromised, tactile and innocuous information traveling to lamina III and IV gains access to the pain-processing centers in lamina I and II. Thus, touch is processed as pain, thereby providing a rational explanation for the generation of allodynia. This idea is supported by the observation that impediment of inhibitory transmission in the spinal cord with bicuculline and/or strychnine produces allodynia and hyperalgesia in uninjured animals (Yaksh, 1989; Sherman and Loomis, 1994; Loomis et al., 2001).
Anomalous distribution of tactile information is reinforced by increased excitatory transmission between deep and superficial laminae. This involves transient expression of the vesicular glutamate transporter vesicular glutamate transporter 3 by a discrete population of neurons that receive direct low-threshold primary afferent input (Fig. 1C). The circuit extends to nociceptive lamina I projection neurons and includes lamina II calretinin neurons (Peirs et al., 2015).
In addition to being a substrate for allodynia, injury-induced ongoing aberrant activity in Aβ fibers (Devor, 2009), including large-diameter cutaneous afferents (Cummins et al., 2000), would be expected to drive persistent activation of the superficial laminae that may contribute to chronic, stimulus-independent pain.
1. Mechanosensitive Ion Channels as Therapeutic Targets
Because light touch instigates allodynia, there is interest in pharmacological manipulation of normally innocuous modalities as a means to alleviate aspects of human neuropathic pain (Eijkelkamp et al., 2013). Transduction of light touch by mechanoreceptors is effected by the mechanosensitive ion channel, Piezo 2 (Ranade et al., 2014), which is modulated by stomatin-like protein 3 (STOML-3) (Wetzel et al., 2007, 2017). Interference with STOML-3 function has been proposed as a novel therapeutic approach for neuropathic pain. Recently, a small molecule blocker of STOML-3 oligomerization was identified. This substance termed OB-1 by the authors was shown to attenuate mechanoreceptor sensitivity and to increase paw withdrawal threshold in mice subject to CCI (Wetzel et al., 2017).
2. Potentiation of GABA or Glycine as Therapeutic Targets
Some of the anticonvulsant agents used in pain management are thought to act, at least in part, by augmentation of GABA function (Table 1). Given the established role of GABAergic and glycinergic dysfunction in the etiology of allodynia (Yaksh, 1989; Sherman and Loomis, 1994; Loomis et al., 2001), restoration of this function represents an attractive therapeutic approach (Zeilhofer et al., 2012a, 2015). Augmentation of glycinergic function may be achieved by pharmacological manipulation of the glial and neuronal glycine transporters GlyT1 and GlyT2 or direct stimulation of glycine receptors (Zeilhofer et al., 2017).
The collapse of the chloride gradient in response to CCI-induced release of BDNF from spinal microglia has already been alluded to (Coull et al., 2003, 2005; Beggs et al., 2004; Lavertu et al., 2014). Because this reflects downregulation of the K+/Cl− cotransporter KCC2, there is considerable interest in developing pharmacological agents that will restore KCC2 function (Gagnon et al., 2013). These authors identified an arylmethylidine family of compounds and found that the analog CLP257 was KCC2 selective and that it restored impaired Cl− transport and Cl− gradients in neurons with diminished KCC2 activity. It also alleviated allodynia in a rat model of neuropathic pain with an oral efficacy equivalent to PGB, thus validating KCC2 as a therapeutic target for neuropathic pain and perhaps for other central nervous system (CNS) disorders (Gagnon et al., 2013).
It is well established that GABAA receptor function is augmented by benzodiazepines, but their use as antiallodynic agents in the clinic is limited by their sedative effects (Besson et al., 2013). It is known, however, that different subtypes of GABAA receptor localize with different cell types and terminals in the spinal dorsal horn. In particular, GABAA receptors containing the α2 subunit associate with C fibers (Paul et al., 2012), and this has led to the notion that benzodiazepines with high affinity for α2 may be effective in treating neuropathic pain (Besson et al., 2013; Paul et al., 2014; Ralvenius et al., 2016). One such agent, clobazam (Besson et al., 2013), which acts through an active metabolite (Ralvenius et al., 2016), is effective in relieving allodynia in the CCI model (Besson et al., 2013). Moreover, preliminary clinical trials with this agent are showing encouraging results (Besson et al., 2015).
E. Other Spinal Mechanisms of Central Sensitization
1. Long-Term Potentiation and Memory Processes
Transmission at CNS synapses is characteristically plastic. Repeated activation of presynaptic fibers can lead to short-term potentiation (lasting for less than half an hour), early-phase long-term potentiation (LTP; lasting for up to 3 hours), and late-phase LTP, which depends on protein synthesis and can last indefinitely (Luo et al., 2014). Given that peripheral nerve injury increases activity of both primary afferent fibers and dorsal horn neurons, these processes are doubtlessly engaged and serve to reinforce central sensitization (Ji et al., 2003; Sandkuhler, 2007; Fenselau et al., 2011; Ruscheweyh et al., 2011; Luo et al., 2014). Mechanistically, spinal LTP involves presynaptic mechanisms (Luo et al., 2014) as well as opening of T-type [voltagae-gated Ca2+ channels type 3 (Cav3)] calcium channels and activation of NMDA receptors (Ikeda et al., 2003, 2006; Zhuo, 2016b). These receptors have long been implicated in the etiology of neuropathic pain (Woolf and Thompson, 1991; Kerr et al., 1999; Latremoliere and Woolf, 2009; Salter and Pitcher, 2012; Hildebrand et al., 2016), and this may account for the effectiveness of the NMDA receptor blocker, ketamine, in the management of chronic pain (Hewitt, 2000; Niesters et al., 2014; Maher et al., 2017) and the observation that T-type calcium channel blockers mibefradil and ethosuximide can reverse neuropathic pain in an animal model (Dogrul et al., 2003). Physiological blockade of NMDA channels with Mg2+ has been shown to be effective in reducing signs of pain in animal models (Brill et al., 2002; Rondon et al., 2010), and there is some evidence that it may play a role in the treatment of human neuropathic pain (Pickering et al., 2011).
Central sensitization in the dorsal horn shares many mechanistic parallels with memory formation (Sandkuhler and Lee, 2013), including reconsolidation, a protein synthesis–dependent process by which memories become labile after reactivation and susceptible to erasure (Nader et al., 2000; Debiec et al., 2002). Thus, following lasting hyperalgesia induced by injection of capsaicin or complete Freund’s adjuvant into mice hindpaw, a second capsaicin injection administered within 2 hours of the first suppressed hyperalgesia by a mechanism independent of protein synthesis (Bonin and de Koninck, 2014). It remains to be determined whether this phenomenon can be exploited therapeutically in management of neuropathic pain.
2. Role of Astrocytic Glutamate Transporter (Excitatory Amino Acid Transporter 2)
Studies using various animal models have also reported that downregulation of the astrocytic excitatory amino acid transporter, excitatory amino acid transporter 2 (EAAT2), correlates with development of signs of neuropathic pain (Cata et al., 2006; Weng et al., 2006). Both partial sciatic nerve ligation and CCI significantly reduce expression of EAAT2 in rat dorsal horn (Sung et al., 2003), whereas spinal EAAT2 gene transfer via recombinant adenovirus significantly decreases mechanical hyperalgesia and allodynia (Maeda et al., 2008). Decreased removal of extracellular glutamate following EAAT2 downregulation may lead to activation of extrasynaptic NMDA receptors (Nie and Weng, 2009). The signal responsible for EAAT2 downregulation remains to be established. It is unlikely to be BDNF as this neurotrophin upregulates EAAT2 in other brain regions (Rodriguez-Kern et al., 2003).
Although riluzole, a positive regulator of glutamate transporters, significantly reduces thermal hyperalgesia and mechanical allodynia in CCI (Sung et al., 2003), this effect may reflect other actions of the drug such as interaction with Na+ channels (Hebert et al., 1994; Munro et al., 2007).
F. The Pain Matrix
Neuronal plasticity during neuropathic pain is by no means limited to the dorsal horn and periphery (West et al., 2015; Zhuo, 2016a,b; Bannister and Dickenson, 2017). Multiple changes are observed in the pain matrix that includes the medial prefrontal cortex, nucleus accumbens, anterior cingulate cortex, insula, amygdala, periaqueductal gray, locus coeruleus, and rostroventral medulla (Schweinhardt and Bushnell, 2010; von Hehn et al., 2012; Zhang et al., 2015c; Peirs and Seal, 2016; Tan et al., 2017; Taylor et al., 2017). For example, synapses in the anterior cingulate cortex are altered after peripheral nerve injury, and LTP of glutamatergic transmission appears in the insula (Zhuo, 2016a). There is also an increase in feed-forward GABAergic inhibition in the medial prefrontal cortex (Zhang et al., 2015c) that is thought to contribute to the emotional aspects of neuropathic pain.
Brain regions that comprise the pain matrix were identified by functional neuroimaging (Apkarian et al., 2011). Interestingly, areas activated by acute pain do not correspond exactly to those activated in chronic pain (Apkarian et al., 2011; von Hehn et al., 2012). The “matrix is reloaded!”
G. Changes in Descending Control Mechanisms
In addition to altering pain perception, changes in the pain matrix can also alter control of spinal dorsal horn neurons as a result of alterations in descending control mechanisms (Porreca et al., 2002; Ossipov et al., 2010). The well-established role of serotonergic and noradrenergic pathways in controlling spinal nociceptive processing (Ossipov et al., 2010) helps to explain the effectiveness of tricyclic antidepressants and related monoamine uptake inhibitors in pain management (Dworkin et al., 2007; Kuner, 2010; Finnerup et al., 2015; Bannister and Dickenson, 2017). Descending inhibition of nociceptive processing is mediated via α2 adrenoceptors and 5HT7 receptors, whereas serotonergic activation of metabotropic 5HT2 receptors and ionotropic 5HT3 receptors facilitates transmission (Millan, 2002; Bannister et al., 2015, 2017a; Bannister and Dickenson, 2017).
Although intrathecal administration of the α2 receptor antagonist, yohimbine, hastens the onset of cold allodynia and heat sensitivity following tibial nerve transduction, the drug loses its sensitizing effect once these phenomena have fully developed. These findings have been interpreted as a loss of descending noradrenergic control once allodynia and hyperalgesia have developed (Hughes et al., 2013). Similarly, silencing of locus coeruleus neurons by adenoviral infection of K+ channels attenuates descending inhibition, but this effect disappears once neuropathy is introduced (Howorth et al., 2009).
Experiments with the antagonist, ondansetron, implicate upregulation of 5HT3 receptor function in the generation of neuropathic pain (Bannister et al., 2015). The resulting facilitation of nociceptive processing along with attenuation of the inhibitory actions of noradrenaline thus conspires to promote allodynia and hyperalgesia.
For additional information on mechanisms of central sensitization and allodynia, consult publications by Jürgen Sandkühler (Sandkuhler, 2009; Ruscheweyh et al., 2011; Sandkuhler and Gruber-Schoffnegger, 2012), Clifford Woolf (Ji and Woolf, 2001; Costigan et al., 2009b; Latremoliere and Woolf, 2009), and Allan Basbaum (Basbaum et al., 2009), as well as that by Peirs and Seal (2016). Several additional reviews address supraspinal changes associated with neuropathic pain (Zhuo, 2008, 2016b; Luo et al., 2014) and alterations in descending control mechanisms (Bannister and Dickenson, 2017).
H. Role of Mesolimbic Reward Circuitry
An emerging area in the study of neuropathic pain mechanisms relates to the ability of peripheral nerve injury to impair dopamine release in the reward circuitry associated with mesolimbic system (Taylor et al., 2015, 2017). This may relate to the changes in affect (anxiety, depression) experienced by neuropathic pain patients (Mitsi and Zachariou, 2016). It was also shown recently that peripheral nerve injury selectively increases excitability of the nucleus accumbens indirect pathway spiny projection neurons and alters their synaptic connectivity. In addition, tactile allodynia was reversed by inhibiting and exacerbated by exciting these neurons. This suggests that neurons in the nucleus accumbens not only participate in the central representation of pain, but that they may gate activity in ascending pathways associated with expression of neuropathic pain in higher centers (Ren et al., 2016).
I. Role of Ectopic Activity in Primary Afferent Fibers
The initial inflammatory event at the site of nerve injury is a major trigger for the sequelae that lead to central sensitization (Watkins and Maier, 2002; Scholz and Woolf, 2007; White et al., 2007; Basbaum et al., 2009; Stemkowski and Smith, 2012b; Grace et al., 2014; Schuh et al., 2014; Ko et al., 2016) (Fig. 2A). As mentioned above, processes involved in the onset of neuropathic pain are very different from those involved in its long-term persistence and maintenance (Ji and Woolf, 2001). Classic mediators of inflammation such as IL-1β (Binshtok et al., 2008; Stemkowski and Smith, 2012a,b; Stemkowski et al., 2015), IL-6 (Ko et al., 2016), prostaglandins (Ma and Eisenach, 2002), and tumor necrosis factor (TNF) (Leung and Cahill, 2010) interact with or modulate ion channels on primary afferent neurons to instigate ectopic activity that contributes to spontaneous, stimulus-independent pain (Pitcher and Henry, 2008; Devor, 2009; Devor et al., 2014). Indeed, it has been shown that, following a peripheral nerve injury, there is altered synaptic activity of substantia gelatinosa neurons with little effect on their intrinsic electrophysiological properties, suggesting that changes in the CNS are driven by activity of peripheral neurons (Balasubramanyan et al., 2006; Devor, 2006). In a simple yet elegant series of experiments, it was shown by application of lidocaine to block nerve conduction and subsequent monitoring of spontaneous baseline discharge that the hyperexcited state of dorsal horn neurons is maintained by ongoing, afferent discharges from the peripheral nerve distal to and proximal to the site of injury (Pitcher and Henry, 2008). Increased activity in afferent nerves leads to upregulation of the α2δ-1 subunit of voltage-gated Ca2+ channels (Boroujerdi et al., 2008). This in turn alters Ca2+ channel function in primary afferent terminals and increases transmitter release (see Gabapentinoids and the α2δ Subunits of Voltage-Gated Ca2+ Channels).
Increased discharge in TRPV1-expressing neurons may contribute to central sensitization more indirectly by increasing the permeability of the blood brain and blood spinal cord barrier (Beggs et al., 2010). This permits access of proinflammatory cytokines and immunocompetent cells such as T-lymphocytes and macrophages (Calvo et al., 2012).
1. Changes in Voltage-Gated Na+ Channels
As mentioned, increases in various types of voltage-gated sodium channel current contribute to the increased excitability of DRG neurons seen in neuropathic pain (Cummins and Waxman, 1997; Waxman et al., 2000a,b; Abdulla and Smith, 2001a, 2002; Stemkowski and Smith, 2012b; Bourinet et al., 2014; Waxman and Zamponi, 2014). Genetic studies have confirmed the importance of voltage-gated sodium channels by showing that gain-of-function mutations in Nav1.7, Nav1.8, and Nav1.9 are implicated in peripheral neuropathy disorders in humans by increasing the excitability of nociceptive neurons (Dib-Hajj et al., 2013; Sexton et al., 2017; Yekkirala et al., 2017).
a. Peripheral Na+ channels as drug targets
Pharmacological treatments such as carbamazepine, which are known to target Na+ channels, have been used for many years to treat some types of neuropathic pain (Demant et al., 2014), particularly trigeminal neuralgia (Walsh and Smith, 1968). Several small-molecule blockers with affinity for Nav1.7, 1.8, or 1.9 have been considered. To date, the most promising compounds that are currently in phase 1 or phase 2 clinical trials target Nav1.7 [Table 2; for additional information, see review by Yekkirala et al. (2017)].
Small-molecule peptide blockers of Nav1.7 (for example, µ-TRTX-Hhn1b derived from spider venom or μ-SLPTX-Ssm6a from centipede venom) are also of interest (Yang et al., 2013; Liu et al., 2014). A monoclonal antibody targeting the voltage sensor paddle domain of Nav1.7 is available to allow for greater sodium channel subtype selectivity (Lee et al., 2014). Slow inactivation-specific channel modulators represent another method by which ion channels can be stabilized in their slow inactivated state to act as a brake during periods of neuronal hyperexcitability, and these have been applied to target both sodium and calcium channel currents for treatment of neuropathic pain (Hildebrand et al., 2011).
A recent review drew attention to the possible pharmacological manipulation of β subunits of Na+ channels rather than the pore-forming α subunits (O’Malley and Isom, 2015). This approach may be especially attractive as three different types of β subunits are differentially and selectively expressed in small, medium, and large DRG neurons (Zhao et al., 2011; Ho et al., 2012).
Further to its obvious role in controlling neuronal excitability, the importance of Nav1.7 in normal and pathologic pain processing may derive from its ability to directly or indirectly affect several other processes, including gene expression (Sexton et al., 2017). These authors have brought forward the possibility that upregulation of the penk (preproenkephalin) gene may contribute to the beneficial effects of Nav1.7 blockers. This is supported by the observation that the analgesic effect of a selective Nav1.7 blocker, mu-theraphotoxin-Pn3a (from the tarantula Pamphobeteus nigricolor), is augmented by administration with subeffective doses of opioids or with an enkephalinase inhibitor (Deuis et al., 2017).
In a more straightforward physiologic mechanism, Nav1.7 appears to be necessary for substance P release from primary afferent terminals (Minett et al., 2012).
2. Changes in High Voltage–Activated Ca2+ Channels (Cav1 and Cav2)
Voltage-gated calcium channels are another player in the field of neuropathic pain and pain therapeutics in general (Baccei and Kocsis, 2000; Abdulla and Smith, 2001b; Bourinet et al., 2014; Waxman and Zamponi, 2014; Zamponi et al., 2015; Pan et al., 2016; Zamponi, 2016; Patel et al., 2017). These channels encompass high-voltage–activated (HVA) L-types (Cav1.1, Cav1.2, Cav1.3, and Cav1.4): P/Q-type (Cav2.1), N-type (Cav2.2), and R-type (Cav2.3), as well as T-type [low-voltage–activated (LVA)] Ca2+ channels (Cav3.1, Cav3.2, Cav3.3) (Zamponi et al., 2015; Dolphin, 2016). Influx of Ca2+ through HVA-Ca2+ channels triggers neurotransmitter release from presynaptic vesicles and thereby determines neuronal network excitability. The importance of HVA-Ca2+ channels in neuropathic pain is illustrated by the clinical effectiveness of the N-type Ca2+ channel blocker ziconotide (Zamponi, 2016) and, as will be discussed below, the relationship between HVA-Ca2+ channel function α2δ-1 subunits and the actions of gabapentinoids (Dolphin, 2012b). (See Gabapentinoids and the α2δ Subunits of Voltage-Gated Ca2+ Channels and α2δ Subunits and Neuropathic Pain.)
Voltage-gated Ca2+ channels (VGCC) consist of five subunits, as follows: the α1 pore-forming subunit and auxiliary subunits α2-δ, β, and γ. The main subtype found in presynaptic terminals is Cav2 (Westenbroek et al., 1998; Zamponi et al., 2015). Cav2.1 and Cav2.2 both contain a synaptic protein interaction site (synprint) that interacts with SNARE proteins (syntaxin and SNAP-25) (Rettig et al., 1996; Sheng et al., 1996). By this mechanism, channels can be closely associated with synaptic vesicles that govern release of neurotransmitter.
Because these channels are responsible for depolarization-induced influx of Ca2+ and triggering consequent release of neurotransmitter, blocking or genetically deleting these channels in hyperexcitable nociceptive neurons would be expected to reduce net excitability (Bourinet et al., 2014). For example, N-type VGCC knockout mice exhibit reduced signs of both inflammatory and neuropathic pain (Saegusa et al., 2001).
It is important to note, however, that there is a classic third–fourth power relationship between Ca2+ influx and neurotransmitter release (Dodge and Rahamimoff, 1967). In other words, even if one were to reduce the amount of Ca2+ entering terminals via VGCCs, there would still be sufficient VGCCs expressed to support substantial neurotransmitter release. This is supported by experiments showing that 200 μM Mn2+, a pan-VGCC blocker, produces no change in sEPSC frequency or amplitude of spinal dorsal horn neurons, but can produce a significant reduction of the amplitude of 35 mM K+-evoked Ca2+ responses that reflect activation of voltage-gated Ca2+ channels following the depolarizing action of a high concentration of extracellular K+ (Biggs et al., 2014).
If suppression of Ca2+ entry via presynaptic Cav2 channels has only very modest effects on neurotransmitter release from primary afferents, these observations raise the question of how conotoxin channel blockers actually work. It has, however, long been known that ω-conotoxin GVIA reduces synaptic potentials in the spinal cord (Yu et al., 1992). In the clinical setting, it is also possible that Cav2 channel block may have additional long-term consequences, including effects on gene expression, as has been suggested for Nav1.7 channels (Deuis et al., 2017; Sexton et al., 2017). Effects of Cav2 block may be mediated via long-term changes in intracellular Ca2+ handling by mitochondria (D’Arco et al., 2015). It is also known that nerve injury downregulates HVA calcium channel current (ICa) in DRG neurons (Baccei and Kocsis, 2000; Hogan et al., 2000; Abdulla and Smith, 2001b; Pan et al., 2016). If a similar or greater decrease occurs in primary afferent terminals, this downregulation in combination with channel block may be sufficient to attenuate neurotransmitter release in the dorsal horn.
a. Cav2 channels as therapeutic targets
In view of the inconvenient pharmacokinetics of peptide neurotoxins such as ziconotide (synthetic ω-conotoxin MVIIA), which has to be administered intrathecally (McGivern, 2007), there is considerable interest in developing small-molecule blockers of CaV2 channels (Waxman and Zamponi, 2014; Zamponi et al., 2015; Zamponi, 2016; Patel et al., 2017). Several state-dependent Cav2 blockers are currently under development, including ZC88 (Meng et al., 2008; Zhang et al., 2015b), A-1264087 (Xu et al., 2014; Zhu et al., 2014), and TROX-1 (Abbadie et al., 2010; Swensen et al., 2012; Patel et al., 2015). Oral administration of all of these drugs displays antiallodynic efficacy in rodent models of neuropathic pain (Patel et al., 2017).
N-type Ca2+ channels are also modulated by α2-adrenoceptor agonists such as clonidine (Abdulla and Smith, 1997) and opioids (Seward and Henderson, 1990; Abdulla and Smith, 1998; Altier and Zamponi, 2008). Given their limited clinical efficacy and loss of functional μ-opioid receptors after nerve injury (Abdulla and Smith, 1998; Zhang et al., 1998), actions of opioids are not particularly relevant to discussion of neuropathic pain. In contrast, the α2 adrenoceptor agonist, clonidine, displays antiallodynic actions in a rodent model (Puke et al., 1991), and meta-analysis of clinical trials suggests some clinical efficacy (Giovannitti et al., 2015). These effects may be mediated by restoration of α2 adrenergic modulation of pain processing at the spinal level following peripheral nerve injury (Bannister and Dickenson, 2017) and/or by attenuation of aberrant interactions between sympathetic and sensory nerves in the periphery (McLachlan et al., 1993; Abdulla and Smith, 1997; Yen et al., 2006). N-type Ca2+ channels in DRG are also modulated by neuropeptide Y (NPY), and this effect, which is mediated via Y2 receptor, is increased after nerve injury (Abdulla and Smith, 1999). This has led to the suggestion that Y2 agonists, which suppress excitatory synaptic transmission in substantia gelatinosa, may play a role in the management of neuropathic pain (Moran et al., 2004; Smith et al., 2007). This idea is supported by the observation that intrathecal injection of NPY attenuates mechanical and cold hypersensitivity in the rodent (spared nerve injury) model (Intondi et al., 2008). Spinal galanin receptors may represent an additional drug target (Alier et al., 2008). Both NPY and galanin are found in inhibitory interneurons in dorsal horn (Iwagaki et al., 2013; Boyle et al., 2017; Todd, 2017).
b. Cav1 channels as therapeutic targets
In addition to the established role of changes in N-type Ca2+ channels, certain lines of evidence support an additional contribution of L-type Ca2+ channels in the maintenance of neuropathic pain (Balasubramanyan and Smith, 2005; Fossat et al., 2010; Chang et al., 2015; Radwani et al., 2016). This has led to increased interest in broad-spectrum dihydropyridine-related Ca2+ channel blockers such as M4, which blocks Cav 1.2 (L-type), Cav 2.2 (N-type), and Cav 3.2 and 3.3 (T-type channels) (Gadotti et al., 2015; Zamponi et al., 2015). Possible cardiovascular actions of this type of substance may curtail their further therapeutic development,
3. Changes in LVA Ca2+ Channels (Cav3)
By contrast with the role of P/Q-type (Cav2.1) and N-type (Cav2.2), HVA-Ca2+ channels in neurotransmitter release, T-type, LVA, Ca2+ channels (Cav3.1, Cav3.2, Cav3.3) play an important role in setting neuronal excitability. In view of this, there is considerable interest in targeting these channels in pain management (Altier and Zamponi, 2004; Iftinca and Zamponi, 2009; Todorovic and Jevtovic-Todorovic, 2013; Snutch and Zamponi, 2017). This is especially the case as T-type Ca2+ channel currents are increased in sensory neurons following nerve injury in a model of diabetic neuropathy (Jagodic et al., 2007, 2008). Mechanistically, this may involve upregulation of the deubiquitinase, USP5, by the action of the inflammatory mediator IL-1β. The resultant impairment of Cav3.2 channel ubiquitination would prolong surface expression of T-type LVA Ca2+ channels and thereby promote increased excitability (Stemkowski et al., 2017).
a. Cav3 channels as therapeutic targets
The observation that T-type Ca2+ channel blockers mibefradil and ethosuximide acutely increase withdrawal thresholds in rats subject to nerve injury further supports the suggestion that T-type channels are involved in neuropathic pain expression. Small-molecule T-channel blockers (KYS05090S or ABT-639) also showed promise in preclinical studies (Jarvis et al., 2014; Zhang et al., 2015a; M’Dahoma et al., 2016). Unfortunately, preliminary clinical results with ABT-639 have been disappointing (Patel et al., 2015).
Cannabinoids, which are effective in some neuropathic pain cases (Moulin et al., 2014; Hauser et al., 2017), inhibit recombinant human T-type (Cav 3.1, 3.2) Ca2+ channels (Ross et al., 2008), and intrathecal injection of the cannabinoid receptor 1/cannabinoid receptor 2 (CB2) receptor agonist NMP-7 inhibits injury-induced neuropathic pain in a rodent model. This effect involves CB2 receptors and Cav3.2 channels (Berger et al., 2014). To the best of our knowledge, NMP-7 has not yet progressed to clinical trials, but its preclinical effectiveness led to the development of the derivative [N-((1-(2-(tertbutylamino)-2-oxoethyl)piperidin-4-yl)methyl)-9-pentyl-9Hcarbazole-3-carboxamide], which displays remarkable effectiveness in both inflammatory and neuropathic pain (Bladen et al., 2015).
Several additional small molecules that modulate Cav3.2 channels either directly or via CB2 receptor activation are in early preclinical development. For further information, see review by Snutch and Zamponi (2017).
4. Changes in Hyperpolarization-Activated Cyclic Nucleotide–Gated Channels
HCN channels have emerged as a promising peripheral drug target for neuropathic as well as inflammatory pain (Chaplan et al., 2003; Emery et al., 2011, 2012; Noh et al., 2014; Young et al., 2014; Tsantoulas et al., 2016). HCN2 is expressed in about half of small somatosensory neurons, which are mainly nociceptors, and plays an important role in the control of firing frequency in response to noxious stimuli (Emery et al., 2011). Indeed, deletion of HCN2 in nociceptive neurons prevents the development of inflammatory and neuropathic pain (Emery et al., 2011).
HCN channels are upregulated after nerve injury, and it is thought that they drive spontaneous activity in the DRG (Luo et al., 2007; Emery et al., 2011, 2012; Young et al., 2014) to increase transmitter release from primary afferents (Antal et al., 2004; Papp et al., 2006).
a. HCN channels as therapeutic targets
A blocker of HCN channels, ivabradine, which is also used clinically to treat angina and heart failure, has been shown to be effective in treating signs of neuropathic pain in animal models through peripheral action on small sensory neurons and to decrease firing frequency in cultured DRG neurons from nerve-injured animals (Noh et al., 2014; Young et al., 2014). More recent work has focused on the search for selective HCN2 blockers (Sartiani et al., 2017) that may abrogate hyperexcitability of DRG neurons without affecting the HCN1 channels that are responsible for controlling cardiac rhythmicity (Tsantoulas et al., 2016).
5. Changes in K+ Channels
Attenuation of various types of K+ channel conductance also contributes to the increased excitability of DRG neurons that follows injury or exposure to inflammatory cytokines such as IL-1β (Abdulla and Smith, 2001a,b; Kim et al., 2002; Tan et al., 2006; Stemkowski et al., 2015; Cao et al., 2012; Stemkowski and Smith, 2012a; Tsantoulas et al., 2012; Waxman and Zamponi, 2014; Gonzalez et al., 2017).
Changes in K+ channel currents may also contribute to pain associated with diabetic neuropathy (Cao et al., 2010). This prompted the suggestion that the anticonvulsant retigabine (Passmore et al., 2003) and related Kv7.2–Kv7.3 channel openers (Zhang et al., 2013; Busserolles et al., 2016), as well as facilitators of Na+ activated K+ channel currents (Huang et al., 2013) or two pore K+ channels or KV2 or Kv9.1 channels, may be useful in pain management (Tsantoulas et al., 2012; Yekkirala et al., 2017). Unfortunately, a clinical trial with retigabine failed to meet its primary efficacy end point (Yekkirala et al., 2017). The retigabine analog N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide (ICA-27243) has been reported to attenuate inflammatory pain in animal models (Hayashi et al., 2014), but we are unaware of any clinical trials with this substance. Beyond neuropathic pain arising from peripheral nerve injury, it has been reported that injury to the spinal cord per se can impede K+ channel function in DRG (Ritter et al., 2015), and this may contribute to mechanical allodynia. Altered K+ channel function in DRG may reflect the diffusion of inflammatory mediators from the site of injury within the spinal cord (Carlton et al., 2009).
Changes in K+ channel function may not always involve changes in expression or properties of pore-forming α subunits themselves but may rather result from changes in regulatory subunits. For example, it was recently shown that K+ channel modulatory subunits KChIP1, KChIP2, and DPP10 are coexpressed with Kv4.3 in nonpeptidergic small neurons of the rat DRG. Spinal nerve ligation downregulated all three modulatory subunits, and their knockdown by intrathecal injection of a gene-specific antisense oligodeoxynucleotides evoked mechanical hypersensitivity. Rescue of the downregulated subunits by injection of appropriate cDNA constructs attenuated injury-induced hypersensitivity (Kuo et al., 2017). These findings imply that targeting of regulatory subunits rather than pore-forming subunits of K+ channels may lead the way to new therapeutic approaches. This is in fact analogous to targeting the α2δ-1 subunit of Ca2+ channels by gabapentin to control α subunit function (Dolphin, 2016) and to the suggested targeting of nonconducting β subunits of Na+ channels (O’Malley and Isom, 2015).
6. Changes in Transient Receptor Potential Channels
TRPV1 are nonselective cation channels that belong to the larger group of multifunctional TRP channels (Nilius et al., 2007). They are activated by the vanilloid, capsaicin, and by noxious heat, low pH, osmolarity changes, and arachidonic acid metabolites (Caterina and Julius, 2001). TRPV1 sensitivity to temperature is increased following exposure to nociceptive molecules like nerve growth factor, or agents acting through G protein–coupled receptors such as bradykinin, prostaglandin E2, or angiotensin acting on angiotensin type 2 receptors (AT2) (Yekkirala et al., 2017).
A recent paper addresses the role of the cold-sensing channel, TRP channel type M8 (TRPM8), in cold allodynia (Gonzalez et al., 2017). Nerve injury does not appear to directly upregulate this channel but rather decreases expression of an excitability brake potassium current (Kv1.1–1.2) in cold-sensitive DRG neurons. Cold-induced currents through an unchanged population of TRPM8 are thus more effective in promoting depolarization and increased action potential discharge.
a. Transient receptor potential type V1 channels as therapeutic targets—capsaicin and transient receptor potential channel type V1 antagonists
TRPV1 is upregulated in DRG following partial nerve injury or spinal nerve ligation (Hudson et al., 2001; Fukuoka et al., 2002), and TRPV1 antagonists have been shown to attenuate injury-induced hyperalgesia (Basso and Altier, 2017). These findings implicate TRPV1 channels in the etiology of neuropathic pain, and it is possible that other TRP channels such as TRPM8 and TRP channel type A1 also play a role (Basso and Altier, 2017). Because TRPV1 antagonists are effective in animal models, it seems paradoxical that capsaicin patches are somewhat effective in management of neuropathic pain (Finnerup et al., 2015). One explanation may be that the Ca2+ influx induced by TRPV1 activation destroys nociceptor terminals or that the receptors become desensitized and no longer respond to inflammatory mediators. Given the demonstration of interruption of pain memory in animal models by capsaicin (Bonin and de Koninck, 2014), perhaps some of its actions in the clinic reflect the presence of a reconsolidation phenomenon in human neuropathic pain.
b. Transient receptor potential type V1 channels as therapeutic targets—angiotensin receptor type 2 receptor antagonists
The observation that activation of the AT2 can enhance TRPV1 channel function has brought forward the idea that it may serve as a therapeutic target in control of neuropathic pain (Smith et al., 2013; Ink and Schatman, 2017; Yekkirala et al., 2017). The AT2 receptor antagonist EMA401 produces dose-dependent antinociception in mouse neuropathic pain models and inhibits capsaicin-induced calcium fluxes in human and DRGs (Anand et al., 2013). The results of phase I and II clinical trials in patients suffering from postherpetic neuralgia are encouraging as EMA401 produces significant analgesia (Rice et al., 2014; Rice and Smith, 2015). The tolerability of EMA401 may reflect the fact that cardiovascular, renal, and vasoconstrictor actions of angiotensin are mediated by the angiotensin receptor type 1.
J. Neuroimmune Interactions in Neuropathic Pain
In addition to the obvious importance of neurons and the accepted role of microglia in the initiation of neuropathic pain (Coull et al., 2005; Lu et al., 2009; Beggs and Salter, 2012b, 2013), the role of other immunocompetent cells, such as astrocytes, endothelial cells, perivascular macrophages, infiltrating T cells, and satellite glial cells of DRG neurons, must not be overlooked (DeLeo et al., 2007; Scholz and Woolf, 2007; Costigan et al., 2009a; Beggs et al., 2010; Calvo et al., 2012; Grace et al., 2014; Vicuna et al., 2015; Dodds et al., 2016; Ji et al., 2016; Lim et al., 2017; Mikuzuki et al., 2017). This follows the recognition that immune cells express many of the receptors and transduction mechanisms that are activated by injury to neurons (Chiu et al., 2012). The idea of immune system involvement in chronic pain came about from observing classic signs of a systemic sickness response (malaise, lethargy, depression, anxiety) in chronic pain patients. This idea was later corroborated by findings showing that elevated levels of IL-1β in peripheral nerves could both mediate and induce hyperalgesia (Grace et al., 2014). It was also demonstrated that IL-1β, in addition to its proinflammatory actions, can directly activate nociceptors to generate action potential firing, increased membrane availability of Cav3.2 (T-type) channels (Stemkowski et al., 2017), and hyperalgesia (Binshtok et al., 2008; Stemkowski and Smith, 2012a; Stemkowski et al., 2015). As already mentioned, cytokines such as IL-1β may be responsible for increased expression of CSF-1 in primary afferent neurons (Lim et al., 2017). This in turn is released into the spinal cord, where it promotes BDNF release from microglia and increased dorsal horn excitability (Guan et al., 2016; Okubo et al., 2016) (Fig. 2A).
In addition to altering neuronal activity via the release of BDNF, reactive microglia release cytokines and chemokines such as fractalkine and other inflammatory mediators that trigger peripheral immune cell infiltration and astrogliosis. In addition, damaged sensory neurons release CCL2 (also known as monocyte chemoattractant protein 1) as well as neuregulin 1 and fractalkine. The net effect of complex interactions between invading and resident immunocompetent cells and numerous signaling molecules is the generation of classic inflammatory mediators such as IL-1β, IL-6, and IL-18, and TNF. These influence neuronal activity by facilitation of spinal LTP (Gruber-Schoffnegger et al., 2013), direct excitatory actions on neurons (Binshtok et al., 2008; Gustafson-Vickers et al., 2008; Kawasaki et al., 2008; Stemkowski and Smith, 2012a; Stemkowski et al., 2015), increased release of glutamate from primary afferents (Yan and Weng, 2013), and decreased release of GABA from dorsal horn interneurons (Zhang and Dougherty, 2011).
It is noteworthy that generation of mediators such as IL-1β and TNF may not be entirely maladaptive as cytokines have been implicated in functional recovery of damaged nerves (Nadeau et al., 2011) and neuroinflammation may assist in remyelination (Goldstein et al., 2016).
A further development in the area of neuroimmune interactions related to pain is the observation that bacterial infections can cause pain via pathogens interacting directly with and activating nociceptors (Chiu et al., 2013; Ji et al., 2016).
A full description of neuroimmune interactions in neuropathic pain is beyond the scope of the present review. For further information, the reader is referred to reviews by Linda Watkins and others (DeLeo et al., 2007; Milligan and Watkins, 2009; Grace et al., 2014; Dodds et al., 2016; Mifflin and Kerr, 2017). Although targeting the immune system may seem a rational basis for developing antiallodynic drugs (Leung and Cahill, 2010), to the best of our knowledge, no suitable small molecules have yet been identified (Yekkirala et al., 2017). This lack of success may be attributed, at least in part, to the notion that neuroimmune interactions trigger the onset of neuropathic pain, whereas other mechanisms, more relevant to the clinical presentation, are responsible for its long-term maintenance.
K. Adenosine A3 Receptors as a Therapeutic Target
The idea that the most effective agents in neuropathic pain management likely exert actions through multiple targets has already been alluded to. Adenosine A3 receptor agonists represent one such class of agents as they interact with several of the pathophysiological mechanisms outlined above (Janes et al., 2016). A3 receptors are found on immune cells, astrocytes (Bjorklund et al., 2008; Janes et al., 2015), microglia (Hammarberg et al., 2003), and both peripheral and central neurons (Janes et al., 2016). In addition, the A3 receptor agonist MRS5698 attenuates KCC2 downregulation and restores Cl− gradients in rodent CCI models (Ford et al., 2015), a process that may involve attenuation of BDNF signaling (Janes et al., 2016). A3 receptor activation also attenuates neuroinflammatory responses, astrocyte reactivity, and cytokine release (Janes et al., 2015, 2016). Efficacy of A3 receptor agonists such as IB-MECA and MRS5698 in relief of allodynia in rodent models of neuropathic pain has been convincingly demonstrated (Ford et al., 2015; Little et al., 2015). Because cardiovascular actions of adenosine are mediated via A1 and A2 adrenoceptors (Sawynok, 2016), A3 agonists show considerable promise as therapeutic agents (Janes et al., 2016). The results of clinical trials are thus eagerly awaited.
III. Current Therapeutic Management of Neuropathic Pain: Focus on Gabapentinoids
Whereas opioids are highly effective in many forms of nociceptive pain (Yekkirala et al., 2017), an equivalent panacea for the management of neuropathic pain is yet to appear (Patel et al., 2017). As mentioned above, loss of opioid effectiveness after nerve injury may reflect downregulation or attenuated signaling via μ opioid receptors (Abdulla and Smith, 1998; Zhang et al., 1998; Kohno et al., 2005). This underlines the urgent need to develop new drugs. Despite the extensive investigation of signaling mechanisms associated with neuropathic pain and the new data available, with the possible exception of cannabinoids (Hauser et al., 2017; Yekkirala et al., 2017), essentially no new evidence-based therapeutic approaches have gained widespread recognition since the introduction of gabapentinoids in the 1990s. As already mentioned, Table 2 lists a variety of agents or viable candidates that are currently under investigation and that have either not yet reached clinical trials or have progressed successfully to phase 1 and/or phase 2 trials.
GBP and PGB, as well as tricyclic antidepressants (such as low-dose amitriptyline) and serotonin–noradrenaline reuptake inhibitors (such as venlafaxine), thus still retain their status as first-line treatments (Finnerup et al., 2015) (Table 1). Given their therapeutic importance and the paucity of information regarding their exact mechanism of action (Dolphin, 2016), the remainder of the review will discuss the current understanding of gabapentinoid action in the light of recent advances in the understanding of the etiology of neuropathic pain. We have already commented that “the most used drugs are the least well understood.”
A. Gabapentinoids and the α2δ Subunits of Voltage-Gated Ca2+ Channels
GBP was originally used as an adjunct or monotherapy for partial and general tonic–clonic seizures, but was later approved as a treatment of postherpetic neuralgia (Taylor et al., 2007). Both PGB and GBP are used to treat various forms of neuropathic pain, including fibromyalgia (Moore et al., 2014). Some reports suggest they may be beneficial in postoperative pain (Dauri et al., 2009; Clarke et al., 2012), as well as in management of neurogenic overactive bladder (Carbone et al., 2006) and migraine (Calandre et al., 2010; Pizzolato et al., 2011; Cain et al., 2017). An enacarbil derivative of GBP with improved oral bioavailability is available (Cundy et al., 2004), and this has been approved for use in postherpetic neuralgia and restless legs syndrome.
Despite their structural similarity to GABA, neither GBP nor PGB binds to GABAA or to GABAB receptors (Lanneau et al., 2001; Moore et al., 2002; Sutton et al., 2002; Li et al., 2011). Gabapentinoids also do not affect GABA uptake, synthesis, or metabolism (Taylor et al., 2007; Taylor, 2009). Gee et al. (1996) isolated and sequenced a protein that bound GBP from porcine brain, which was identified as the α2δ-1 subunit of voltage-gated calcium channels (VGCCs). Gabapentinoids are thus referred to as α2δ ligands. Because partial purification of α2δ subunits increases their apparent apparent affinity for GBP (Davies et al., 2006), it has been suggested that the drug competes for binding of an endogenous ligand, which may be a small molecule such as leucine, isoleucine, or valine (Hendrich et al., 2008).
The importance of the α2δ-1 subunit for gabapentinoid action is supported by the observation that gabapentinoid binding is reduced and inhibition of allodynia is also lost in a R217A-α2δ-1 loss-of-function mutant mouse (Field et al., 2006). These mice do, however, exhibit allodynia following peripheral nerve injury. Structure activity studies of PGB analogs also identified a correlation between α2δ-binding affinity (labeled GBP displacement) and therapeutic effectiveness (Belliotti et al., 2005).
The α2δ subunits are encoded by one of four mammalian genes, CACNA2D1–CACNA2D4. Although the α2δ subunit is encoded by a single gene, it is expressed as two individual proteins (α2 and δ), which then reassemble and associate with the pore-forming α subunit of the VGCC (Dolphin, 2012b, 2013, 2016).
1. Isoforms and Structure of α2δ Subunits
The four mammalian genes, CACNA2D1, 2, 3, and 4, encode the four known isoforms of the α2δ subunit: α2δ-1, α2δ-2, α2δ-3, and α2δ-4 (Dolphin, 2013, 2016). All isoforms have a similar structure (Dolphin, 2012b), and a signal sequence at the N terminus of the nascent polypeptide directs α2δ into the lumen of the endoplasmic reticulum. The subunit is thus expressed extracellularly once transported to the plasma membrane. The protein sequence of all α2δ isoforms contains a Von Willebrand Factor A (VWA) domain that plays an important role in protein–protein interactions. Such interactions may be affected by a five-residue sequence comprising the metal ion-dependent adhesion site (MIDAS) motif (Whittaker and Hynes, 2002). The nature of a short hydrophobic domain at the C terminus of α2δ subunits predicts that they are glycosylphosphatidylinositol anchored (Davies et al., 2010). This means that the mature protein appears on the extracellular surface, making it potentially susceptible to extracellular modulation (Davies et al., 2010). However, recent data suggest that gabapentinoids interact with α2δ at an intracellular site prior to its trafficking to the cell surface (Lana et al., 2016). As will be discussed below, this is supported by the observation that GBP has to gain access to the neuronal cytoplasm via the system-L neutral amino acid transporter to exert its action (Hendrich et al., 2008; Biggs et al., 2014, 2015).
Another cache domain containing the RRR (triple arginine repeat) is positioned adjacent to the VWA domain in α2δ-1. This sequence may be important for gabapentinoid binding (Field et al., 2006). The structure of α2δ-1 is illustrated schematically in Fig. 5A.

(A) Diagram to illustrate the essential structural features of the α2δ-1 protein. (B) Diagram to illustrate slow and fast transport processes associated with α2δ-1. When levels of α2δ1 are normal, its accumulation in nerve terminals is controlled by transport along primary afferent fibers from cell bodies to the dorsal horn, a distance of about 7.5 mm in a rat. When levels of α2δ-1 are elevated after nerve injury, rapid cycling in nerve terminals over distances of <1 μm determines level at release sites. Gabapentinoids that inhibit transport of α2δ-1 thus work slowly when α2δ-1 levels are normal, but rapidly when its levels are elevated in neuropathic pain.
2. Tissue Distribution of α2δ Subunits
The expression of α2δ subunits is fairly ubiquitous (Gong et al., 2001). Whereas the α2δ-1 subunit is expressed in skeletal, cardiac, and smooth muscles as well as in the CNS, peripheral nervous system, and endocrine tissues, the α2δ-2 subunit is expressed predominantly in the CNS, especially the cerebellum (Dolphin, 2012b, 2013). The α2δ-3 subunit is expressed in the CNS, whereas α2δ-4 is expressed primarily in the retina and in endocrine tissues. In neurons, the α2δ-1 transcript preferentially colocalizes with excitatory neurons, and the α2δ-2 mRNA colocalizes with GABAergic inhibitory neurons (Cole et al., 2005). This is congruent with the observation that overexpression of α2δ-1 increases excitatory synaptic transmission with minimal effect on inhibitory synaptic transmission in the spinal dorsal horn (Zhou and Luo, 2015). Gabapentin binds with greater affinity to the α2δ-1 than to the α2δ-2 subunit, with no binding affinity for α2δ-3 (Marais et al., 2001).
B. α2δ Subunits and Neuropathic Pain
It is now well established that α2δ-1 protein expression is increased in rodent models of neuropathic pain (Luo et al., 2002; Xiao et al., 2002; Boroujerdi et al., 2008, 2011; Bauer et al., 2009). After nerve ligation in nerve-injured (but not sham-operated) rats, a 17-fold increase in the α2δ-1 protein in ipsilateral L5/L6 DRGs was observed. No increase in expression levels of other subtypes of the α2δ subunit (α2δ-2, -3, and -4) following nerve injury has been reported. Another spinal nerve ligation study showed that injury-induced discharges that contribute to the onset of neuropathic pain moderate the expression of α2δ in the spinal dorsal horn (Boroujerdi et al., 2008, 2011). Local application of lidocaine blocked both injury-induced discharges and the upregulation of α2δ in both dorsal spinal cord and dorsal root ganglia.
In mice, α2δ-1 gene deletion results in deficits in behavioral signs of mechanical and cold sensitivity and delayed mechanical hypersensitivity in response to peripheral nerve injury (Patel et al., 2013). This correlates with the observation that sensory neurons in α2δ-1 knockout mice show diminished ability to generate repetitive action potentials (Margas et al., 2016). In addition, mice that overexpress α2δ develop mechanical allodynia similar to nerve injury models, and this has been shown to be mediated by a presynaptic mechanism involving an increase in excitatory synaptic transmission, as is the case with nerve injury (Nguyen et al., 2009; Zhou and Luo, 2014, 2015). Inhibitory transmission is unaffected. It is clear therefore that the α2δ-1 subunits of VGCCs play an important role in the onset of neuropathic pain.
C. Physiologic Roles of α2δ Subunits
Mutation or dysfunction of α2δ subunits has been associated with convulsive and neuropsychiatric disorders as well as with night blindness and cardiovascular dysfunction (Heyes et al., 2015; Dolphin, 2016). These observations relate directly to the use of gabapentinoids as anticonvulsants (Taylor et al., 2007; Offord and Isom, 2015). For the purpose of the present review, we will focus on the physiologic role of α2δ subunits as they pertain to nociception and neuropathic pain.
1. Rapid Trafficking of Ca2+Channel α Subunits to the Plasma Membrane
Upregulation of α2δ subunits increases the expression of Cav1 and Cav2 α subunits at the plasma membrane as well as calcium current density (Yasuda et al., 2004; Canti et al., 2005; Tran-Van-Minh and Dolphin, 2010; Dolphin, 2016). The MIDAS motif in the VWA domain of α2δ-1 and α2δ-2 is responsible for both effects (Canti et al., 2005; Hoppa et al., 2012; D’Arco et al., 2015). In the case of Cav1 channels, this involves interaction with extracellular site on the α1 subunit (Walsh et al., 2009). Altered surface expression of Ca2+ channels has been proposed to exert a secondary effect on mitochondrial Ca2+ buffering (D’Arco et al., 2015).
2. Slower Trafficking of Ca2+ Channel α Subunits to Primary Afferent Terminals
In addition to controlling trafficking of Ca2+ channel α subunits to the cell surface, α2δ subunits are thought to interact with proteins involved in trafficking of membrane protein cargos (Dolphin, 2012b, 2016). In the case of primary afferent neurons, α2δ is implicated in the relatively slow trafficking of α subunits of HVA-Ca2+ channels from their site of synthesis in the cell body, along primary afferent axons, to their terminals in the spinal dorsal horn (Bauer et al., 2009). It is tempting to speculate that attenuation of this slow trafficking process by gabapentinoids corresponds to reports of their slow onset of action in neuropathic pain patients (Cheshire, 2002; Stacey et al., 2008; Sharma et al., 2010), but, as discussed in Rate of Onset of Gabapentinoid Effect, we think this is unlikely to be the case.
3. Regulation of High-Voltage–Activated Ca2+ Channel Function
Because α2δ does not affect single-channel conductance, increases in whole-cell HVA ICa observed following increased α2δ expression may be a consequence of more channels being inserted into the plasma membrane (Dolphin, 2016). However, in the case of α2δ-1 effects on Cav2.2 channels in N2a cells, the increase in current is 10-fold, whereas the increase in protein surface expression is <twofold (Hoppa et al., 2012; Cassidy et al., 2014). This implies that α2δ has additional effects on channel function. In rat DRG neurons, we found that PGB decreased the rate of inactivation of HVA ICa (mainly Cav2.2 and Cav1.2) in medium and small IB4+ and IB4− neurons, but not in non-nociceptive large neurons (Biggs et al., 2014). This is consistent with the the generalization that α2δ-1 may serve to increase the rate of inactivation (Dolphin, 2016). Further attempts to elucidate these mechanisms in terms of voltage dependence and rate of activation and inactivation have led to conflicting findings. This is because the effects observed depend on which α2δ subunits–Cav channel α subunit combination is studied. Effects may also be contingent on the presence of β subunits (Dolphin, 2016).
It was shown recently, however, that the post-translational cleavage of α2δ-1 into the disulfide-linked polypeptides α2-1 and δ is an essential step that permits voltage-dependent activation of plasma membrane N-type Ca2+ channels (CaV2.2), and that uncleaved α2δ inhibits native calcium currents (Kadurin et al., 2016). These authors showed that voltage-dependent activation of CaV2.2 channels is promoted following cell surface proteolytic cleavage of α2δ, and that this effect was independent from the role of α2δ trafficking. Trafficking of CaV2.2 channel complexes into neuronal processes was not supported by uncleaved α2δ. This mechanism may maintain immature calcium channels in an inhibited state such that proteolytic processing of α2δ permits their voltage-dependent activation. This may allow selective trafficking of mature calcium channel complexes into neuronal processes (Kadurin et al., 2016).
4. Setting Neurotransmitter Release Probability
Experiments on hippocampal neurons have shown that, in addition to setting presynaptic HVA-Ca2+ channel abundance, α2δ subunits also directly control neurotransmitter release probability. It has been suggested that α2δ subunits configure presynaptic Ca2+ channels to directly drive exocytosis. This process involves interaction affected via the MIDAS domain on α2δ (Hoppa et al., 2012). It may be of special relevance to gabapentinoid action, as their ability to control Ca2+ channel abundance may not fully account for its ability to control neurotransmitter release from primary afferents (Biggs et al., 2014).
For additional information on α2δ subunits and VGCC, see reviews by Bauer et al. (2010), Dolphin (2012b, 2013, 2016), and Zamponi et al. (2015).
D. How Do the Gabapentinoids Work?
1. Evidence for an Intracellular Site of Action
As already mentioned, GBP is thought to exert its actions by entering the neuronal cytosol through the system-L-neutral amino acid transporter prior to binding with α2δ-1. Structure-activity studies have revealed that PGB analogs that strongly bind α2δ, but which are not substrates for the transporter, lack therapeutic efficacy (Belliotti et al., 2005). This may, however, reflect failure of transport of the analogs into the brain. Stronger support for an intracellular site of action comes from the observation that GBP inhibition of HVA ICa expressed in tsA-201 cells is prevented when the transporter is inhibited by 2-(-)-endoamino-bicycloheptene-2-carboxylic acid (BCH). GBP effectiveness was also increased in Xenopus oocytes when the system-L-neutral amino acid transporter was coexpressed with HVA-Ca2+ channels (Hendrich et al., 2008). Additional support for an intracellular site of action comes from the observation that GBP application ipsilateral to CCI alleviates allodynia and hyperalgesia, whereas contralateral application does not (Shahid et al., 2017).
One potential concern with these findings is that BCH binds to α2δ-1, albeit with a 10-fold lower affinity than GBP (Gong et al., 2001). Our recent findings argue against the possibility that BCH prevents the effect of GBP by attenuating its binding to α2δ-1. We first established that application of GBP for 5 days reduced the excitability of spinal cord neurons maintained in organotypic culture, and that this effect was blocked by BCH (Biggs et al., 2014). We next used capsaicin to open TRPV1 channels and showed that GBP could enter the cytoplasm of primary afferent neurons via the open channel pore (Biggs et al., 2015). We used this method of GBP delivery to circumvent its entry via the transporter. GBP delivered via TRPV1 channels decreased spinal cord activity, but this effect was not blocked by BCH. This indicates that the weak binding of BCH to α2δ cannot account for its ability to attenuate GBP effects. Had this been the case, BCH would have attenuated the effect of GBP regardless of its route of access.
More recent work from our laboratory lends support to the idea that gabapentinoids work intracellularly (Alles et al., 2017). Part of this study involved in vivo administration of GBP and study of substantia gelatinosa neurons ex vivo from animals that had received GBP. These were nerve-injured animals and GBP effectively attenuated mechanical allodynia. Compared with controls, synaptic transmission was impaired in spinal cord slices obtained from animals that had received GBP, and liquid chromatography with mass spectrometry measurements confirmed the continued presence of the drug. Had the drug worked extracellularly to block allodynia in vivo, it would have been washed away by the time spinal cord slices were studied and no persistent effect of the drug would be seen. This supposition is supported by detailed quantitative arguments outlined in our paper (Alles et al., 2017). These observations are congruent with those of Lana et al. (2016), who showed that GBP displacement of thrombospondin 4 binding (see below) is effected at an intracellular site on α2δ-1.
2. A Role of Thrombospondins?
Thrombospondins (TSP) 1–4 are endogenous ligands that bind to α2δ-1 (Eroglu et al., 2009; Lana et al., 2016; Park et al., 2016b). They are large extracellular matrix proteins that are upregulated by peripheral nerve injury (Pan et al., 2015). Because α2δ-1 has been implicated in synaptogenesis (Eroglu et al., 2009; Bauer et al., 2010), aberration of this process following nerve injury and the actions of thrombospondin 4 on α2δ-1 may contribute to central sensitization (Li et al., 2014; Crosby et al., 2015; Park et al., 2016a,b). GBP disrupts the interaction between α2δ-1 and the synaptogenic domain of TSP2 and thereby disrupts synaptogenesis in vitro. It has been argued, however, that this effect may be of limited relevance to the therapeutic action of GBP, as any synaptic sprouting and remodelling associated with onset of neuropathic pain would have taken place before the initiation of therapy (Dolphin, 2016). Despite this, recent findings suggest that increased levels of TSP4 produced by nerve injury may contribute to hypersensitivity of peripheral sensory systems by decreasing HVA ICa and increasing LVA ICa in DRG neurons by interaction with α2δ-1. These effects are blocked by GBP (Pan et al., 2016).
3. Rapid and Slow Actions in the Spinal Dorsal Horn
The idea that α2δ participates in two types of trafficking processes in primary afferent neurons has already been alluded to. This idea is supported by studies in hippocampal neurons, in which Hoppa et al. (2012) posited that α2δ subunits function through two mechanisms: “A trafficking step from the cell soma and a local step at the presynaptic terminal allowing synapses to have increased exocytosis with decreased Ca2+ influx.” We suggest that when α2δ-1 levels are elevated, either in neuropathic pain models or by genetic modulation (Luo et al., 2001, 2002; Bauer et al., 2009), it is rapidly and locally trafficked from the endoplasmic reticulum to the plasma membrane within primary afferent nerve terminals (Bauer et al., 2010; Alles and Smith, 2016; Alles et al., 2017). The presence of high levels of α2δ-1 in nerve terminals facilitates interaction of the α subunits of HVA-Ca2+ channels with the neurotransmitter release machinery. This interaction occurs via the MIDAS motif within the VWA domain of α2δ-1 (Hoppa et al., 2012).
This process is interrupted by gabapentinoids within 30 minutes of i.p. injection and accounts for the rapid antiallodynic effect seen in nerve-injured animals (Hunter et al., 1997; Field et al., 2006; Narita et al., 2012; Kumar et al., 2013; Alles and Smith, 2016; Alles et al., 2017). This idea is supported by the observation that a rapidly developing effect of PGB in rats subject to spared nerve injury was not associated with reduced accumulation of α2δ-1 in the superficial dorsal horn (Yang et al., 2014a). The immunohistochemical methods used in this paper would not distinguish between endoplasmic reticulum and cell surface–expressed α2δ-1.
By contrast, slowly developing actions of gabapentinoids seen in uninjured dorsal horn neurons in vitro (Moore et al., 2002; Hendrich et al., 2012; Biggs et al., 2014) may reflect interruption of trafficking of α2δ subunits from cell bodies to nerve terminals (Alles and Smith, 2016). The concept of actions of gabapentinoids on injured nerves and slower actions on uninjured nerves is illustrated schematically in Fig. 5B.
4. Differential Effects of Gabapentinoids on Excitatory and Inhibitory Processes in the Substantia Gelatinosa
As mentioned already, peripheral nerve injury increases excitatory synaptic transmission between primary afferent fibers and excitatory neurons in substantia gelatinosa (Balasubramanyan et al., 2006) (Fig. 4A). These neurons usually display a delayed or transient firing pattern in response to depolarizing current (Yasaka et al., 2010; Punnakkal et al., 2014). Acute treatment with GBP (Alles et al., 2017) restores synaptic transmission to the level seen in uninjured animals (Fig. 4A). As mentioned above, Fig. 4B shows that nerve injury also reduces excitatory synaptic transmission to inhibitory neurons (Balasubramanyan et al., 2006). Inhibitory neurons in substantia gelatinosae usually display a tonic firing pattern in response to depolarizing current commands (Yasaka et al., 2010). Interestingly, GBP appears to increase excitatory drive to inhibitory neurons (Biggs et al., 2014; Alles et al., 2017). The mechanism of this unexpected effect, which involves an increase in the frequency of sEPSC, remains to be elucidated.
By contrast, GABA/glycinergic inhibitory postsynaptic currents per se seem to be enhanced or unaffected by GBP (Moore et al., 2002). Taken together, these actions culminate in overall decrease in dorsal horn excitability as judged from Ca2+ responses to 35 mM K+ (Biggs et al., 2014; Alles et al., 2017).
Figure 3B illustrates the effect of gabapentin on the electophysiological footprint produced in the substantia gelatinosa by sciatic CCI. The majority of the changes produced by gabapentin are the opposite of those seen with CCI (Fig. 3A). Thus, GBP almost entirely eliminates the electrophysiological footprint of CCI in the substantia gelatinosa (Fig. 3C).
5. Supraspinal Actions of Gabapentinoids
In addition to its spinal actions, supraspinal mechanisms clearly contribute to the therapeutic effects of gabapentinoids (Bannister et al., 2017b). This is hardly surprising given the ubiquitous distribution of α2δ subunits throughout the nervous system (Dolphin, 2012a,b, 2013). Systemically administered GBP (100 mg/kg) inhibits descending excitatory pronociceptive serotonergic (5HT3-mediated) pathways from the brain to spinal cord in neuropathic rats (Suzuki et al., 2005). The effect of GBP was antagonized by a 5HT3 antagonist, which suggests that this could be a more predominant effect compared with a peripheral effect for an acute mechanism of cell-type–specific action of systemically delivered GBP. It has also been shown that GBP can increase extracellular glutamate release in the locus coeruleus via astrocyte-dependent mechanisms involving glutamate-transporter-1 (GLT-1; also known as EAAT2) to activate descending noradrenergic inhibition to reduce nociception (Suto et al., 2014).
Another study using 8F-fluorodeoxyglucose-positron emission tomography supports a central contribution to the antiallodynic action of acute GBP (Lin et al., 2014). It was shown that spared nerve injury induced increases of glucose metabolism in thalamus, cerebellar vermis, and medial prefrontal cortex and that these changes were attenuated by acute GBP treatment. In a functional magnetic resonance imaging study, acute 100 mg/kg GBP was shown to evoke changes in multiple nociceptive brain regions, including the thalamus and periaqueductal gray in anesthetized naive rats (Governo et al., 2008). More recently, it was shown that PGB can reduce neuronal hyperexcitability that accompanies mechanical and heat-evoked responses in the ventral posterior thalamus of spinal nerve–ligated rats (Patel and Dickenson, 2016b). In addition, Alles et al. (2017) demonstrated that GBP can reduce neuronal hyperexcitability in the primary somatosensory cortex immediately following systemic injection in neuropathic rats, and that these effects correlate with effects on synaptic transmission in the spinal dorsal horn and antiallodynic actions of GBP.
6. Additional Sites of Gabapentinoid Action
The ubiquitous distribution of α2δ subunits throughout the nervous system (Dolphin, 2012a,b, 2013) brings forward the possibility that they interact with downstream effectors other than the α subunits of voltage-gated Ca2+ channels. This would predict additional mechanisms of gabapentinoid effectiveness. For example, gabapentin has been reported to shift the voltage dependence of HCN4 channels to more negative potentials (Tae et al., 2017), but may increase hyperpolarization-activated cation current in hippocampal neurons (Surges et al., 2003). It has also been reported to interact with NMDA and AMPA receptors, as well as KATP channels (Cheng and Chiou, 2006). It remains to be determined whether these actions proceed via interactions with α2δ-1 or whether other molecular processes are engaged. Some of the interactions of gabapentinoids with ion channels may reflect simple channel block.
E. Doses of Gabapentinoids Used in Animal Studies and Relationship to Clinical Studies
In early studies of GBP action and interaction with α2δ, seemingly high drug concentrations of drugs were used. For example, some of the results obtained by Hendrich et al. (2008) used 1 mM GBP. These authors argued that the actions of GBP may reflect antagonism of positive modulators of α2δ function, namely isoleucine and leucine, which were present at 800 μM in their culture medium, as well as valine, which was present at 400 μM. Despite this, they were also able to observe some effects with more reasonable concentrations of GBP (100–200 μM). We have routinely used 100 μM gabapentin in our in vitro studies (Biggs et al., 2014, 2015; Alles and Smith, 2016; Alles et al., 2017), and this is within the range of plasma drug concentrations measured in patients: 70–120 (Johannessen et al., 2003) or 34.5–127.9 μM (Berry et al., 2003).
IV. Basic Science of Gabapentinoid Action and Observations in the Clinic
A. Inconsistency of Gabapentinoid Effectiveness
In laboratory models of neuropathic pain, every rat or mouse that is tested responds to the antiallodynic action of gabapentinoids (Kimura et al., 2016). This differs from the clinical picture, in which as few as 35% of patients report appreciable pain relief (Moore et al., 2011). There may be many reasons for this discrepancy. One possibility may relate to decreased clinical effectiveness with time. This is frequently observed but poorly documented (Cheshire, 2007). It is not known whether it reflects tachyphylaxis or tolerance as a result of repeated administration of drug or whether the pain pathophysiology changes with time. Loss of gabapentinoid effectiveness has, however, been replicated in an animal model of poststroke pain and was also seen in animals subject to spared nerve injury (Kimura et al., 2016; Yang et al., 2016). In the first case, this was attributed to downregulation of α2δ-1 with time despite the persistence of pain (Yang et al., 2016). In the second case, loss of gabapentin efficacy was associated with downregulation of GLT-1 (better known as EAAT2) in locus coeruleus (Kimura et al., 2016). It had been established previously that GBP increases glutamatergic transmission within the locus coeruleus by a GLT-1/EAAT2-dependent mechanism to stimulate descending noradrenergic inhibition of spinal nociceptive circuits (Suto et al., 2014). Interestingly, this loss of gabapentinoid effect in the spinal nerve ligation model could be restored using the histone deacetylase inhibitor and anticonvulsant agent, valproic acid (Kimura et al., 2016). This effect was attributed to increased GLT-1/EAAT2 expression. Other histone deacetylase inhibitors such as JNJ-26481585 produce robust upregulation of α2δ-1 in mice in vivo (Capasso et al., 2015). It is therefore possible that upregulation of α2δ-1 also contributes to the restoration of GBP effectiveness by valproate described by Kimura et al. (2016). This may go along with the idea that long-term persisting neuropathic pain may be α2δ-1 independent, and that high levels of α2δ-1 in nerve terminals are required for gabapentin effectiveness (Alles and Smith, 2016; Yang et al., 2016). There is, however, a problem with the idea that if long-term neuropathic pain is truly α2δ-1 independent, why should its upregulation and its binding to GBP have any effect at all? Nevertheless, the obvious clinical implication is that loss or absence of gabapentinoid effect in the clinic may be restored or established by treatment with valproate, a clinically approved anticonvulsant agent.
B. Improving Gabapeninoid Effectiveness
Another emerging approach to pain management is to introduce drugs into the neuronal cytoplasm via the open pore of TRP channels (Binshtok et al., 2007, 2009). This may be achieved by direct activation of TRPV1 by capsaicin or indirect activation of other drug-permeable channels via Toll-like receptor 5 (TLR5) (Peirs and Seal, 2015; Xu et al., 2015). The TRPV1 channel pore is large enough to permit permeation of a quaternary derivative of lidocaine known as QX-314. This substance does not have a local anesthetic when applied extracellularly, as it is membrane impermeable. It is effective when introduced into the cytoplasm via the TRPV1 channel, and this allows selective targeting of classic C fiber nociceptors in nociceptive pain. Because TLR5 is expressed on medium to large A fibers, including Aβ mechanoreceptors in both mice and humans (Xu et al., 2015), this may allow selective pharmacological blockade of transmission in fiber types that mediate allodynia. We recently noted that GBP is able to permeate TRPV1 channels activated by capsaicin, and this led to increased effectiveness (Biggs et al., 2015). Perhaps topical application of GBP (Shahid et al., 2017) combined with a TLR5 agonist such as flagellin (Xu et al., 2015) may facilitate its access to α2δ-1 and thereby increase its effectiveness in the clinic.
C. Rate of Onset of Gabapentinoid Effect
In animal models of neuropathic pain, gabapentinoids consistently produce an antiallodynic effect within 30 minutes of administration (Hunter et al., 1997; Field et al., 2006; Narita et al., 2012; Kumar et al., 2013; Alles and Smith, 2016; Alles et al., 2017). We contend this rapidity reflects gabapentin interaction with upregulated α2δ-1 and increased trafficking of VGCC from endosomes to the plasma membrane within the confine of the nerve terminal (Fig. 5B; see Rapid Trafficking of Ca2+ Channel α Subunits to the Plasma Membrane). Effects of gabapentinoids in animal models of anxiety and epilepsy are similarly rapid and are α2δ-1 mediated (Vartanian et al., 2006; Lotarski et al., 2011, 2014). Although this could mean α2δ-1 is upregulated in these conditions, we could find no evidence to support this. There is evidence, however, that various mutations in the Cacna2d2 gene that codes for α2δ-2 exhibit an epileptic phenotype.
Gabapentinoids may thus require a pathologic situation to work in neuropathic pain, but not necessarily in epilepsy and anxiety. One possible explanation for this is that Ca2+ channels are rapidly turning over at central synapses involved in anxiety and epilepsy and are thus sensitive to GBP. By contrast, Ca2+ channel turnover in uninjured primary afferents may be relatively slow and thus insensitive to gabapentinoids. The idea that gabapentinoids target Ca2+ channel populations that are rapidly turning over has been used to explain their lack of effect on cardiac and skeletal muscle channels (Dolphin, 2016).
Despite their rapid efficacy in animal models of neuropathic pain (Hunter et al., 1997; Field et al., 2006; Narita et al., 2012; Kumar et al., 2013; Alles and Smith, 2016; Alles et al., 2017), several reports indicate that GBP effects in clinical pain management take a day or more to develop (Cheshire, 2002; Stacey et al., 2008; Sharma et al., 2010; Rauck et al., 2013).
One factor that may contribute to this discrepancy in time course is the difference in evaluating drug effects in rodents and people (Yekkirala et al., 2017). Even if a human patient exhibited an increased withdrawal threshold to von Frey filament immediately after GBP, it might take several days for them to state improvement on a pain questionnaire; despite a rapid antiallodynic effect, it may take a longer period of time for other manifestations of neuropathic pain such as causalgia and spontaneous pain to be altered. It should also be noted that it is often necessary to titrate appropriate dosage in human patients so that the absence of obvious effectiveness may reflect the failure to achieve an effective concentration of drug in plasma and cerebrospinal fluid.
We therefore think it unlikely that slowly developing action of gabapentinoids in inhibiting transport of Ca2+ channel α subunits from the cell body to nerve terminals in animal models in vitro (Fig. 5B; see also Rapid and Slow Actions in the Spinal Dorsal Horn) has any direct bearing on their antiallodynic action in the clinic.
D. Animal Models of Antiallodynic Effectiveness
The discrepancy between observations in people and in animal models may start to be addressed as more refined models for evaluating pain are developed (Mogil and Crager, 2004; Mogil, 2009). Indeed, recent review has stated that current models of animal testing are predicted to fail in modeling human pain and drug effectiveness (Yekkirala et al., 2017). Supraspinal processing of nociceptive information may be more akin to the human sensation of pain than tests of mechanical, cold, or thermal allodynia. Human pain may be better modeled and evaluated in animals by using operant models such as conditioned place aversion (King et al., 2009; Zhang et al., 2015c; Bannister et al., 2017b) or evaluation of spontaneously emitted behaviors such as facial grimacing (Langford et al., 2010) or examination of changes in social interaction (Mogil, 2009).
V. Conclusions
There is an urgent need to develop new pharmaceutical agents for the treatment of neuropathic pain. Extensive investigation of the etiology of neuropathic pain has identified several potential drug targets, and some promising new compounds are in development.
Results from clinical trials have in general been disappointing. Many substances identified in preclinical studies have failed in phase 1 or phase 2 trials because of lack of efficacy or the presence of dose-limiting untoward effects. Experience suggests that highly specific agents that target one receptor or channel never progress to the clinic, and this may reflect the diverse etiologies of pain seen in the human population. By contrast, drugs that interact with multiple targets such as broad-spectrum dihydropyridine Ca2+ channel blockers (Gadotti et al., 2015) may be more effective. Interestingly, A3 adenosine receptor antagonists and gabapentinoids interact with clearly defined targets, A3 receptors and α2δ-1, yet consequences are wide ranging. Even carbamazepine, which has been used for many years to treat trigeminal neuralgia (Cheshire, 2007), affects both Na+ channels and GABA function. Ion channel function is also affected by the noradrenaline serotonin uptake inhibitor amitriptyline (Wolff et al., 2016), which retains its status as a first-line treatment of neuropathic pain (Finnerup et al., 2015; Gilron et al., 2015).
Despite their inconsistent effectiveness, gabapentinoids remain a first-line treatment option. Because these drugs are expected to retain this status for the immediate future, therapeutic management may be improved in the light of new information regarding their mechanism of action. Might rational combinations of gabapentinoids with drugs such as valproate, TLR5 agonists, or TRP channel openers improve their therapeutic effectiveness?
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Alles, Smith.
Footnotes
↵1 Current affiliation: Molecular Nociception Group, Wolfson Institute for Biomedical Research, University College London, London, United Kingdom.
This work was supported by grants from Pfizer Canada (Neuropathic Pain Research Awards), Paralyzed Veterans of America, and Natural Sciences and Engineering Research Council.
Abbreviations
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AT2
- angiotensin receptor type 2
- BCH
- 2-(-)-endoamino-bicycloheptene-2-carboxylic acid
- BDNF
- brain-derived neurotrophic factor
- Cav1, Cav2, Cav3
- voltagae gated Ca2+ channels types 1, 2 and 3
- CB2
- cannabinoid receptor 2
- CCI
- chronic constriction injury
- CCL
- chemokine (C-C motif) ligand
- CNS
- central nervous system
- CSF-1
- colony-stimulating factor 1
- DRG
- dorsal root ganglion
- EAAT2
- excitatory amino acid transporter 2
- GBP
- gabapentin
- GLT-1
- glutamate-transporter-1
- HCN
- hyperpolarization-activated cyclic nucleotide gated
- HVA
- high voltage activated
- IB4
- isolectin B4
- ICa
- calcium channel current
- IL
- interluekin
- KCC2
- potassium-chloride exporter 2
- LTP
- long-term potentiation
- LVA
- low voltage activated
- mEPSC
- miniature excitatory postsynaptic current
- MIDAS
- metal ion-dependent adhesion site
- Nav1.7, Nav1.8, Nav1.9
- voltage gated sodium channels types 1.7, 1.8 and 1.9
- NMDA
- N-methyl D-aspartate
- NPY
- neuropeptide Y
- P2X4R
- purinergic ionotropic receptor 2X4
- PGB
- pregabalin
- sEPSC
- spontaneous excitatory postsynaptic current
- STOML-3
- stomatin-like protein 3
- TLR5
- Toll-like receptor 5
- TNF
- tumor necrosis factor
- TrkB
- tropomyosin receptor kinase B
- TRP
- transient receptor potential
- TRPM8
- TRP channel type M8
- TRPV1
- TRP channel type V1
- TSP
- thrombospondin
- VGCC
- voltage-gated calcium channel
- VNUT
- vesicular nucleotide transporter
- VWA
- Von Willebrand Factor A
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
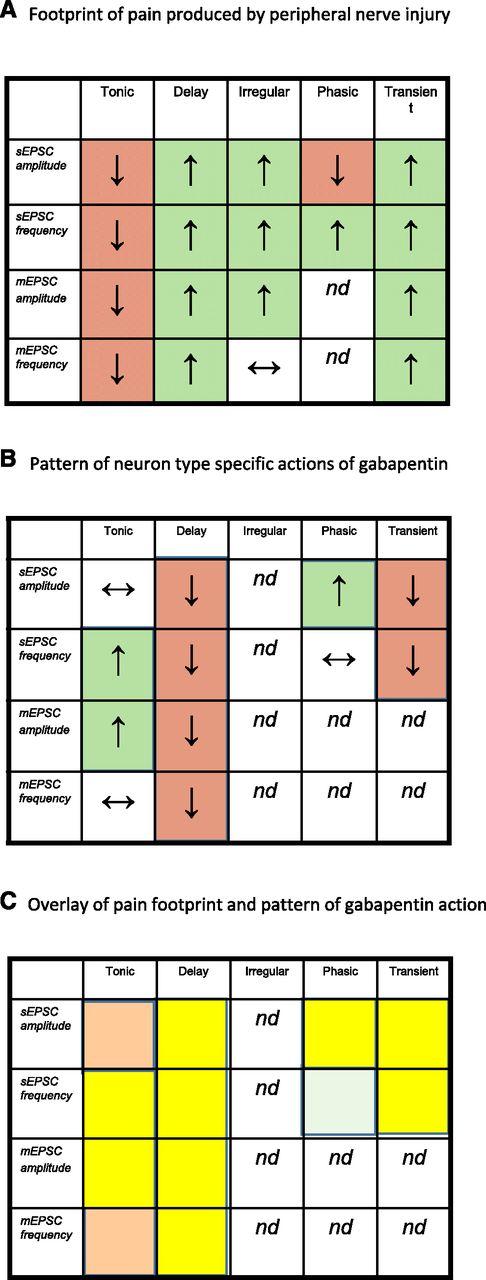{kind=link}
{kind=link}
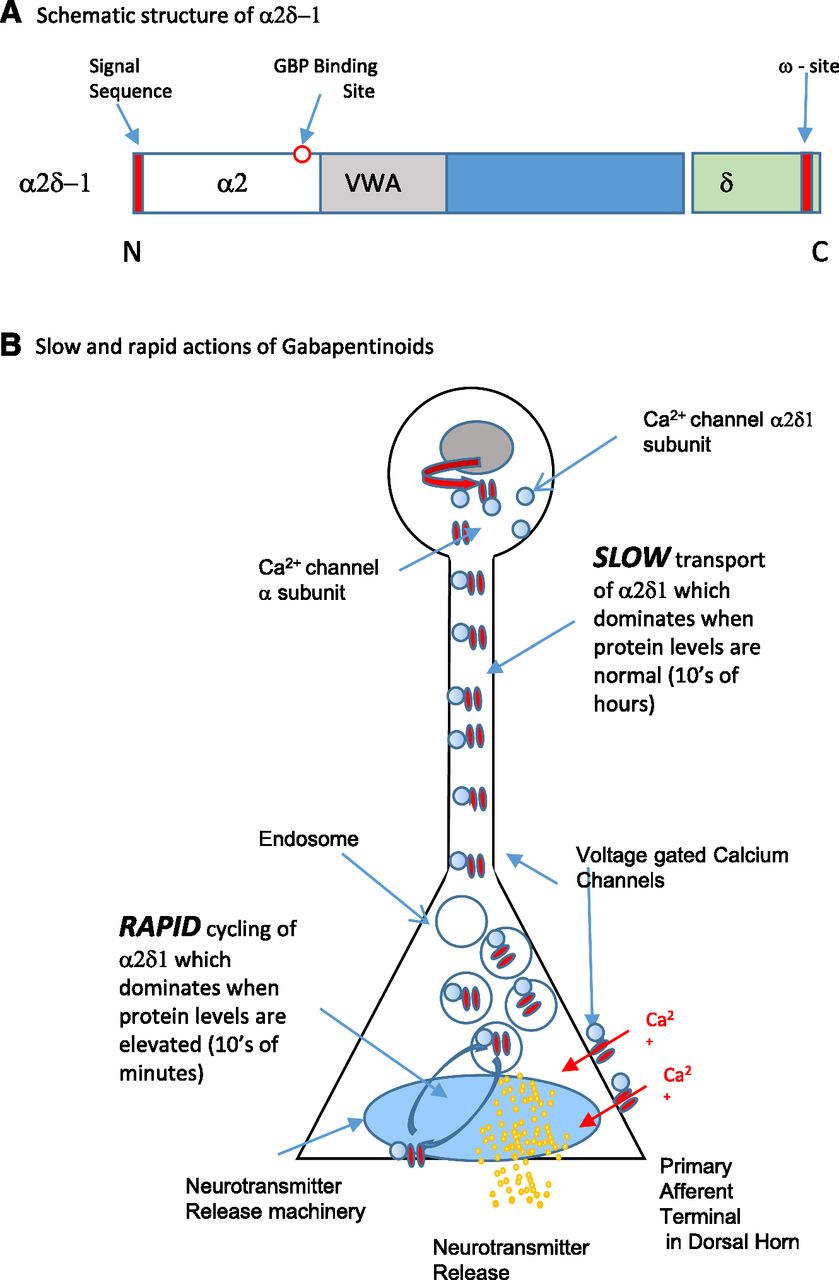{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Mechanisms of Neuropathic Pain and Potential Therapeutic Targets
- III. Current Therapeutic Management of Neuropathic Pain: Focus on Gabapentinoids
- IV. Basic Science of Gabapentinoid Action and Observations in the Clinic
- V. Conclusions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters