


Abstract
Central nervous system (CNS) barriers predominantly mediate the immune-privileged status of the brain, and are also important regulators of neuroimmune communication. It is increasingly appreciated that communication between the brain and immune system contributes to physiologic processes, adaptive responses, and disease states. In this review, we discuss the highly specialized features of brain barriers that regulate neuroimmune communication in health and disease. In section I, we discuss the concept of immune privilege, provide working definitions of brain barriers, and outline the historical work that contributed to the understanding of CNS barrier functions. In section II, we discuss the unique anatomic, cellular, and molecular characteristics of the vascular blood–brain barrier (BBB), blood–cerebrospinal fluid barrier, and tanycytic barriers that confer their functions as neuroimmune interfaces. In section III, we consider BBB-mediated neuroimmune functions and interactions categorized as five neuroimmune axes: disruption, responses to immune stimuli, uptake and transport of immunoactive substances, immune cell trafficking, and secretions of immunoactive substances. In section IV, we discuss neuroimmune functions of CNS barriers in physiologic and disease states, as well as pharmacological interventions for CNS diseases. Throughout this review, we highlight many recent advances that have contributed to the modern understanding of CNS barriers and their interface functions.
I. Introduction
A. The Blood–Brain Barrier and Immune Privilege
The central nervous system (CNS) has traditionally been viewed as an immune-privileged area in that it is protected against the immune events of the periphery. This immune privilege was once considered absolute with its violation only occurring in disease states, resulting in dire consequences for the CNS. Current appreciation is that the immune privilege is real, but relative. Both past and current thinking ascribes this immune privilege to the BBB. By its ability to prevent (past thinking) or to control and modulate (current thinking) the impact of peripheral immune events on the CNS, the BBB acts to protect the brain from peripheral immune events. However, whereas the barrier aspects of the BBB induce immune privilege, it is also the BBB that makes such protection relative. The BBB makes immune privilege relative by possessing ways in which it controls the interplay of CNS and peripheral immune events. Such interplay relies on the transfer of immune elements (substances or cells) between the CNS and blood; such a transfer can be termed a neuroimmune axis. Five neuroimmune axes can be currently identified that involve the BBB and a sixth that does not but is mediated by afferent and efferent nerve activities (Goehler et al., 1999; Romeo et al., 2001; Kelley et al., 2003; Kenney and Ganta, 2014; Kanashiro et al., 2016). The five pathways involving the BBB are as follows: 1) BBB disruption; 2) modulation of barrier and interface functions (other than BBB integrity) by immune substances; 3) transport, penetration, and uptake of neuroimmune-related substances; 4) immune cell trafficking between blood and brain; and 5) immune secretions of the barrier cells. Most likely, these axes work together rather than independently, and it is one of the great promises of the field, as well as one of its greatest challenges, that it will elucidate the mechanisms of neuroimmune integration that underlie such diverse phenomena as sleep, responses to sepsis, mindfulness, and depression. Below, we consider each of the five axes involving the BBB.
This review will first consider the concepts, components, functions, and interactions that form the basis of the BBB field that are germane to neuroimmunology, then discuss how the BBB defines and participates in the five known neuroimmune axes, and finally discuss how the BBB in general and how these axes in particular are involved in CNS diseases, drug delivery, and therapy.
B. Working Definitions of Brain Barriers and Interfaces
BBB is a term that refers to one function of a highly specialized cellular interface between the blood and the CNS parenchyma. The barrier function of this interface prevents unregulated diffusion of circulating substances into the brain, which is critical for maintaining a CNS milieu that supports neuronal function and survival. Generally, the BBB refers to the vascular barrier where capillary endothelial cells are the interface between the blood and brain parenchyma. Specialized vascular CNS barriers that have distinct features from brain parenchymal vessels include the blood–retinal barrier, the blood–nerve barrier, the blood–labyrinth barriers, and the blood–spinal cord barrier (Neuwelt et al., 2008). Other cellular barriers/interfaces such as the choroid plexus epithelial cells of the blood–cerebrospinal fluid (CSF) barrier (BCSFB) and tanycytes that are located along the ventricular boundaries of circumventricular organs (CVOs) may be considered arms of the BBB as well because they also prevent unregulated leakage of blood components into the CSF and adjacent brain interstitial fluid (ISF) (Ghersi-Egea et al., 1996). Furthermore, these interfaces have many nonbarrier functions that are essential in supporting CNS homeostasis. The nonbarrier functions include regulating the transport of circulating substances into the brain, removing potentially harmful substances from the brain, secreting molecules that signal to cells in the brain parenchyma, and responding to stimuli that arise within both the brain and blood compartments. In the remainder of this section, we will provide an overview of the historical work that defined the barrier functions of brain interfaces, discuss aspects of their barrier and interface functions that contribute to CNS homeostasis, and relate each blood–brain interface to one another with regard to their unique structures and functions in neuroimmune communication.
C. Historical Work: Defining the Brain Barriers
Evidence in support of a BBB dates back to works published in the late 19th/early 20th century. At the time, the field of medicinal chemistry was in its infancy, and Paul Ehrlich, who would later make many seminal contributions to the field (Bosch and Rosich, 2008), was investigating the selectivity of dyes for different cells and tissues. As part of this work, Ehrlich (1885) noted that certain water-soluble dyes that were parenterally injected into animals stained peripheral tissues and the choroid plexus, but did not stain the brain or spinal cord parenchyma. Ehrlich (1906) posited that the reduced CNS staining was due to a low affinity of the dyes for CNS tissue. However, other groups who were investigating bile acids (Biedl and Kraus, 1898) and sodium ferrocyanide (Lewandowski, 1900) found that the toxicities of these substances were much more potent when introduced directly into the cerebrospinal fluid versus systemically. Both groups interpreted their findings to mean that brain capillaries had unique properties that blocked the transfer of certain molecules from blood to brain. Goldmann (1909, 1913) later conducted critical experiments showing that Ehrlich’s trypan blue dye, which did not bind CNS tissue when injected parenterally, did stain CNS tissue when injected into the CSF. This finding demonstrated that exclusion of trypan blue from the CNS was not due to reduced binding affinity to CNS tissue, which had been suggested by Ehrlich (1906). However, Goldmann (1913) supposed in this work that the choroid plexus was the predominant barrier site of the CNS, supplying nutrients to the CNS in a fashion analogous to the placenta. The term BBB (barrière hémato-encéphalique) was first used in a publication by Stern and Gautier (1921); although many attribute the first use of this term (Blut-Hirnschranke) to Lewandowski, this term was not used in his original publication (Saunders et al., 2014).
Following these early studies, controversy surrounded the true nature of the BBB. Early ultrastructural studies of the brain using electron microscopy used methods of tissue preservation that led to the belief that the CNS had essentially no extracellular space (Bradbury, 2000). Therefore, some adopted the belief that the limited CNS penetration of aqueous solutes was due to tightly packed neuronal and glial membranes and a lack of aqueous medium for diffusion (Davson and Spaziani, 1959; Bradbury, 2000). However, it was then demonstrated that extracellular markers injected in ventricular CSF did penetrate the brain and spinal cord, and therefore, CNS tissue contained extracellular fluid permissive to solute diffusion (Davson and Segal, 1969). Based on these findings, it was posited that a BBB to such solutes injected in blood must exist (Davson et al., c). Later that decade, Vanharreveld et al. (1965) showed that the brain extracellular space could be visualized by electron microscopy when modified techniques were used for tissue preservation. This report was followed by the eminent findings of Reese and Karnovsky (1967), who used electron microscopy to explore the subcellular features of brain capillaries. They found that the brain’s limited uptake of peroxidase, which had been shown previously (Straus, 1958), could be attributed to two specialized features of the brain endothelium: uniquely impermeant tight junctions (TJs) that were present at contacts between capillary membranes, and markedly reduced endothelial vesicles (Reese and Karnovsky, 1967). In a later study, Brightman and Reese (1969) explored the distribution of peroxidase injected into CSF of mice, chickens, and goldfish to determine which cell types of the CNS expressed peroxidase-impermeant TJs. Although peroxidase did diffuse through gap junctions that were present at contacts of astrocytic endfeet, it did not permeate the junctions between brain endothelial cells (BECs) or choroid plexus ependymal cells. This work highlighted that TJs were a unique feature of BECs, as well as epithelial cells of the choroid plexus that conferred barrier properties (Brightman and Reese, 1969). The tight barriers of the brain vasculature were further exemplified by Crone and Olesen (1982) and by Butt et al. (1990), who showed that brain vessels have exceptionally low ion permeabilities due to very high transendothelial electrical resistance (TEER), averaging over 1000 Ω/cm2 in frog and mammalian pial vessels. As pial vessels lack astrocytes, TEER is estimated to be much higher in brain parenchymal vessels that are ensheathed by astrocyte endfeet, which contribute to the BBB phenotype (Abbott et al., 2006). By determining the CNS permeability/surface area coefficients for K, Na, and Cl in vivo, Smith and Rapoport (1986) estimated TEER of the parenchymal vessels to be approximately 8000 Ω/cm2, which compares to that of a cell membrane.
II. Features and Functions of the Blood–Brain Barrier and Blood–Brain Interface
A. Specialized Features That Confer Barrier Functions
Barrier functions are largely thought of as those related to the exclusion of blood-borne substances from the brain. Such exclusion is critical to the CNS being an immune-privileged tissue. The specialized features that underlie those functions include TJs, mechanisms that limit macropinocytosis, efflux mechanisms, and enzymatic activities.
1. Tight Junctions
TJs are macromolecular complexes that prevent the paracellular diffusion of solutes across brain barriers. They communicate with adherens junctions (AJs), which are another junctional complex that contributes to the tightness of the barrier (Wolburg and Lippoldt, 2002). AJs are located at the basal membrane and are comprised of cadherins, which associate with each other in the extracellular space, and catenins, which link the cadherins to the cytoskeleton (Huber et al., 2001). TJ proteins at the vascular BBB include occludin, claudins, junctional adhesion molecules (JAMs), zonula occludens (ZO), and cytoplasmic accessory proteins such as cingulin (Hawkins and Davis, 2005). Occludins, claudins, and JAMs are membrane-spanning proteins that heterotypically interact in the intercellular space between adjacent capillary membranes. ZOs and cingulin are adaptor proteins that link TJ membrane proteins to the cytoskeleton and contribute to junction stability (Abbott et al., 2010). TJ stability is regulated by calcium, phosphorylation, cyclic AMP, and G proteins (Huber et al., 2001), and decreases in TJ expression as well as TJ mislocalization and post-translational modifications can cause increases in paracellular permeability of solutes across the BBB (Deli et al., 2005). Importantly, TJs are not only a diffusion barrier for transcellular passage of circulating substances into the CNS, but also for the lateral diffusion of membrane proteins (Abbott et al., 2010). Therefore, TJs also contribute to the membrane polarity of BECs. Later in this review, we will compare TJ organization in the different arms of the BBB as well as the contributions of TJs to the neuroimmune axes of the BBB.
2. Reduced Macropinocytosis
Reese and Karnovsky (1967) were the first to note that BECs had relatively low numbers of vesicles compared with endothelial cells in the periphery. In contrast, epithelial cells of the choroid plexus do have vesicles that largely reside at their apical membranes (Johanson et al., 2011). Recent work has begun to identify the molecular mechanisms that suppress brain endothelial pinocytic vesicle formation. In a study that aimed to characterize gestational development of the BBB, Ben-Zvi et al. (2014) found that the BBB tightening that occurred around E15.5 in mice was associated with upregulation of major facilitator superfamily domain-containing 2A (MFSD2A) in BECs. In the same study, it was shown that MFSD2A knockout mice had a leaky BBB, although the morphology of the brain vasculature and TJs did not appear to be affected. Instead, MFSD2A knockout mice had increased luminal, abluminal, and cytoplasmic vesicles in their BECs, which facilitated the fluid-phase uptake of solutes such as peroxidase and dextrans, which are usually excluded from the CNS. In a parallel study, it was realized that MFSD2A knockout mice had cognitive and behavioral symptoms that resembled omega-3 fatty-acid deficiency (Nguyen et al., 2014). Using a lipidomics approach, this group revealed that docosahexaenoic acid (DHA), an omega-3 fatty acid that is important for CNS development and cognition, was reduced in the CNS of mice lacking MFSD2A. They further demonstrated that MFSD2A was a transporter for lysophosphatidylcholine (LPC)-DHA, as well as LPC-oleate and LPC-palmitate at slightly lower capacities (Nguyen et al., 2014). Andreone et al. (2017) then showed that the DHA-transporting function of MFSD2A also facilitated DHA enrichment of BEC membranes, which inhibited caveolin-1–induced formation of vesicles. In mice lacking MFSD2A, caveolin-1 knockout inhibited the increased formation of brain endothelial vesicles as well as leakage of the BBB (Andreone et al., 2017). Therefore, BECs acquire a specialized lipid composition during embryonic development that inhibits caveolae-mediated fluid-phase transcytosis.
3. Efflux Transporters
Efflux transporters at brain barriers facilitate the passage of substances in the brain-to-blood or CSF-to-blood direction. A subset of efflux transporters, namely members of the ATP-binding cassette (ABC) protein family, confers barrier functions by limiting the brain uptake of endogenous macromolecules and xenobiotics. ABC transporter subtypes, their localization, and functions in the CNS have been extensively reviewed elsewhere (Hartz and Bauer, 2011). Some of the most studied ABC transporters at the BBB include P-glycoprotein (Pgp/ABCB1), multidrug resistance protein (MRPs/ABCC), and breast cancer resistance protein (BCRP/ABCG2) (Qosa et al., 2015). ABC transporters have a broad substrate spectrum, including some phospholipids, sphingolipids, aldosterone, and amyloid β (Aβ) for Pgp; glutathione, glutathione-conjugated leukotrienes and prostaglandins, and glucoronidation and sulfation products for MRP-1; and bile acids and estrones for BCRP (Qosa et al., 2015). ABC transporters also efflux a broad range of xenobiotic substrates, including opioids, antibiotics, antiretroviral drugs, chemotherapeutics, and others (Qosa et al., 2015). The expression and function of efflux transporters at the BBB are dynamically regulated, for example, at the transcriptional level by nuclear receptors (Chan et al., 2013), and at post-translational levels by vascular endothelial growth factor (VEGF) (Hawkins et al., 2010). Aspects of ABC transporter modulation regarding immune-regulated functions are discussed in greater detail later in this review.
4. Metabolic Enzymes
The BBB is also an enzymatic barrier, and expresses phase I and phase II enzymes that contribute to the metabolism and elimination of biomolecules and drugs from the body. BECs contain monoamine oxidase and catechol-O-methyltransferase, which metabolize adrenaline, noradrenaline, and dopamine, as well as 4-aminobutyrate aminotransferase, which metabolizes GABA (Lasbennes et al., 1983; Spatz et al., 1986). Therefore, enzymatic barriers inhibit the transport of many neurotransmitters in the blood-to-brain and brain-to-blood directions. Furthermore, the toxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine when systemically administered is inversely associated with monoamine oxidase expression in BECs (Kalaria et al., 1987; Riachi and Harik, 1988). The cytochrome P450 enzyme CYP1B1 is expressed in human BECs (Dauchy et al., 2008; Shawahna et al., 2011), and its expression can be regulated by environmental toxicants such as 2,3,7,8-tetrachlorodibenzo-p-dioxin through the aryl hydrocarbon receptor (Jacob et al., 2015). Glutathione S-transferases are also expressed in human brain capillaries (Shawahna et al., 2011).
B. Specialized Features That Confer Interface Functions
Brain barriers do much more than divide the CNS from the peripheral circulation. They are also critical in CNS homeostasis, nutrition, and brain–body communication. These features are essential to the existence of some of the neuroimmune axes. Features that aid in these functions include transcellular diffusion and transport via solute carriers, receptor-mediated transcytosis, and adsorptive endocytosis.
1. Transcellular Diffusion
Early work by Davson demonstrated that lipid solubility and size determined partitioning of substances from blood into CSF and brain tissue (Davson, 1955; Davson and Smith, 1957). It is now appreciated that small, lipophilic molecules can cross the BBB by passive transmembrane diffusion. Passive transmembrane diffusion is a nonsaturable mechanism by which most drugs that can enter the CNS do so. The degree to which lipid-soluble compounds may cross the BBB is often determined by their partitioning into aqueous versus nonpolar medium, such as water and octanol (Oldendorf, 1974; Levin, 1980). However, as substances that passively diffuse across the BBB must traverse the luminal membrane, cytosol, and then the abluminal membrane prior to reaching the CNS, there is a limit to the lipophilicity of a substance that favors diffusion across the BBB versus sequestration within the cell membrane (Banks, 2016). Transcellular diffusion of substances across the BBB is also affected when the test substance is an efflux transporter substrate. In this case, CNS uptake is much lower than what would be predicted based on size and lipophilicity.
2. Blood–Brain Barrier Transport via Solute Carriers
Solute carrier proteins are integral membrane proteins that permit the directional or facilitated diffusion of aqueous molecules across cell membranes. The initial studies that identified carrier-mediated transporters at the BBB investigated nutritional substrates of the brain. D-glucose was the first substance demonstrated to have a saturable transport system (Crone, 1965). In this study, Crone used an indicator diffusion technique (IDT) to quantify first-pass uptake of glucose by the brain. The IDT involves coinjecting a radioactive test substance and a capillary-impermeant tracer (e.g., Evan’s blue albumin), which estimates the dilution of the injected substance in blood. The substances are injected into the carotid artery, and then venous blood from the superior sagittal sinus is immediately sampled to determine the percent loss of injected substance. Crone used the IDT to assess the brain uptake of D-glucose during hypo- and hyperglycemic states, and found that CNS uptake of the glucose tracer was highest when blood glucose was low, and lowest when blood glucose was high, indicating that glucose used a saturable transport system (Crone, 1965). Subsequently, Oldendorf (1971) used a different technique to assess the brain uptake of glucose and amino acids. The method, called the brain uptake index (BUI), is carried out by coinjecting a highly brain-penetrant radioactive standard along with a radioactive test substance into the carotid artery, and then immediately removing and counting brain tissue for uptake of the test substance with reference to the standard. The BUI is therefore also a first-pass measurement of brain uptake. In support of the findings by Crone (1965), Oldendorf’s BUI method also demonstrated saturable transport of glucose, as well as amino acids. Importantly, Oldendorf (1971) also conducted cross-inhibition studies and found that amino acids with similar chemical properties often used a common transporter. It is now understood that there are three broad classes of amino acid transporters with many subtypes in each class (Hawkins et al., 2006; Abbott et al., 2010). Glucose transport across the BBB is mediated by GLUT1 (Dick et al., 1984; Pardridge et al., 1990; Boado and Pardridge, 1994), and GLUT1 expression is considered to be a hallmark of BBB endothelial cells (Cornford et al., 1993, 1994), as its expression is absent from endothelial cells within brain regions that lack an endothelial BBB (Rahner-Welsch et al., 1995). In addition to glucose and amino acids, solute carriers transport nucleosides, ions, prostaglandins, and many other small polar molecules (Bito et al., 1976; Abbott et al., 2010). Therefore, one function of solute carriers is to provide the brain with the essential circulating substrates for energy generation, protein and nucleic acid synthesis, and maintenance of pH and electrolytes. At the choroid plexus epithelium, solute carriers are necessary for the production of CSF from plasma ultrafiltrate (Johanson et al., 2011). Solute carrier proteins may be present on the luminal and/or abluminal membrane of brain endothelial and epithelial barrier cells, and the directionality of their transport may be in the blood-to-brain or brain-to-blood direction, depending on their orientation within the membrane (Abbott et al., 2010).
3. Blood–Brain Barrier Transport via Receptor-Mediated Transcytosis
Another mechanism of transport across the BBB is receptor-mediated transcytosis (RMT), which is thought to be the predominant mode of transport of larger macromolecules such as peptides and proteins across the BBB (Bickel et al., 2001). RMT is energy dependent, saturable, and depends on vesicular pathways. RMT may be in the blood-to-brain direction, or brain-to-blood direction. For ligands to completely cross the BBB via RMT, they must first bind their transporter at either the luminal or abluminal endothelial membrane. Second, they must be internalized in a vesicle, which may be clathrin or cavaeolae dependent (Georgieva et al., 2014). Third, they must be routed from the luminal to abluminal membrane or vice versa, which may involve subcellular trafficking through organelles such as endosomes, or the Golgi (Bickel et al., 2001). Furthermore, ligands must escape vesicular routing to and degradation by the lysosome. Ligands may become dissociated from their receptors during subcellular routing, due to the slightly acidic pH of the endosome (Bickel et al., 2001). Fourth, the ligand must be exocytosed to the opposite side of the membrane and released into brain interstitial fluid (Bickel et al., 2001). This final step also requires the dissociation of the ligand from its transporter. RMT at the BBB can be regulated at the level of transporter expression, localization, and conformation, as well as by concentrations of other molecules that might compete with the ligand for transport, or that may sequester the ligand from interacting with its transporter at the BBB.
4. Blood–Brain Barrier Transport via Adsorptive Transcytosis
Adsorptive endocytosis is a receptor-independent mode of vesicular transport across the BBB and involves interactions of cationic protein residues with the anionic glycocalyx, which lines the lumial surface of endothelial cells, or membrane glycoproteins (Broadwell et al., 1988; Villegas and Broadwell, 1993). Conferring a positive charge to proteins that typically do not cross the BBB, such as albumin (Griffin and Giffels, 1982; Kumagai et al., 1987), enhances their uptake by BECs by adsorptive mechanisms. Adsorptive endocytosis may be saturable (Kumagai et al., 1987), but may also be induced by compounds such as wheat germ agglutinin and the human immunodeficiency virus (HIV) coat proteins TAT and gp120 (Mann and Frankel, 1991; Banks et al., 1998a). Adsorptive endocytosis increases in vitro following lipopolysaccharide (LPS) or cytokine treatment (Schenk and de Vries, 2016).
C. From Brain Barriers to Brain Interfaces: Components of the Neurovascular Unit
Brain barriers are uniquely poised to communicate signals between the CNS and peripheral compartments. Communication is not only achieved through transporters, but also from molecules that are produced and secreted by cells of the brain interfaces. These secreted substances can engage autocrine targets, and/or signal to other cells of the neurovascular unit (NVU), and distal cells in the brain and periphery. BBB secretions may be constitutive or inducible, and are also polarized in that they may be released into either blood or brain compartments (Banks, 2016). Finally, cells of the BBB respond to signals that arise from the CNS or blood compartments, which may stimulate alterations in their barrier, transport, and secretory functions (Verma et al., 2006; Krasnow et al., 2017). How these interface functions contribute to the neuroimmunomodulatory activities of brain barriers will be discussed in section II of this review. First, we consider the individual components and their functions (Fig. 1).

The neurovascular unit. The BBB is in contact and communicates with other cells of the CNS as well as circulating immune cells and peripheral tissues through the endocrine-like secretions of the latter. Differences occur in NVU function regionally as well as among the anatomic areas in which barrier cells are located. As an example of the latter, immune cell trafficking occurs largely at the postcapillary venule. Endothelial cells, astrocyes, pericytes, neurons, and macrophages/microglia, as well as the extracellular matrix and glycocalyx are part of the NVU. There is renewed interest in mast cell functions, and the cellulis incompertus represents cell types yet to be discovered that participate in the NVU. Not drawn to scale.
1. Endothelial Cells
In addition to their barrier, transport, and interface functions, BECs contribute to the specialized phenotypes of other cells of the NVU. Endothelial cells induce astrocyte differentiation in vitro via leukemia-inhibitory factor production (Mi et al., 2001). They influence the localization of the water channel aquaporin 4 on the plasma membrane of astrocyte endfeet and stimulate the upregulation of antioxidant enzymes within astrocytes (Abbott, 2002). Endothelial cells secrete factors such as transforming growth factor (TGF)-β and platelet-derived growth factor subunit B (PDGF-B) and signal through Tie2 and sphingosine-1 phosphate, which maintain pericyte functions (Armulik et al., 2005). Neuroimmune functions of BECs are discussed extensively in later sections.
2. Brain Pericytes
Pericytes have important functions in the development and maintenance of the vascular BBB. Of the cells of the NVU, pericytes are the most closely apposed to capillary endothelial cells; they share a basement membrane and make direct contact with BECs via peg and socket as well as gap junctions (Dore-Duffy and Cleary, 2011). Brain pericytes are derived from the mesoderm and neuroectoderm (Winkler et al., 2011) and undergo proliferative expansion and recruitment to the developing neurovasculature during embryonic development and the early postnatal period (Daneman et al., 2010). Pericyte attachment to BECs during embryonic development facilitates BBB tightening by downregulating genes that are associated with pinocytic vesicle formation and immune cell recruitment (Daneman et al., 2010; Ben-Zvi et al., 2014). PDGF-B produced by brain capillaries signals to platelet-derived growth factor receptor β (PDGFRβ) on brain pericytes and regulates pericyte proliferation, attachment to endothelial cells, and survival. The absence of PDGF-B or PDGFRβ is lethal in mice (Leveen et al., 1994; Kaminski et al., 2001), whereas mice with partial PDGF-B or PDGFRβ deficiency survive into adulthood, but have reductions in capillary-associated pericytes (Armulik et al., 2010; Bell et al., 2010; Daneman et al., 2010). Pericyte deficiency induced by a PDGF-B mutation results in leakage of intravascular markers of different sizes into the CNS, indicative of BBB disruption. Astrocyte associations with capillaries were also altered in this model; however, TJ protein expression and localization were relatively unaffected (Armulik et al., 2010). Mice lacking one copy of PDGFRβ have an age-dependent loss in pericytes of about 20% by 1 month of age, and 60% by 14–16 months (Bell et al., 2010). BBB disruption is evident by 1 month and worsens with age. In this model, synaptic deficits and impaired learning and memory are evident by 6–8 months, but precede neuroinflammation, which does not significantly increase until 14–16 months of age. Pericytes are also important for the induction of the BBB phenotype in vitro, as pericyte coculture with BECs increases the integrity of the barrier (Nakagawa et al., 2007).
Pericytes also have dynamic functions in the NVU. Pericytes are multipotent stem cells that can differentiate into cells of neural lineage (Dore-Duffy et al., 2006). They may also adopt a contractile phenotype that contributes to the regulation of cerebral blood flow (Hall et al., 2014). Pericytes contribute to the neuroimmune response and are potent modulators of BBB function due to their proximity to endothelial cells. Pericytes secrete cytokines and chemokines constitutively in culture and upregulate cytokine and nitric oxide production in response to LPS (Fabry et al., 1993; Kovac et al., 2011). They present antigen in response to interferon (IFN)-γ, which may contribute to T-cell activation (Balabanov et al., 1999). They also enhance the transcytosis of HIV-1 free virus and neutrophils across in vitro BEC monolayers in the presence of an immune stimulus (Dohgu and Banks, 2013; Pieper et al., 2013). In response to injuries that are associated with neuroinflammation such as hypoxia (Gonul et al., 2002) and traumatic brain injury (Dore-Duffy et al., 2000), pericytes dissociate from the brain vasculature and migrate away from the vessels within 1–2 hours following the insult (Dore-Duffy et al., 2000; Gonul et al., 2002). In contrast, a systemic inflammatory insult such as intraperitoneal LPS results in pericyte detachment from the basal lamina between 6 and 24 hours, which coincides with reactive microgliosis and BBB disruption (Nishioku et al., 2009). Pericytes that leave the basement membrane and enter brain parenchyma have been reported to adopt a phenotype similar to that of infiltrating macrophages (Guillemin and Brew, 2004). In summary, pericytes may contribute to the neuroimmune response as follows: 1) causing a leaky BBB, either by secreting endothelial-disrupting factors, or by physical disassociation; 2) facilitating the transport of immune cells and pathogens into the brain; and 3) propagating neuroinflammation by stimulating both resident and recruited immune cells.
3. Astrocytes
Astrocytes are the most abundant brain cell type and regulate a number of physiologic processes in the CNS that include neurotransmission, synaptic plasticity, functional hyperemia, and convective flow of brain interstitial fluid (Sofroniew and Vinters, 2010). Astrocytes are also integral in the induction and maintenance of the mature BBB phenotype (Abbott et al., 2006). Their endfeet surround brain capillaries, arterioles, and venules. At capillaries, the astrocytic endfeet are located on the CNS side of the basement membrane that ensheaths the endothelial cells and pericytes (Abbott et al., 2006). These endfeet are in close proximity to the endothelial cells (Thal, 2009) and therefore positioned for crosstalk that promotes the phenotypic specialization of both cell types. The contribution of astrocytes to BBB formation during embryonic development is thought to be negligible in rodents, as astrocytes appear immediately after birth and do not begin to ensheath brain vessels until the first postnatal week (Daneman et al., 2010). In contrast, radial glia, which are precursors for neurons and astrocytes, do form endfeet around capillaries in fetal baboons and humans (Bass et al., 1992; Bertossi et al., 1999). Therefore, there may be species differences in the contribution of astrocytes or their precursors to the embryonic BBB. Astrocytes do contribute to BBB functions during postnatal development and throughout adulthood. In vitro, astrocytes strengthen the barrier properties of BECs and also enhance expression of BBB transporters, such as Pgp and Glut1, and enzymes of the metabolic barrier (Abbott et al., 2006). The close proximity of astrocytes and endothelial cells at capillaries likely favors BBB tightening, as in vitro studies have demonstrated that in vitro BEC barriers are tightest when astrocytic processes contact the endothelial monolayer (Abbott, 2002). However, secreted factors from astrocytes also contribute to the BBB phenotype. BECs likewise contribute to the organization of aquaporin 4 and potassium channels on astrocytic endfeet (Abbott, 2002), which regulate water and ion exchange in the CNS (Stokum et al., 2015). Therefore, communication between endothelial cells and astrocytes is important for both the barrier and interface functions of BECs.
Astrocytes are immune-active cells and were the first cell type in the CNS shown to express class II major histocompatibility complex upon IFN-γ stimulation in vitro (Wong et al., 1984). More recent work has reviewed aspects of astrocyte responses to systemic inflammation; additionally, CNS injury in the context of novel subsets of reactive astrocytes and their functions are beginning to be characterized (Liddelow and Barres, 2017). In response to systemic inflammatory insults such as sepsis, astrocytes upregulate their production of proinflammatory cytokines and chemokines, as well as VEGF (Bellaver et al., 2017). Inflammatory lesions and interleukin (IL)-1β can induce astrocyte production of VEGF-A and thymidine phosphorylase, which downregulate TJ protein expression in BECs (Chapouly et al., 2015). Astrocytes also upregulate cytokines and chemokines in response to proinflammatory stimuli and in disease states (Dong and Benveniste, 2001; Norden et al., 2016). Astrocytic inflammatory responses are differently regulated in comparison with those of microglia; for example, astrogliosis following induction of systemic inflammation is inhibited by indomethacin, whereas microgliosis is not (Banks et al., 2015). Furthermore, in response to a systemic inflammatory insult, astrocytes adopt a delayed proinflammatory phenotype in comparison with that of microglia (Norden et al., 2016). Astrocytes may also function in the resolution of neuroinflammation, as they can downregulate microglial activation by secreting TGF-β (Vincent et al., 1997).
4. Neurons
The brain is extensively vascularized—the mean distance of a neuronal cell body to a capillary is approximately 15 μm in mice (Tsai et al., 2009) and 30 µM in nonhuman primates (Mabuchi et al., 2005). Therefore, each neuron receives and can regulate its own blood supply from an adjacent capillary. Neurons regulate their blood supply through communication with astrocytes, which facilitate dilation of arterioles in response to neuronal glutamate release (Zonta et al., 2003). Some studies suggest that capillary pericytes contribute to functional hyperemia; however, the relative contribution of pericytes versus mural cells of arterioles to neurovascular coupling has been disputed (Fernandez-Klett et al., 2010; Winkler et al., 2011; Fernandez-Klett and Priller, 2015). Neuronal activity also contributes to the NVU architecture by promoting neurovascular density and branching during adulthood (Lacoste et al., 2014), but, during neonatal development, excessive sensorimotor stimulation and repetitive neural activation result in reduced microvascular density (Whiteus et al., 2014).
Neurons exhibit pleiotropic responses to inflammatory stimuli. Cytokines such as tumor necrosis factor (TNF)-α, via its interaction with astrocytes, and fractalkine, which is expressed by neurons and activates the microglia fractalkine receptor, can both stimulate synaptic activity (Prieto and Cotman, 2017). Furthermore, cytokines such as IL-1β, IL-6, and IL-18 are upregulated in the brain following long-term potentiation (LTP) induction in awake rats (del Rey et al., 2013). IL-1β at physiologic concentrations promotes LTP, but at higher concentrations can inhibit LTP and impair learning and memory (Ross et al., 2003; Prieto et al., 2015). This function of IL-1β may be potentiated with aging (Prieto et al., 2015). TNF-α is not required for learning and memory, but its overexpression by glia can impair memory and synaptic plasticity (Donzis and Tronson, 2014).
5. Microglia and Perivascular Macrophages
Microglia are resident macrophages of the CNS and rapidly respond to CNS insults. Their lineage is unique from recruited brain macrophages, in that microglia derive from the yolk sac during development, whereas recruited brain macrophages derive from bone marrow (Alliot et al., 1999). During development, microglia associate with the brain vasculature and contribute to angiogenesis (Arnold and Betsholtz, 2013). In adult brains, microglia remain closely associated with the neurovasculature. Their production of proinflammatory mediators such as cytokines, chemokines, nitric oxide, prostaglandins, matrix proteases, and reactive oxygen species can have profound effects on cells of the NVU and BBB integrity (da Fonseca et al., 2014). There are also populations of brain macrophages that reside in the perivascular space and are thought to derive from bone marrow (Hickey and Kimura, 1988), although more recent works suggest that their recruitment to the brain under physiologic conditions is rare (Prinz et al., 2011). The perivascular macrophages are thought to protect the brain during infection (Polfliet et al., 2001) and prevent deposition of protein aggregates such as Aβ peptide within the perivascular space (Lai and McLaurin, 2012). However, perivascular macrophages may also contribute to neurovascular pathologies associated with increases in Aβ peptide levels in the brain (Park et al., 2017). An important protective function of microglia is their ability to rapidly migrate to sites of brain injury and alter their morphology to form a specialized phagocytic network that prevents diffusion of harmful substances into the brain parenchyma (Roth et al., 2014). Microglia also contribute to the resolution of inflammation in the brain after injury (Cherry et al., 2014).
6. Mast Cells
Mast cells are granulocytes that originate from bone marrow and circulate as precursor cells. Upon recruitment to tissues, mast cells complete their differentiation according to their local environment (Silver and Curley, 2013). Mast cells can be recruited to the CNS (Silverman et al., 2000; Nautiyal et al., 2011), where they reside in perivascular spaces of some brain regions, as well as in the choroid plexus and meninges (Silver and Curley, 2013). Mast cells are important mediators of peripheral IgE-mediated allergic responses; however, they also have emerging functions in the CNS. Granules of mast cells contain bioactive mediators that include histamine, serotonin, serine proteases, and heparin. Mast cells can also synthesize prostaglandins, cytokines, growth factors such as nerve growth factor, reactive oxygen species, and substance P in response to stimuli (Silver and Curley, 2013). Therefore, mast cells likely play important roles in regulating neurotransmission as well as BBB function. Induction of mast cell degranulation causes BBB disruption that is localized to brain regions enriched in mast cells, such as the medial habenula in doves (Zhuang et al., 1996). Mast cells are thought to contribute to CNS dysfunction in conditions such as stress (Theoharides et al., 1995), postoperative cognitive dysfunction (Zhang et al., 2016), and rodent models of multiple sclerosis (Costanza et al., 2012). However, mast cells may also regulate physiologic aspects of behavior, as mice that lack functional mast cells in the CNS display increased anxiety-like behaviors (Nautiyal et al., 2008). Mast cells are also a predominant source of brain histamine (Goldschmidt et al., 1985). Serotonin derived from mast cells has been implicated in promoting hippocampal neurogenesis and learning and memory (Nautiyal et al., 2012).
7. Extracellular Matrix
Within the NVU, basement membranes composed of extracellular matrix (ECM) are present between endothelial cells and pericytes, and also along the astrocytic endfeet (McConnell et al., 2017). Microvascular basement membranes are composed of laminins, collagen IV, fibronectin, glycosaminoglycans including hyaluronan, chondroitin sulfate-rich proteoglycans, and glycoproteins that contribute to the resiliency of the microvasculature (del Zoppo and Mabuchi, 2003; Lennon and Singleton, 2011; Reed et al., 2017). The ECM functions as a cellular scaffold that is generated during development, and endothelial adhesion to the matrix is mediated by integrins (del Zoppo and Mabuchi, 2003). Integrins are unique receptors that can respond to and relay both intracellular and extracellular signals (Shen et al., 2012). Blocking the function of β1 integrin in BECs reduces the expression of the TJ protein claudin 5 and causes BBB disruption (Osada et al., 2011). The ECM is also a barrier to leukocyte and erythrocyte entry into brain parenchyma during inflammation and hemorrhage (del Zoppo and Mabuchi, 2003; del Zoppo, 2009). Components of the ECM can be degraded by matrix metalloproteinases (MMPs), which contribute to BBB disruption and leukocyte trafficking during neuroinflammation (Rosenberg, 2002). MMPs and their modulatory effects on the BBB have recently been reviewed elsewhere (Rempe et al., 2016). Hyaluronan and its fragments bind to Toll-like receptors, influencing the neuroimmune environment (Jiang et al., 2011).
8. Glycocalyx
The glycocalyx lines the luminal surface of endothelial cells, including those of the brain. It is a gel-like layer estimated to be approximately 5 μm thick, and is predominantly composed of heparin sulfate proteoglycan, chondroitin sulfate, hyaluronan, and glycoproteins (Kolářová et al., 2014). The glycocalyx begins to form during brain neovascularization during early embryonic development and matures postnatally (Vorbrodt et al., 1990). The glycocalyx has important barrier functions in preventing direct exposure of plasma components to the endothelial luminal membrane surface (Vorbrodt, 1989), and it also functions as a mechanosensor and relays signals of sheer stress to the endothelium (Tarbell, 2010). Degradation of the glycocalyx occurs during inflammation, which is associated with increased passage of solutes across the endothelial barrier, and increased leukocyte adhesion to the endothelium (Kolářová et al., 2014; Varatharaj and Galea, 2017).
D. Arms of the Blood–Brain Barrier and Their Neuroimmune Functions
The barriers formed by the components above and the resulting mechanisms by which they form neuroimmune axes can be categorized into three main arms: the vascular BBB, the choroid plexus, and the tanycytic barrier. The unique cellular and anatomic features of these barriers with reference to their neuroimmune functions are discussed below.
1. The Vascular Blood–Brain Barrier
The vascular BBB is a broadly applied term that most often refers to the capillaries within the brain parenchyma. However, vascular BBBs extend to pre-and postcapillary arterioles and venules, respectively (Bechmann et al., 2007). Vascular BBBs are also present in the spinal cord, retina, nerves, and the inner ear, and the structural and functional organization of these has been reviewed elsewhere (Choi and Kim, 2008). Brain capillaries exhibit functional heterogeneity within different anatomic locations, as certain brain regions are more vulnerable to disruption during neuroinflammation (Banks et al., 2015), and transport rates of immunoactive substrates also vary depending on brain region (Moinuddin et al., 2000; Banks et al., 2001c; Erickson et al., 2014). Therefore, the heterogeneity of the vascular BBB imparts some anatomic specificity to the neuroimmune response.
In brain capillaries, the perivascular space between the endothelial/pericyte basement membrane and astrocytic endfeet is small (Thal, 2009). The minimal distance between capillary endothelial cells and the brain parenchyma makes them ideally positioned for secreting or transporting molecules into the CNS (Bechmann et al., 2007). In contrast, precapillary arterioles and postcapillary venules have a lamina media, and the basement membranes of this layer and astrocytic endfeet form a perivascular space (Thal, 2009). The brain CSF/ISF flows along this perivascular space, which facilitates the clearance of solutes from the brain parenchyma (Iliff et al., 2012). A subset of brain macrophages also resides in perivascular spaces (Bechmann et al., 2007), and these macrophages are positioned to respond to antigens and endogenous immune signals that are carried by the perivascular bulk flow of CSF, as well as those that are transported or secreted by the endothelium. Transmigration of leukocytes across the vascular BBB occurs at postcapillary venules (Owens et al., 2008). Some leukocytes that cross the BBB reside in the perivascular space, whereas others completely transmigrate across the glia limitans and enter brain parenchyma (Bechmann et al., 2007).
BECs actively respond to immune stimuli and are an active interface in neuroimmune communication. Mechanisms by which BECs contribute to neuroimmune axes will be discussed in section II of this review.
2. The Blood–Cerebrospinal Fluid Barrier
Epithelial cells of the choroid plexus are the primary cellular components that predominantly comprise the blood–CSF barrier/interface. Tanycytes (see below) and the arachnoid membrane also interface with the CSF, but are structurally and anatomically distinct from the choroid plexus epithelium (CPE). The choroid plexus is a specialized structure within all four brain ventricles. Choroid plexuses extend from the ependymal lining of the ventricles, and consist of a single layer of cuboidal epithelial cells that reside on a basement membrane. CPE cells have microvilli and cilia on their apical surface, which contacts the CSF (Damkier et al., 2013). These apical projections provide a large surface area for secretory activity, and also have functions in regulating and sensing pH, osmolarity, and ion balance in CSF (Damkier et al., 2013). The basal and lateral membranes of CPE cells are relatively flat, except at lateral membrane contacts near the basal end that assume a folded labyrinth structure that is thought to contribute to a paracellular diffusion barrier (Damkier et al., 2013). A plexus of leaky blood vessels is located on the basal side of the CPE cells. This vascular plexus is devoid of astrocytes, and secretions of proteins such as VEGF from the basal side of CPE cells may contribute to the fenestrated vascular phenotype (Esser et al., 1998). Leakage of blood components into the CSF is prevented by the presence of TJs that are present in close proximity to the apical surface of CPE cells (Johanson et al., 2011). TJ proteins expressed by CPE cells include claudins 1, 2, and 11. CPE TJs are thought to be more leaky than those of the vascular BBB, and this has been attributed to the presence of claudin-2, which can form diffusive channels within the junction (Amasheh et al., 2009; Rosenthal et al., 2010; Johanson et al., 2011). However, the BCSFB is still relatively impermeable as small molecules such as ascorbic acid and ions require solute carriers for their passage into CSF (Johanson et al., 2011).
The choroid plexus is the major site of CSF production in the brain. Humans produce about 500–600 ml CSF per day, with approximately 80% of CSF being produced by the choroid plexus and the remainder derived from brain ISF (Damkier et al., 2013), as there is no barrier that prevents mixing of brain ISF with brain CSF in the adult (Ghersi-Egea et al., 1996). The CSF provides a nutritive and homeostatic milieu for the brain and circulates through the ventricular, subarachnoid, and paravascular spaces in the CNS, as well as through brain tissue, where it mixes with the brain ISF (Hladky and Barrand, 2014). This flow pathway contributes to the homeostatic milieu of the ISF and also acts as a sink that prevents the localized buildup of solutes in the parenchyma (Oldendorf and Davson, 1967; Johanson et al., 2011). CSF exits the brain via arachnoid granulations into venous sinuses and in spinal nerves, as well as via perineural spaces that penetrate the cribriform plate (Hladky and Barrand, 2014). CSF turns over in the human brain approximately three times per day (Damkier et al., 2013), which is governed by CSF production, convective forces that facilitate bulk flow, intracranial pressure, and patency of resorption sites (Pollay, 2010). CPE cells express a variety of ion transporters, solute carriers, and water channels that contribute to CSF production by enabling passage of water and solutes from the plasma ultrafiltrate across the epithelium and into the ventricular space (Damkier et al., 2013). CPE cells also express efflux transporters that inhibit the buildup of potentially harmful substances in CSF. These include ABC transporters such as MRP-1 (Gazzin et al., 2008) and Pgp (Pascale et al., 2011), as well as monoamine transporters such as SLC29A4, which facilitates histamine efflux from CSF into blood (Usui et al., 2016), and the organic cation transporter 3, which clears creatinine from CSF (Tachikawa et al., 2008). CPE cells also express receptors that have been implicated in transcytosis, including low-density lipoprotein receptor-related protein-1 (LRP-1), -2, (LRP-2/megalin), and the receptor for advanced glycation endproducts (RAGE) (Zlokovic et al., 1996; Fujiyoshi et al., 2011; Pascale et al., 2011).
The choroid plexus is an immune-active tissue. It is a site of leukocyte trafficking and immune surveillance (Baruch and Schwartz, 2013; Schwartz and Baruch, 2014), and CPE cells upregulate their expression of proinflammatory cytokines, c-fos, cell adhesion molecules, and major histocompatibility complex antigens following systemic inflammatory stimuli (Vallieres and Rivest, 1997; Endo et al., 1998; Wolburg et al., 1999; Engelhardt et al., 2001; Marques and Sousa, 2015). Structural changes in CPE cells, such as swelling of the apical microvilli, increases in numbers of dark, electron dense epithelial cells, and altered mitochondria, occur during inflammation (Engelhardt et al., 2001). Inflammation also impairs CSF turnover (Erickson et al., 2012b), which influences the clearance of solutes from the CNS.
The arachnoid epithelium comprises another component of the BCSFB. Arachnoid epithelial cells express TJ proteins such as claudin 11 (Brochner et al., 2015) and form a size-selective diffusion barrier in vitro (Lam et al., 2012). Arachnoid epithelial cells also express high levels of the efflux transporters Pgp and BCRP at their apical membranes (Yasuda et al., 2013), which are exposed to plasma ultrafiltrate from the leaky vessels of the dura matter. Therefore, the activity of these transporters would prevent the transport of substances from blood into CSF in the subarachnoid space. BCRP expression was also noted at the basal membrane of arachnoid epithelial cells, suggesting that a second function of BCRP is to facilitate the transport of substances into CSF (Yasuda et al., 2013).
3. Tanycytic/Ependymal Barriers of Circumventricular Organs
The CVOs of the brain are important interfaces for humoral communication with the CNS. Within CVOs, the brain capillaries are leaky, lack TJ proteins, and are usually fenestrated, allowing for communication of circulating molecules with resident neurons and glia. The CVOs are generally classified as those having sensory functions, which are the subfornical organ, organum vasculosum of the lamina terminalis (OVLT), and area postrema, and those having secretory functions, which include the neurohypophysis, median eminence (ME), and pineal gland (Miyata, 2015). The subcomissural organ is also considered a CVO, but its capillaries are not leaky and express TJ proteins (Petrov et al., 1994; Langlet et al., 2013); however, they lack GLUT1, which is typically expressed in BBB capillaries (Rahner-Welsch et al., 1995). The choroid plexus (CP) is also sometimes considered a CVO, but has specialized properties in that the CP has an epithelial barrier and is located within the brain ventricles (Miyata, 2015). Neuronal and glial populations within sensory CVOs can detect and respond to changes in circulating components such as electrolytes, glucose, cytokines, and hormones. In the sensory CVOs, neuronal cell bodies and dendrites are exposed to blood-derived exudates from leaky capillaries, but project their axons outside of the CVOs to brain regions protected by a BBB (Rodriguez et al., 2010). In contrast, secretory CVOs receive axonal projections from neuronal cell bodies that are located outside of the CVO. These axons release peptides into the bloodstream that can signal to distal organs and elicit physiologic responses such as changes in blood pressure (Mimee et al., 2013). Thus, the CVOs facilitate bidirectional communication between the brain and periphery and regulate vital physiologic functions that include fluid balance, metabolism, reproduction, and immune responses (Ferguson, 2014).
Although serum components can freely diffuse into and within CVOs, tanycytic barriers prevent their diffusion into CSF and adjacent regions of the brain. Tanycytes are specialized ependymal cells that differentiate from radial glial cells beginning in the last few days of prenatal development and continue to mature postnatally (Edwards et al., 1990). Tanycytes are morphologically distinct from cuboidal ependymal cells that line the ventricles in that they lack cilia, and instead have long, unipolar projections that are proximal to the fenestrated CVO capillaries (Rodriguez et al., 2010). Tanycyte structure and functions have been best characterized in the ME (Mullier et al., 2010; Rodriguez et al., 2010), although their barrier functions appear to be similar in other CVOs (Langlet et al., 2013). Four types of ME tanycytes have been described, which include α1, α2, β1, and β2. Barrier properties are ascribed to the β1 and β2 tanycytes, which are located at the lateral extensions and floor of the infundibular recess, respectively (Rodriguez et al., 2010). The basal processes of β1 tanycytes form bundles with axons that define the boundary of the arcuate nucleus, which has an intact BBB, and the ME (Rodriguez et al., 2010). TJs and AJs are present between contacts of adjacent tanycyte processes as well as the axons they surround, and the anatomic location of these β1 projections defines the diffusion barrier of i.v. injected substances such as Evan’s blue dye between the ME and arcuate nucleus (Rodriguez et al., 2010). In contrast, the β2 tanycytes express TJ proteins, including ZO-1, occludin, claudin-1, and claudin-5 (Mullier et al., 2010), at their apical contacts between cell bodies lining the ventricle (Rodriguez et al., 2010). These junctions form a barrier that prevents diffusion of blood components into the CSF, and similar TJ organization of tanycytic blood–CSF barriers has been characterized in the subfornical organ, OVLT, and area postrema (Langlet et al., 2013). Tanycytes of the ME also have important interface functions. They are thought to participate in the regulation of hypothalamic pathways that control energy balance through glucose sensing and leptin transport into CSF (Balland et al., 2014; Elizondo-Vega et al., 2015).
The sensory CVOs are important interfaces for neuroimmune communication. Neuroimmune functions of CVOs were first demonstrated by the pioneering work of Blatteis et al. (1983) in the 1980s, in context of the febrile response, which was diminished by OVLT ablation. It was later shown that cells within CVOs rapidly upregulate proinflammatory cytokines following systemic application of LPS, whereas the brain parenchymal inflammatory response occurs as a second wave (Quan et al., 1998). The localized inflammation in CVOs may disrupt the tanycytic blood–CSF barrier, as increased paracellular permeability between junctions of tanycytes lining the third ventricle has been observed in response to LPS (Liu et al., 1996).
III. The Neuroimmune Axes
A. Axis 1: Blood–Brain Barrier Disruption
Strictly speaking, disruption is usually considered a pathologic condition. However, there is a perception that, even under physiologic conditions, barrier function may slightly vary. As such, a role for neuroinflammation in the physiologic regulation of barrier tightness may emerge, and so this section is presented as a neuroimmune axis. Furthermore, the term “disruption” as it pertains to brain barriers is often not clearly defined. In the strictest sense, disruption refers to loss of barrier function resulting from loss of TJ function, reinstitution of macropinocytosis or fenestrae, or development of cannulae/vesiculo-tubular structures (Lossinsky and Shivers, 2004), thus allowing leakage of normally restricted substances, such as serum proteins, across barrier cells. But it is often used much more loosely to describe an alteration in endothelial or epithelial function permitting leukocyte entry into brain parenchyma, loss of Pgp function, or dysfunctions resulting in some form of enhanced passage. Notably, the latter are distinct but possibly interrelated molecular processes. In this work, we will use “disruption” in its strictest sense and explore inflammatory influences on immune cell trafficking and Pgp function in separate sections.
The experimental determination of BBB disruption and the subsequent leakage under various neuroinflammatory states are often accomplished by measuring the CNS entry of serum proteins or i.v. injected tracer, as well as assessment of TJ protein expression (Saunders et al., 2015). In living humans, the CSF/serum albumin ratio is also a common indicator used to infer BBB leakage, and technical aspects of this approach and others that have been used to detect BBB disruption in humans are critically appraised elsewhere (Erickson and Banks, 2013). BBB leakage is observed in diverse pathologic states in which distinct modes of BBB disruption may be apparent. In this study, we describe three mechanisms by which inflammatory conditions may contribute to a leaky BBB (Fig. 2).

Axes 1 and 3: disruption, transport, and penetration. Major influx mechanisms are transcellular diffusion and saturable transport. Influx is countered by efflux (transcellular diffusion, saturable transport, reabsorption of CSF) and enzymatic activity at the BBB. Disruption can be by way of transcellular/transcytotic or paracellular mechanisms. Endothelial damage and hemorrhage are not depicted. The extracellular pathways are relatively inefficient routes of CNS uptake vs. saturable transport and used by substances that include albumin, immunoglobulins, erythropoietin, and soluble receptors.
1. Disruption of Paracellular Tight Junctions
BBB leakage between endothelial cell contacts can occur when TJ proteins decrease in expression, mislocalize, or are posttranslationally modified (Luissint et al., 2012). In many instances, inflammatory mediators can modulate TJ proteins and induce paracellular BBB leakage. For example, an injection into brain parenchyma of IL-1β results in the loss of occludin and ZO-1 expression at endothelial cells, which coincides with paracellular leakage of an intravascular tracer and neutrophil recruitment to vessels where TJs are absent (Bolton et al., 1998). TGF-β1, which plays classic roles in the resolution of inflammation, also increases tyrosine phosphorylation of VE-cadherin and claudin-5 and downregulates claudin-5 expression (Shen et al., 2011; McMillin et al., 2015). The chemokine C-C motif chemokine ligand (CCL) 2, which attracts monocytes, causes loss of TJ protein expression, as well as TJ protein redistribution that is mediated by caveolae (Stamatovic et al., 2005, 2006, 2009). Enzymatic degradation of TJ proteins at the BBB can also occur in response to neuroinflammatory insults. MMP inhibition or knockout prevents the degradation of TJs and BBB disruption in the acute phase following brain ischemia-reperfusion injury (Asahi et al., 2001; Yang et al., 2007). Protective factors have also been identified that preserve TJ protein expression at the brain endothelium. These include IL-25, netrin-1, and annexin A1, which are expressed by BECs, and sonic hedgehog, which is secreted by astrocytes (Sonobe et al., 2009; Alvarez et al., 2011; Cristante et al., 2013; Podjaski et al., 2015). IL-1β can decrease sonic hedgehog expression (Wang et al., 2014b). Other inflammatory mediators of increased paracellular BBB permeability include bradykinin, histamine, serotonin, arachidonic acid, and ATP (Abbott, 2000). Clearly, TJs may become dysfunctional under a number of different proinflammatory states. However, other routes of BBB dysfunction that result in leakiness also occur in response to inflammation and are discussed below.
2. Transcytotic Vesicular Pathways
Ultrastructural studies of the BBB dating as far back as the 1970s have revealed that damaged BECs can form patent vesicular channels that permit the passage of large molecular tracers such as horseradish peroxidase into the CNS (Lossinsky and Shivers, 2004). Such vesicular structures have been described in cerebral edema, traumatic brain injury, and sepsis (Castejon, 1980, 1998; Esen et al., 2012). In some instances, the formation of transcellular channels occurs in the absence or independently of ultrastructural changes in TJs (Lossinsky and Shivers, 2004; Esen et al., 2012; Goncalves et al., 2017). Despite the potential contribution of vesicles and transendothelial channels to BBB leakage, the molecular underpinnings of their formation remain relatively understudied. One protein that is ubiquitously expressed in peripheral endothelial cells is plasmalemma vesicle-associated protein (Plvap), which is sometimes referred to as MECA-32. Plvap associates with endothelial fenestrae and contributes to fenestrae formation in peripheral vessels (Herrnberger et al., 2012a,b). Plvap is suppressed in brain endothelium (Hallmann et al., 1995), but is upregulated in diseases that are associated with BBB disruption, such as Alzheimer disease (AD) or multiple sclerosis (MS) (Engelhardt et al., 1994; Yu et al., 2012). Furthermore, Mfsd2a (described above in Features and Functions of the Blood–Brain Barrier and Blood-Brain Interface) is downregulated in a mouse model of intracerebral hemorrhage that, in part, mediates BBB disruption (Yang et al., 2017). Therefore, formation of endothelial fenestrations may be an important contributor to BBB leakage in some disease states.
3. Endothelial Cell Damage and Hemorrhage
Cerebral microbleeds (CMB) are associated with neurovascular insults such as ischemia-reperfusion injury, intracranial hemorrhage, cerebrovascular diseases, and following traumatic brain injury (Kleinig, 2013). CMBs are also observed in sepsis patients (Correa et al., 2012), and systemic inflammation is higher in patients with CMBs (Miwa et al., 2011). CMBs are visualized as hemodesmerin deposits, which are iron-rich breakdown products of hemoglobin (Kleinig, 2013). Recent work using animal models of subchronic systemic inflammation and ischemia-reperfusion injury has demonstrated that BBB disruption and neuroinflammation can be associated with subsequent development of microbleeds (Krueger et al., 2015; Sumbria et al., 2016). In C57BL6/J mice treated with three repeated doses of LPS, CMBs became evident 2 days after the final injection and persisted by day 7. Furthermore, CMBs significantly correlated with markers of neuroinflammation following LPS treatment (Sumbria et al., 2016). In a rodent ischemic-reperfusion injury model, it was observed that leakage of albumin within the ischemic area occurs in the absence of changes in TJ or AJ protein-staining patterns (Krueger et al., 2015). However, structural alterations to the endothelial surface were evident and indicated regions where the endothelium was damaged or absent; these damaged endothelial cells colocalized with albumin extravasation. Ultrastructural analysis revealed that, at early stages of damage, endothelial cell edema occurs without apparent extravasation of intravascular tracer. Influx of tracer into the brain parenchyma only became apparent after complete loss of endothelial cell integrity, and influx of red blood cells into the CNS occurred following basement membrane degradation (Krueger et al., 2015). Neutrophil-derived proteases such as MMP9 and elastase have been shown to contribute to the breakdown of the ECM and destruction of the endothelium in ischemia-reperfusion injury (Gidday et al., 2005; Stowe et al., 2009; Ikegame et al., 2010; Turner and Sharp, 2016). Intracerebral injection of neutrophil elastase causes endothelial swelling and focal necrosis of blood vessels, as well as focal hemorrhages and leukocyte cuffing of the vessels (Armao et al., 1997).
The existence of these three modes of BBB disruption necessitates a careful interpretation of TJ protein expression data. The absence of apparent changes in TJ proteins does not necessarily mean that the BBB is intact, as leakage may occur via vesicles, transcellular channels, or damaged endothelial cell membranes. Conversely, a decrease in TJ protein expression may reflect BBB damage that is more severe than paracellular leakage, such as endothelial cell degeneration where the whole cell is lost in addition to the TJ. The works that have highlighted these varied modes of BBB disruption also suggest that different therapeutic approaches to protect the BBB may need to be considered based on which mode of BBB leakage predominates.
B. Axis 2: Modulation of Barrier and Interface Functions by Immune Substances
The BBB has many functions other than that of forming a barrier between the peripheral circulation and the CNS. It broadly serves other roles, including that of regulating the homeostatic environment of the CNS, supplying the nutritional needs of the CNS, and being the center point in the humoral-based communications between the CNS and peripheral tissues. One of the main ways in which the BBB fulfills these functions is through the possession of various transport systems. These transporters have in common that they are self-saturable. As described above, transporters can be variously classified as energy requiring (active transport) or not energy requiring (facilitated diffusion). Active transport can be unidirectional, transporting a substance against its concentration gradient, whereas facilitated diffusion is bidirectional with net transport being in the direction of higher to lower concentration. As a rough guide, substances that are the major ligand for a blood-to-brain (influx) transporter can enter the brain at rates that are 10–100 times faster than if they were to depend on nonsaturable mechanisms, and substances transported out of the CNS (efflux transporters) accumulate at rates 1/10 or so lower than would be expected from their transcellular diffusion. Several important transporters are modulated by neuroinflammation and neuroimmune substances (Fig. 3). Pgp is a major efflux transporter for small, lipid-soluble molecules. It resides in the luminal membrane of BECs and the ependymal cells forming the choroid plexus. As discussed above, its ligands represent a diverse group of substances and include protease inhibitors, opiates (endogenous and exogenous; peptides and small molecules), anti-epileptics, cyclosporins, glucocorticoids, aldosterone, dexamethasone, and calcium channel blockers (Begley, 2004). Its activity explains why certain substances do not accumulate in the brain in sufficient quantities to produce an effect on the CNS. As such, it can be viewed either as protecting the brain from xenobiotics, including drugs, that would otherwise produce significant CNS side effects or as a major obstacle to the development of CNS therapeutics. Pgp function is modulated by inflammation with the main effect in vivo being a downregulation of its transport function. Details of immune regulation of Pgp function are detailed later in section IV of this review.

Axis 2: modulation of barrier/interface function. Immunoactive (IA) substances work through four main pathways to alter BBB functions. (A) IA substances act on a peripheral cell that then releases a substance that acts on the barrier. Example: LPS acts on a peripheral cell inducing it to release nitric oxide, and the nitric oxide then acts on BECS to alter insulin transport. (B) IA acts on the BEC to induce an alteration mediated through intracellular machinery. Example: TNF alteration of Pgp function, which is mediated through a pathway involving nitric oxide and endothelin-1. (C) IA acts directly at a BEC receptor or transporter. Example: IL-1ra blocks BBB transport of IL-1β. (D) IA acts on barrier cell receptor/transporter (i) inducing barrier cell secretion that acts in autocrine fashion to affect barrier function (example: LPS induces BEC to secrete IL-6 and granulocyte-macrophage colony-stimulating factor, which mediates LPS-induced increase in HIV-1 passage across the BBB) or (ii) to induce barrier cell to communicate with another CNS cell whose release modifies barrier cell activity (example: presence of pericyte enhances LPS-induced increase in HIV-1 passage across the BBB).
Influx transport can also be affected by inflammation. The Na-K-Cl cotransporter at the BEC, important in cerebral ionic homeostasis, is modulated by IL-6 secreted from astrocytes (Sun et al., 1997). Insulin is transported across the BBB and acts in the brain, having effects on cognition and feeding (Banks et al., 2012b). CSF/serum ratios of insulin are reduced in AD, and delivery of insulin to the brain of AD patients can improve cognitive functions (Craft et al., 1998, 1999, 2012). LPS acts indirectly through a nitric oxide–dependent pathway to increase BBB transport of insulin (Xaio et al., 2001).
Some of the many and diverse effects of LPS on barrier functions are directly mediated by the presence of Toll-like receptors on barrier cells. The expression by BECs of at least some of these receptors are themselves regulated by oxidative stress and TNF-α (Nagyoszi et al., 2010). The bacterial cell wall components LPS and muramyl dipeptide also regulate the BEC expression of nucleotide-binding oligomerization domain (NOD) and NOD-like receptors (NLRs), intracellular sensors of pathogen and damage/danger-associated molecules (Nagyoszi et al., 2010). Expression of NLRs and of NODs, the domain of NLRs that binds glycopeptides such as N-acetylglucosamine, is upregulated as well by inflammatory cytokines, including IFN-γ, TNF-α, and IL-1β (Nagyőszi et al., 2015).
The response of the brain barriers to neuroimmune stimuli can be modulated, reversed, or blocked by a number of agents as well. BECs express cannabinoid type 2 receptors. Agonists of these receptors prevented 32 of 33 genes from being upregulated by TNF-α and diminished TNF-induced BBB disruption and macrophage migration (Persidsky et al., 2015). Prostaglandins, as evidenced by the effects of treatment with indomethacin, can block, enhance, or have no effect on the actions of LPS on BBB functions (Guillot and Audus, 1990; Minami et al., 1998; Xaio et al., 2001).
C. Axis 3: Transport, Penetration, and Uptake of Neuroimmune-Related Substances
Blood-to-brain entry has been assessed for some neuroimmune substances. Several cytokines have been shown to cross the BBB by way of saturable transport systems and antibodies and soluble receptors can enter the brain by way of the extracellular pathways.
Many cytokines are transported across the BBB in the blood-to-brain direction (Fig. 2). Such transport intermingles the peripheral pool of the cytokine with its CNS pool. The transport systems for cytokines are saturable and are selective, perhaps even specific, for a cytokine or family of cytokines. For example, the transport of TNF-α is self inhibited, but not inhibited by IL-6 nor by any of the IL-1s (Gutierrez et al., 1993; Banks et al., 1994). The IL-1s (IL-1α, IL-1β, IL-1 receptor antagonist) both self inhibit as well as inhibit each other’s transport, but have not been found to inhibit the transport of any other cytokine (Banks et al., 1991). Therefore, the IL-1 family either shares a single transporter or a family of closely related transporters. Epidermal growth factor crosses the BBB using a transporter shared with TGF-α (Pan and Kastin, 1999). CCL2 (monocyte chemoattractant protein 1) transport is not shared with CCL3 (macrophage-inhibitory protein 1-α) (Ge et al., 2008). Other cytokines formally demonstrated to be transported across the BBB include ciliary neurotrophic factor and TGF-β2 (Pan et al., 1999; McLennan et al., 2005), but TGF-β1 is not transported across the intact BBB (Kastin et al., 2003). To date, only IL-2 and CCL11 have been found to have a saturable component to their brain-to-blood efflux phase (Banks et al., 2004b; Erickson et al., 2014), although others can enter the circulation with CSF reabsorption (Chen et al., 1997; Chen and Reichlin, 1998). It has also been suggested that the duffy antigen chemokine receptor on brain endothelial cells contributes to brain efflux of CCL2 and CCL5 following a neuroinflammatory stimulus (Minten et al., 2014).
Little is known about the cellular biology of cytokine transport across the BBB or what circulating factors may affect such transport. Cytokine-induced neutrophil chemoattractant 1 enters the brain by a nonsaturable mechanism, presumably that of transcellular diffusion (Pan and Kastin, 2001a). At a mol. wt. of 7800 Da, this would be the largest known substance to use this pathway, which essentially involves the molecule first partitioning into the lipids of the barrier cell membrane and eventually back into the aqueous environment of brain interstitial fluid or CSF. Erythropoietin enters the CNS by the nonsaturable process of the extracellular pathways, although it is unclear the degree to which this underlies its many neuroprotective effects (Brines et al., 2000; Banks et al., 2004a). In general, the extracellular pathways account for little of the uptake for cytokines that use a saturable transporter to cross the BBB (Plotkin et al., 1996). For most endogenous biologics studied to date, the protein responsible for transport is not the same as that used by the cell for receptor functions (Pan and Kastin, 1999), although there seem to be more exceptions to this rule for cytokines than for other biologics (Pan and Kastin, 2002; Pan et al., 2006a; Ge et al., 2008). CCL2 transport is caveolae dependent (Ge et al., 2008); IL-2 blood-to-brain transport is inhibited, in addition to other mechanisms, by protein binding (Banks et al., 2004b); and CCL11 (eotaxin-1) binds to cellular components in blood that slows its early-phase entry (Erickson et al., 2014).
Most cytokine transport studies have been done in the mouse, but transport of IL-1α and IL-6 has been shown to occur in rats and of IL-1β and IL-6 in fetal sheep (Luheshi et al., 1994; Plotkin et al., 2000; Threlkeld et al., 2010; Sadowska et al., 2015), indicating that cytokine transporters are expressed early in development and across species. An in vitro study using porcine BBB endothelial cells found transport of IL-1 and IL-1 receptor antagonist, further supporting the idea that cytokine BBB transporters occur in multiple species (Skinner et al., 2009).
The transport of cytokines has been shown to affect brain function in several experimental designs (Table 1). The first such investigation showed that human IL-1α crossed the BBB at the posterior division of the septum to induce cognitive dysfunction (Banks et al., 2001a). This finding is consistent with IL-1α being the chief mediator of sickness behavior, including the cognitive dysfunction of sickness behavior (Larson and Dunn, 2001; Kelley et al., 2003). Injection into the posterior division of the septum of blocking antibodies that were specific for human IL-1α, but unable to block mouse IL-1α, was able to prevent the cognitive impairment induced by the i.v. injection of human IL-1α. Thus, it was demonstrated that the IL-1α injected into the bloodstream must be crossing the BBB at the posterior division of the septum to induce cognitive dysfunction. Fibroblast growth factors have been shown to cross the BBB (Cuevas et al., 1996; Hsuchou et al., 2013) so that peripherally administered fibroblast growth factors promote neurogenesis and protect against stroke and traumatic brain injury (Cuevas et al., 1998; Wagner et al., 1999; Sun et al., 2009).
CNS effects of cytokines dependent on their transport across the BBB
Two studies with TNF-α have shown that its transport across the BBB is needed for the peripherally administered cytokine to induce its CNS effects. Qin et al. (2007) showed that TNF-α crossing the BBB acts on microglia to induce brain inflammation, including the release of more TNF, resulting in apoptosis of dopaminergic cells in the substantia nigra. Tangpong et al. (2006, 2007) in a mouse model of chemobrain showed that TNF crosses the BBB to release additional TNF from brain stores of TNF, thus augmenting the toxicity of blood-derived TNF (Joshi et al., 2010). This model of dual action of a blood-derived cytokine acting both directly on a CNS target tissue as well as augmenting its levels by inducing release from CNS sources may apply generally to cytokines that cross the BBB. Indeed, in the IL-1α studies considered above, blocking antibodies specific for mouse IL-1α were partially effective in preventing cognitive impairment. Ogawa et al. (2016) found that peripherally administered orexin improved survival from endotoxic shock. Because they found that orexin crossed the BBB, confirming previous work by Kastin and Akerstrom (1999), and because orexin administered into the CNS produced many of the effects of peripherally administered orexin, they concluded that peripheral orexin crossed the BBB to induce its effects via the CNS.
The rate of cytokine transport is not homogenous throughout the brain but varies tremendously across regions. Uptake by the CVOs, areas of the brain in which the capillaries form either no or a less efficient barrier, takes up an estimated sevenfold more cytokine than the adjacent barrier areas (Plotkin et al., 1996; Maness et al., 1998). Interestingly, CVO uptake was also saturable and, although entering at a much higher rate than the rest of the brain, only accounted for 5% of whole brain uptake (Plotkin et al., 1996). Diffusion from the CVO to the adjacent brain region is either highly restricted because of the diffusional limitations of Brownian motion or absent because of a tanycytic barrier between the CVO and adjacent brain tissue (Maness et al., 1998).
Cytokine transport is also heterogeneous for those brain regions with a complete BBB. TNF transport varies by 10-fold between the region with the highest uptake, the hypothalamus, and lowest, the parietal cortex (Banks et al., 2001c). For IL-1, the posterior division of the septum has a very high uptake, whereas the midbrain and striatum did not transport IL-1 (Moinuddin et al., 2000). TGF-β2 is transported at its highest rate into the hypothalamus and lowest into the cerebral hemispheres (McLennan et al., 2005). The spinal cord can also take up cytokines with transport rates varying among the cervical, thoracic, and lumbar regions (Pan et al., 1997b).
Cytokine transport rates are altered by physiologic and pathologic events. IL-1 and TNF transport varies diurnally with transport into the spinal cord varying 10-fold for IL-1 (Banks et al., 1998b; Pan et al., 2002). TNF transport is increased by traumatic brain injury, spinal cord injury, in experimental autoimmune encephalomylelitis (EAE), and stroke (Pan et al., 1996, 1997a, 2003a, 2006b; Pan and Kastin, 2001b). In contrast to TNF, IL-15 transport across the BBB is decreased in EAE (Hsuchou et al., 2009). Transport of leukemia-inhibitory factor is increased by spinal cord injury, neuroinflammation, and TNF (Pan et al., 2006a, 2008). Chronic opiate exposure and withdrawal are associated with the induction of blood-to-brain transport of IL-2 (Lynch and Banks, 2008), whereas the efflux of IL-2 and influx of IL-1α and TNF are unaffected. The response of the BBB to injury often shows heterogenous regional and temporal changes in transport rates as illustrated by TNF (Pan et al., 1997a, 2003b; Pan and Kastin, 2001b).
Soluble receptors, as exemplified by that for IL-6, can cross the BBB to influence behavior. Patel et al. (2012) showed that peripherally administered soluble IL-6 receptor interacts with Gp130 in some brain regions to induce stereotypic behaviors.
Some immune substances are able to enter the brain by the extracellular pathways (Broadwell and Sofroniew, 1993). The extracellular pathways are a major way in which that small amount of albumin found in the CSF enters the CNS. The extracellular pathways result in a unique distribution in brain, including that of tracking along the penetrating arterioles. Thus, the distribution of substances entering the CNS via the extracellular pathways is distinct from the patterns seen for substances entering via the vascular route or the choroid plexus. To take advantage of the extracellular pathways, a substance must have a very long half-life in blood and, because very small amounts enter by this route, it must be potent. Pharmacologic substances that have been shown to enter the CNS by this pathway include antibodies, soluble receptors, and erythropoietin (Banks et al., 2002; Banks, 2004).
D. Axis 4: Immune Cell Trafficking between Blood and Brain
In most tissues, blood-borne leukocytes survey parenchymal spaces and are actively recruited upon infection or injury. CNS barriers confer specialization to mechanisms of leukocyte surveillance and recruitment to the brain (Fig. 4). Surveillance by blood-borne leukocytes is thought not to occur in the healthy brain parenchyma (Filiano et al., 2017) or to occur at low levels (Banks et al., 2012), but the chorioid plexus and meninges are surveyed by blood-borne populations of T-cells. Leukocyte counts of up to 5000/ml may be observed in healthy human CSF (Seehusen et al., 2003), and about 80% of CSF leukocytes are CD4+ central memory T-cells (Engelhardt and Ransohoff, 2005). T-cells within the choroid plexus and meningeal compartments contribute to host defense and mediate responses to injury, and they also regulate physiologic CNS functions (Shechter et al., 2013; Raposo et al., 2014; Filiano et al., 2017). For example, mice that lack T-cells or receive antibodies that block T-cell migration have impaired learning and memory and social behavior (Filiano et al., 2017). Because meningeal T-cells do not actively survey the brain parenchyma under physiologic conditions, it is believed that soluble factors secreted by T-cells within the meninges may be carried by the CSF/ISF to brain regions that contribute to rodent behaviors. One candidate T-cell–derived regulator of behavior is IFN-γ. Mice lacking IFN-γ are less social and have aberrant hyperconnectivity in their prefrontal cortex (Filiano et al., 2016). Furthermore, administration of IFN-γ in CSF can rescue social deficits in mice lacking T-cells (Filiano et al., 2017).
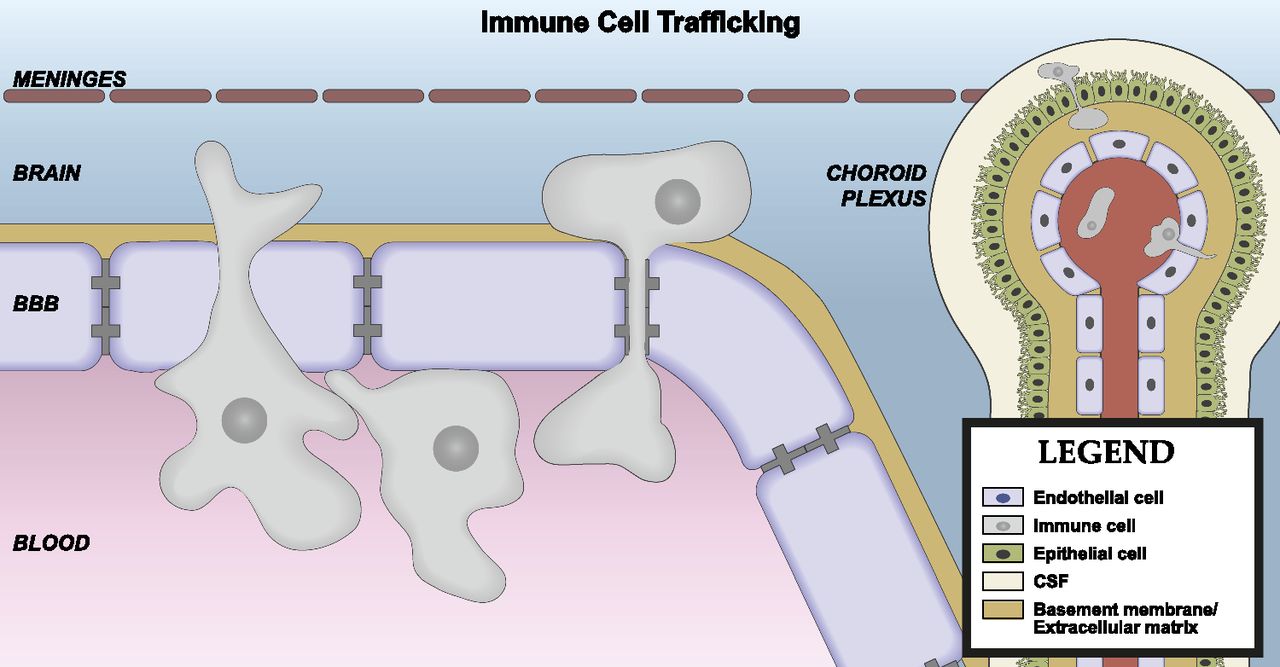
Axis 4: immune cell trafficking. Immune cell passage across the BBB is a highly regulated process and can occur at the vascular BBB, chroroid plexus, or meninges. Passage can be between or across the BEC and involves steps that include capture, rolling, arrest, crawling, and diapedesis.
Leukocyte recruitment to CSF compartments through the BCSFB, as well as to brain parenchyma via parenchymal blood vessels, relies on a multistep extravasation process that varies depending on the basal expression of adhesion molecules. The typical process of leukocyte extravasation into tissues involves an initial capture of the circulating leukocyte, followed by rolling on the endothelial surface, arrest, crawling, and diapedesis across the endothelium (Vestweber, 2015). Additionally, to enter the brain parenchyma, leukocytes must penetrate the basement membrane along the glia limitans that lines the paravascular space (Engelhardt and Coisne, 2011). The initial capture and rolling steps are mediated by the interactions of selectins on the endothelial cell surface with glycoproteins on the leukocyte surface (Vestweber, 2015). Peripheral and meningeal endothelial cells, as well as endothelial cells within the choroid plexus stroma, express selectins that are retained in intracellular vesicles called Wiebel–Palade bodies (Engelhardt and Coisne, 2011). These vesicles are routed to the endothelial lumen in response to inflammatory mediators, enabling the rapid upregulation of selectins at the luminal membrane (Metcalf et al., 2008). The initial capture of CD4+ T-cells that reside in the CSF is mediated by P-selectins expressed on blood vessels in the choroid plexus and subarachnoid space (Kivisakk et al., 2003). In contrast, endothelial cells of the brain parenchyma do not express selectins in their Weibel–Palade bodies and require de novo synthesis of selectins to enable the capture and crawling of leukocytes (Engelhardt and Coisne, 2011). Activated lymphocytes can cross the naive BBB, but they do so by a very inefficient process of abrupt arrest on the endothelial cell surface that is mediated by interactions between endothelial cell adhesion molecules and integrins on the lymphocyte surface (Vajkoczy et al., 2001). Therefore, leukocyte trafficking to the inflamed CNS tends to be delayed when compared with other tissues.
The regulation of leukocyte trafficking to the CNS is multifaceted and depends on the activation state of the leukocytes and the endothelial interface, as well as the inflammatory milieu of the CNS and peripheral compartments. IL-1β is an important mediator of neuroinflammation, including leukocyte trafficking to the CNS (Bolton et al., 1998; Ching et al., 2007; McKim et al., 2017). BECs are the predominant expressors of type 1 IL-1 receptor (IL-1R1) in the CNS, and endothelial IL-1R1 is an important contributor to IL-1β–mediated leukocyte influx (Ching et al., 2007; Liu et al., 2015; McKim et al., 2017). The endothelial-specific knockdown of IL-1R1 abrogates leukocyte influx induced by centrally injected IL-1β (Ching et al., 2007; Liu et al., 2015). However, the effects of centrally expressed IL-1β on leukocyte trafficking can be diminished by systemic inflammation. For example, systemic administration of LPS, when given within 2 hours of an intracerebroventricular injection of IL-1β, inhibits leukocyte recruitment to the CNS by preventing the upregulation of selectins on BECs (Ching et al., 2006). Functional consequences of this pathway have been demonstrated in a rodent model of stress-induced anxiety, in which the interaction of IL-1β–expressing monocytes with endothelial cells in threat-appraisal centers of the CNS mediates anxiety behaviors (McKim et al., 2017). In contrast, lymphocytes from chronically stressed mice have antidepressant effects (Brachman et al., 2015).
Diapedesis of leukocytes in CNS tissue may occur via paracellular or transcellular routes, depending on cell type and disease state (Millan et al., 2006; Gorina et al., 2014; Abadier et al., 2015). There is crosstalk between leukocytes and tight junctional complexes at the BBB that may affect both paracellular permeability and the leukocyte’s ability to traverse the endothelial barrier. For example, an antibody that blocks JAMs also inhibits monocyte transmigration to the CSF and brain parenchyma in experimental meningitis (Del Maschio et al., 1999). Intracellular adhesion molecule 1 activation stimulates VE cadherin phosphorylation and cytoskeletal rearrangement in BECs, which facilitates diapedesis (Turowski et al., 2005; Turowski et al., 2008). However, in EAE, a model of MS, transcellular diapedesis of mononuclear cells across cerebral endothelial cells leaves TJs morphologically intact (Wolburg et al., 2005). Therefore, the loss of paracellular BBB integrity may not be requisite for diapedesis in all cases.
Recent work has identified the choroid plexus as an important site for leukocyte trafficking following CNS injury, despite its distal location. For example, in spinal cord injury, anti-inflammatory macrophages are recruited to the site of injury via the choroid plexus (Shechter et al., 2013). In this model, it was shown that CPE cells upregulate their expression of adhesion molecules that facilitate the extravasation of recruited macrophages into CSF. Furthermore, blocking the flow of CSF to the injury site using Matrigel also inhibited trafficking of the macrophages to the site of injury (Shechter et al., 2013). In a distinct model of permanent middle cerebral artery occlusion, it was shown that about two-thirds of T-cells that infiltrate the peri-infarct area are recruited from the choroid plexus stroma (Llovera et al., 2017). Interestingly, this study noted that the apical region of the CP stroma was physically connected to penetrating vessels from the brain parenchyma, and that the apparent routes of T-cell trafficking from CP stroma to the corpus callosum and finally to the peri-infarct region with fast kinetics were suggestive of a direct pathway of migration that did not involve homing through the CSF. Furthermore, injection of Matirgel into the ventricle, which blocks CSF-mediated routes of T-cell homing, had no effect on T-cell trafficking to the peri-infarct region in this model (Llovera et al., 2017). These results highlight a possible novel route of T-cell trafficking to brain parenchyma where T-cells may bypass brain barriers.
The functional consequences of leukocyte trafficking to the CNS have been studied in a variety of disease states, and these, along with other aspects of BBB dysfunction in disease, will be discussed in section IV below.
E. Axis 5: Immune Secretions of the Barrier Cells
The barrier cells that form the vascular BBB, the choroids plexus, and the tanycytic barriers are in constant crosstalk with other cells of the CNS. The concept of this crosstalk is embodied in the term NVU; participating cells include pericytes, astrocytes, microglia, neurons, and mast cells, as discussed above. The crosstalk, among other functions, serves to inform the barrier cells of the needs of the CNS so that the barrier cells can adapt their functions to meet those needs. Many of the agents used to conduct this crosstalk are neuroimmune mediators, including cytokines, prostaglandins, and nitric oxide. Furthermore, the barrier cells not only respond to these and other neuroimmune agents, but also secrete them (Fig. 5). Neuroimmune substances, then, are intimately involved in NVU crosstalk and the resulting characteristics of the barriers.

Axis 5: immune secretions by barrier cells. Secretions can be from either the luminal or abluminal surface (cell A). Secretions can be constitutive or induced by immune modulators (cell B). Barrier cells can receive immune modulator input from one surface (e.g., abluminal) and respond by secreting from the opposite surface (e.g., luminal), forming an axis that transmits neuroimmune communication across the BBB (cell C).
Most of the work examining the ability of barrier cells to secrete neuroimmune substances has been in vitro and has been conducted with BECs. Early work showed that BECs could secrete IL-6, prostaglandins, and nitric oxide constitutively and in response to stimulation (Fabry et al., 1993; Mandi et al., 1998; Reyes et al., 1999; McGuire et al., 2003). Later work found that many other cytokines are secreted by barrier cells. Major principles can now be elucidated for this area, including the following: 1) constitutive secretion can be modulated by immune stimulants, such as LPS, viruses and viral proteins, and bacteria (Hofman et al., 1999; Didier et al., 2002); 2) barrier cells can respond to immune stimulation acting at either their CNS or blood side; 3) coculture with other components of the NVU such as astrocytes and pericytes can modulate secretion of cytokines and other neuroimmune substances; and 4) secretion is bipolar in the sense that cytokines can be secreted from the luminal (i.e., into the blood) or the abluminal (i.e., into the brain) side of the BBB.
The combination of axis 2 and axis 5 means that a barrier cell can receive immune stimulation for one side (e.g., the blood or luminal side) and release cytokine from the other side (e.g., the brain or abluminal side) (Verma et al., 2006). As such, this forms a type of neuroimmune axis in which information from one side of the BBB is transferred to the other.
There are several examples in which barrier secretions respond to immune stimulations. In response to LPS, BECs release granulocyte-macrophage colony-stimulating factor and IL-6, which then act in an autocrine fashion to increase transport of HIV-1 across the BBB. Pericytes secrete substances, possibly keratinocyte chemoattractant and CCL2, that further enhance this effect of LPS (Dohgu and Banks, 2013). Adiponectin can act at the luminal membrane of the BEC to modify BEC secretion of IL-6 into the brain.
The combination of axis 2 and axis 5 is also a basis for interactions between the BBB and other elements of the NVU. Most of these have been elucidated in vitro and some are complex, involving bidirectional communications among several cell types. For example, McCarthy and Kosman (2014) have elucidated a pathway involving immune regulation of BBB transport of iron by astrocytic ceruloplasmin. In this scenario, LPS acting at the luminal surface of BECs elicits secretion of IL-6 and IL-1β from their abluminal surface; these cytokines then stimulate glial/astrocytic secretion of ceruloplasmin, which then acts at the abluminal surface of the BEC to faciliate BBB transport of iron (McCarthy and Kosman, 2014). Insulin transport across the BBB also shows a complex interplay of cellular crosstalk. An in vivo study showed that animals treated with LPS have an increased rate of transport of insulin across the BBB that is mediated by nitric oxide. However, this effect is not direct. LPS treatment increases activity of all three nitric oxide isoenzymes, but nitric oxide generated from neuronal nitric oxide synthase inhibits insulin function. LPS also stimulates endothelial nitric oxide synthase and neuronal nitric oxide synthase, which indirectly through an unknown cell and unknown mediator increase insulin transport across the BBB (Banks et al., 2008). Other examples of crosstalk include the ability of Aβ peptide to induce release of chemokines from monocytes that then act on BECs to increase immune cell trafficking (Fiala et al., 1998) and astrocytic release of TNF, inducing BECs to secrete nitric oxide (Shafer and Murphy, 1997).
IV. Physiologic Conditions, Disease States, and Pharmacologic Agents
The interplay of the BBB and neuroimmunology has the potential to both provide a substrate for diseases of the CNS and also for pharmacologic intervention. There is an increasing number of conditions and treatments that involve the BBB and neuroimmunology. Below, we consider some examples from a rapidly growing field.
A. Sickness Behavior
Sickness behavior is a conglomeration of host responses to infection or inflammation and includes lethargy; sleepiness; depression/anhedonia; anorexia; anxiety/irritability; decreased social interactions; loss of interest in sex, grooming, and the environment; and cognitive changes, including deceased concentration, learning, and memory (Dantzer and Kelley, 2007). Sickness behavior, at least short-term, is adaptive in that it reorganizes the host’s priorities to cope with infection.
Sickness behavior is not only an important adaptive response to illness, but also important as a model of psychoneuroimmune processes. Sickness behavior can be induced by a wide range of immune stimulants, but IL-1 seems to be especially efficient at inducing the complete range of behaviors (Bluthe et al., 1992; Larson and Dunn, 2001). A role for the BBB has been elucidated for several aspects of sickness behavior (Fig. 6). As discussed above, transport of IL-1 to the posterior division of the septum is an important step in decreased learning and memory (Banks et al., 2001a). Activation of BEC cyclooxygenase (COX)-1 or COX-2 with subsequent production of prostaglandin E2 is important in activation of the hypothalamic-pituitary-adrenal (HPA) axis, fever, and malaise/discomfort, the latter mediated through the striatum and GABAergic inhibition of dopaminergic cells (Engstrom et al., 2012; Fritz et al., 2016; Nilsson et al., 2017). Transport of kynurenine into the brain by the amino acid transporter LAT-1 is key to the depression-like symptoms of sickness behavior (O’Connor et al., 2009; Walker et al., 2015). By blocking kynurenine transport, leucine can confer antidepressant effects. COX-2 activation in an unknown cell type other than the BEC is involved in anorexia (Nilsson et al., 2017). The mechanisms underlying other aspects of sickness behavior are yet to be explored.

Sickness behavior. IL-1 induces the full spectrum of sickness behavior, but other inflammatory agents can directly or through IL-1 release elicit many aspects as well. IL-1 transport across the BBB of the posterior division of the septum (PDS) acts there to induce cognitive impairments. Prostaglandin E2 (PGE2) produced by stimulation of BEC COX-1 and COX-2 alters the HPA, induces fever, and results in malaise/discomfort, the latter mediated through dopaminergic and GABAergic pathways. COX-2 of non-BEC origin mediates anorexia. Depression-like symptoms are mediated through enhanced activity of indoleamine 2,3-dioxygenase (IDO), resulting in increased blood levels of kynurenine, which crosses the BBB using the large-neutral amino acid transporter (LAT-1). Kynurenine entry into brain is opposed by a brain-to-blood efflux transporter. Taste aversion is COX-2 independent; mechanisms of many other sickness behaviors are yet to be fully elucidated.
B. Perinatal Brain Ischemia
Hypoxia-ischemia is a common problem that can occur during gestation, with birth, or in the postnatal period. In the short-term, it can result in seizures and an encephalopathy, and long-term it can result in cerebral palsy, impairments in hearing and vision, cognitive impairments, and problems with attention and irritability. In utero brain ischemia induces proinflammatory events, including increased increases in IL-1β and IL-6 in the brains of both mothers and their fetuses (Sadowska et al., 2012), opening of the fetal BBB, and increased cytokine transport into the fetal brain (Chen et al., 2012; Sadowska et al., 2015; Patra et al., 2017). That these cytokine elevations are causal to brain damage is illustrated by the ability of blocking antibodies directed against IL-1β and IL-6 to reduce cytokine transport across the BBB, reduce cytokine levels in brain, and attenuate or reverse BBB disruption (Chen et al., 2015; Zhang et al., 2015; Patra et al., 2017). Thus, blocking antibodies directed against these cytokines has therapeutic potential.
C. Multiple Sclerosis
MS is a chronic neuroinflammatory disease that affects the brain and spinal cord. The average age of MS onset is 30 years and causes serious physical disabilities in those afflicted. MS presents heterogeneously, with symptoms that may include sensory and visual disturbances, impaired motor functions, pain, fatigue, and/or cognitive dysfunction depending on the afflicted CNS regions (Dendrou et al., 2015). The most common course of MS is relapsing-remitting, in which the presentation of symptoms is followed by partial or complete recovery. However, there are also progressive courses of MS, in which symptoms do not improve and become worse over time. In primary progressive MS, neurologic symptoms worsen from the onset of disease presentation. Secondary progressive MS follows an initial relapsing-remitting course (Lublin et al., 2014). Inflammatory lesions of infiltrating immune cells are a hallmark of MS, and the inflammation and gliosis within these regions contribute to demyelination and axonal degeneration. Although the precise cause of MS is unknown, the disease is thought to arise from an autoimmune response to brain antigens (Dendrou et al., 2015). In human disease, it is unclear whether autoimmunity develops in the CNS or in the periphery. However, in rodent experimental autoimmune encephalomyelitis (EAE), which is a model of human MS, autoimmunity is induced outside of the CNS via either immunization against brain antigen or systemic injection of T-cells that are autoreactive against brain antigens (Baxter, 2007; Lassmann, 2008). In MS and EAE, T-cells infiltrate the brain in early stages of disease (Fletcher et al., 2010; Dendrou et al., 2015).
In EAE models that employ exogenous application of autoreactive T-cells to induce disease, very few of these T-cells initially invade the CNS parenchyma (Engelhardt and Ransohoff, 2012). This is, in part, because the BBB is not fully primed for leukocyte recruitment and so does not efficiently permit T-cell diapedesis. Encephalitogenic T-cell blasts that are injected into naive mice interact with spinal cord white matter microvessels via a direct capture in the absence of rolling that is mediated by α4-integrin and vascular cell adhesion molecule (VCAM)-1 (Vajkoczy et al., 2001). However, T-cell trafficking through the choroid plexus also occurs, and this T-cell founder population is thought to contribute to activation of CNS parenchymal vessels and the subsequent robust recruitment of T-cells across the vascular BBB into the spinal cord parenchyma (Reboldi et al., 2009).
It is also appreciated that CNS autoreactive T-cells rapidly home to peripheral secondary lymphoid organs following peripheral injection, followed by their egress back into the blood and subsequent recruitment to the CNS, where they initiate the symptomatic phase of EAE ( Flugel et al., 2001; Odoardi et al., 2012). Furthermore, T-cells that were reisolated after homing to lymphoid tissues could rapidly cross the BBB and initiate disease sooner in naive recipients (Odoardi et al., 2012), suggesting that homing to peripheral lymphoid tissues contributes to the clinical course of EAE. Notably, this same study found that the bronchus-associated lymphoid tissues (BALT) were a major site of T-cell homing. Although rats express BALT constitutively, in mice and humans, BALT is induced under conditions such as respiratory infections or chronic pulmonary diseases (Hwang et al., 2016). Whether inducible BALT also contributes to MS or EAE is not clear; however, inflammatory stimuli such as infections or toxicants that induce BALT may also be relevant in symptomatic MS (Dendrou et al., 2015).
Pathologic changes at the BBB are also observed in MS lesions, and these include changes in expression of TJ proteins (Plumb et al., 2002; Kirk et al., 2003). Fibrinogen leakage is also evident in MS lesions, and fibrinogen can be a potent activator of microglia (Adams et al., 2007; McQuaid et al., 2009). Proteins implicated in BBB maintenance are also affected during MS. Endothelial IL-25 is downregulated in active MS lesions, indicating a loss of BBB protective functions. In contrast, sonic hedgehog is upregulated in EAE and in active MS lesions, which may be a protective response to counteract leukocyte trafficking and demyelination (Adams et al., 2007).
There are currently many disease-modifying therapies for relapsing-remitting MS, and one recently approved therapy for primary-progressive MS (English and Aloi, 2015; Dargahi et al., 2017). In general, these therapies are immunomodulatory, limit lymphocyte proliferation, inhibit lymphocyte trafficking, and/or protect against oxidative damage (English and Aloi, 2015; Dargahi et al., 2017). Despite their disease efficacy, some MS therapies that deplete T-cells or that inhibit T-cell trafficking are associated with progressive multifocal leukoencephalopathy (PML), which is a rare condition caused by reactivation of the JC virus in the CNS (Misbah, 2017). The association of PML with T-cell deficiency underscores a functional role for T-cells in protecting the CNS from latent viral infections and will be discussed in detail below.
D. Human Immunodeficiency Virus-1 Penetration of the Blood–Brain Barrier and Consequences of Human Immunodeficiency Virus-1 Infection on Blood–Brain Barrier Function
HIV infection continues to be a burden in developed and underdeveloped nations, and it is estimated that over 30 million people worldwide are HIV positive (Hong and Banks, 2015). One complication of HIV infection is CNS dysfunction, termed HIV-associated neurologic disorder (HAND) (Saylor et al., 2016). HAND encompasses diverse clinical symptoms, ranging from mild, asymptomatic neurocognitive impairment to dementia in the most severe cases, and about 50% of HIV-positive patients are afflicted with HAND (Heaton et al., 2010). Prior to the widespread use of highly active antiretroviral therapy (HAART), about 20% of HIV-infected patients eventually developed HIV encephalitis (HIVE) (Gelman, 2015). The major neuropathological changes associated with HIVE included microglia nodules and multinucleated giant cells, which were associated with active HIV infection predominantly in microglia and brain macrophages (Gelman, 2015). However, in the post-HAART era, HIVE is increasingly rare, and neuropathological features of milder forms of HAND include synaptic loss and neuroinflammation, but not overt neurodegeneration (Everall et al., 1999; Gelman, 2015).
HIV establishes reservoirs in the CNS soon after infection (Davis et al., 1992; Persidsky and Poluektova, 2006), and HIV entry into the CNS is thought to occur by free virus as well as infected monocytes or T-cells entering the CNS via hematologic routes across brain barriers (Hong and Banks, 2015). HIV-1 pseudovirus crosses the BBB by adsorptive transcytosis via the mannose-6-phosphate receptor (Banks et al., 2001b, 2005; Dohgu et al., 2012), and vesicular uptake of HIV in BECs has been demonstrated ultrastructurally within minutes of injection (Dohgu et al., 2012). HIV-infected monocytes are also believed to cross the BBB within days of initial infection. HIV-infected monocytes can cross the BBB in vitro, and cell–cell contacts between HIV-infected monocytes and human BECs initiate the upregulation of E-selectin on BECs (Nottet et al., 1996). Furthermore, E-selectin and VCAM-1 are upregulated in brains of AIDS patients with HIVE, and tissue-binding assays revealed that human monocytes preferentially bound endothelial cells in brain tissue regions where E-selectin and VCAM-1 were upregulated in HIVE patient samples (Nottet et al., 1996). Upon CNS entry, HIV is thought to predominantly infect perivascular macrophages during peak viremia (Williams et al., 2001), and subsequently, establish reservoirs in brain macrophages and microglia (Schnell et al., 2011; Joseph et al., 2015). Infected microglia may then propagate neuroinflammation, which subsequently recruits leukocytes to the CNS, including those infected by HIV (Fischer-Smith et al., 2001; Yadav and Collman, 2009; Gray et al., 2013).
Systemic and neuroinflammation may contribute to CNS invasion of HIV. LPS enhances the transport of gp120 and HIV-1, and pericytes contribute to the increased transport of HIV-1 in vitro (Banks et al., 1999; Dohgu and Banks, 2013). The LPS-induced increases in transport of free HIV across the in vitro BBB are mediated by IL-6 and granulocyte cell-stimulating factor (Dohgu et al., 2011). LPS also potentiates the upregulation of VCAM-1 and E-selectin induced by HIV-1–infected monocytes (Nottet et al., 1996). Notably, the activation of circulating immune cells by bacterial antigens may be relevant in the course of HIV, as the acute phase of HIV infection is associated with increased gut permeability, microbial translocation, and increased concentrations of circulating LPS (Douek, 2007). HIV also has modulatory effects on BECs and induces the upregulation and release of BEC endothelin-1 (Didier et al., 2002), a cytokine whose levels in CSF correlate with the degree of HIV encephalopathy (Rolinski et al., 1999). Tat and gp120 induce oxidative stress in BECs (Price et al., 2005), and blood-borne factors in gp120 mice are sufficient to induce BBB disruption (Cioni and Annunziata, 2002). There is also evidence of BBB disruption in HAND. In autopsied brain tissue from HIV-positive patients with encephalitis, there were discontinuities in ZO-1 and occludin staining in regions with astrocytosis and accumulation of HIV-infected macrophages (Dallasta et al., 1999).
As many anti-retroviral therapy (ART) drugs are ABC transporter substrates, altered ABC transporter expression during HIV infection may have important consequences on the pharmacokinetics and pharmacodynamics of ART (Seifert et al., 2017), including their efficacy in suppressing HIV in the CNS. Low-grade, chronic inflammation is evident in HIV-1–infected patients, including those on ART with viral suppression (Seifert et al., 2017), and inflammatory substances such as bacterial cell wall constituents and cytokines are known to alter the expression and functions of ABC transporters at the BBB (see section IV.I., below). Application of the HIV-1 protein Tat to mouse brain microvessels in vitro and to mouse brains in vivo upregulates the expression and function of Pgp (Hayashi et al., 2005). Furthermore, exposure of primary human brain endothelial cells to HIV-1 induces increased expression of Pgp mRNA, but functional increases in Pgp were not evident unless the cells were coexposed to HIV-1 and a HIV protease inhibitor (Roy et al., 2013). Conversely, a nonsignificant decrease in brain microvascular Pgp expression was observed in autopsied brain tissue from AIDS patients with HIVE versus AIDS patients without HIVE (Langford et al., 2004). Disparities in Pgp changes at the BBB observed in cells and mice acutely treated with HIV-1 and its proteins versus autopsied human brain tissue could, in part, be explained by the chronicity of HIV-1–associated inflammation that may contribute differently to transporter expression versus acute exposure to HIV-1. Furthermore, functional changes in Pgp can be regulated post-translationally (Hawkins et al., 2010), and therefore may not always be reflected in the assessment of protein expression. HIV-1 infection is also known to affect ABC transporters, including Pgp, BCRP, and MRPs in other tissue types, including glial cells that may affect ART distribution in the CNS and NVU function (Alam et al., 2016).
In summary, the interaction of HIV with brain barriers is necessary to establish CNS infection early in the course of disease, and subsequent BBB-disruptive and proinflammatory responses to CNS HIV infection contribute to the persistent recruitment of HIV to the CNS and damaging neuroinflammatory responses that contribute to the development of HAND.
E. Amyloid β Peptide and Alzheimer Disease
AD is a progressive neurodegenerative disease characterized by dementia and deposition of Aβ and τ in brain regions that are important for learning and memory [for a recent review, see Masters et al. (2015)]. Aβ accumulation, aggregation, and deposition in the CNS are thought to be a causative pathologic event in AD (Selkoe and Hardy, 2016). The CNS prevents Aβ deposition by a number of mechanisms, including enzymatic degradation, microglial phagocytosis, clearance by CSF bulk flow, and efflux at the BBB. Furthermore, impaired efflux of Aβ across the BBB has been implicated in AD pathogenesis. LRP-1 is an efflux transporter located on brain endothelial cells and choroid plexus epithelial cells whose ligands include Aβ (Shibata et al., 2000; Deane et al., 2004; Donahue et al., 2006; Fujiyoshi et al., 2011). Impaired efflux of Aβ from the CNS occurs in AD patients and in animal models of AD largely because of impaired function of LRP-1 (Erickson and Banks, 2013). There is also an influx transporter for Aβ, the RAGE, which facilitates the passage of Aβ from the blood-to-brain direction and is upregulated in AD (Deane et al., 2003). Additionally, ABC transporters including Pgp and BCRP are thought to contribute to Aβ efflux, either by facilitating transcytosis of Aβ in the brain-to-blood direction across the luminal membrane of brain endothelial cells, or by limiting the influx of Aβ that is circulating in blood (Tai et al., 2009; Hartz et al., 2010; Do et al., 2012). The efflux transporter, Pgp, is downregulated in AD (van Assema and van Berckel, 2016).
AD has several links to inflammation, including being considered a proinflammatory disease and the long-held clinical observation that infections or other inflammatory events can unmask or worsen AD (Holmes, 2013). Systemic inflammation can increase Aβ production, as well as inhibit major Aβ clearance mechanisms, including CSF bulk flow and Aβ efflux across the BBB (Lee et al., 2008; Jaeger et al., 2009b; Erickson et al., 2012b). LRP-1 transport activity is inhibited by inflammation and results in decreased efflux of Aβ peptide from the brain (Erickson et al., 2012b). Decreased efflux of Aβ peptide as shown in LRP-1 knockdown mice results in increased brain levels of Aβ peptide and cognitive impairment (Jaeger et al., 2009a; Storck et al., 2016). Thus, decreasing LRP-1 function could be one mechanism by which inflammation enhances the onset, progression, or unmasking of AD. Systemic inflammation also modulates Pgp expression and function (see Introduction) and increases Aβ influx in the absence of changes in RAGE protein expression in brain microvessels (Jaeger et al., 2009b).
F. Neuromyelitis Optica
Neuromyelitis optica (NMO), like MS, is an autoimmune inflammatory disease resulting in demyelination that affects the optic nerve and spinal cord (Khorooshi et al., 2015). Autoantibodies directed at aquaporin-4, a water channel primarily located on brain ependymal cells and astrocytes, are considered the immediate cause, resulting in astrocytic activation, complement fixation, neuroinflammation with cytokine release, immune cell infiltration, and BBB disruption. Aquaporin-4 is highly expressed in ependymal cells, neuroepithelium denudation occurs in NMO, and anti-aquaporin antibodies are elevated in the CSF, all suggesting that NMO involves the choroid plexus or blood–CSF barrier (Castaneyra-Ruiz et al., 2014). The antibody is an IgG that must reach the brain to induce disease and is ineffective when given peripherally unless the animals already have CNS inflammation resulting from a NMO-like picture (Castaneyra-Ruiz et al., 2014).
Other autoantibodies are found in NMO, including those directed against the myelin oligodendrocyte glycoprotein, N-methyl-D-asparate–type glutamate receptor, glycine receptor, and glucose-regulated protein 78 (GRP78) (Shimizu et al., 2017). GRP78 is a heat-shock protein and expressed in all brain cells, including BECs. Autoantibodies to GRP78 bind to BECs and increase nuclear factor κB (NF-κB) and intercellular adhesion molecule, decrease claudin-5, and increase BBB leakage to macromolecules, including fibrinogen, albumin, and IgG (Shimizu et al., 2017). Anti-GRP78 is also found in diseases that do not have NMO, such as some cancers and systemic lupus erythematosis. Thus, GRP78 autoantibodies are unlikely to cause NMO, but do appear to be altering several aspects of BBB function, including NF-κB–mediated immune functions, immune cell trafficking, and a BBB disruption that is possibly mediated by transcytotic mechanisms.
G. Euthyroid Sick Syndrome
Tanycytes are involved in the mechanism by which systemic inflammation induces euthyroid sick syndrome (Lee and Farwell, 2016), a condition by which the body can decrease its caloric needs in times of illness. Like sickness behavior, euthyroid sick syndrome, also known as nonthyroidal illness syndrome, can be considered adaptive, at least short-term. A hallmark of euthyroid sick syndrome is a low blood level of thyroid-stimulating hormone (TSH) in the face of low blood levels of thyroid hormones, especially of triiodothyronine (T3); this is paradoxic in that low thyroid hormone levels should stimulate TSH. This paradox is evidence of the resistance to the stimulatory effects of TSH and of thyroid-releasing hormone in this condition. Inflammation increases type 2 deiodinase in hypothalamic tanycytes, which is dependent on the RelA subunit of NF-κB (de Vries et al., 2016). This increase in type 2 deiodinase at the tanycyte results in an increase in T3 locally within the hypothalamus, the increased T3 in turn suppressing the expression of thyroid-releasing hormone and its ability to stimulate TSH (de Vries et al., 2014, 2016), and the suppression in TSH, finally resulting in lower levels of thyroid hormones in blood.
H. Chemobrain
Chemotherapy-induced cognitive dysfunction (CICD) or “chemobrain” is a syndrome of cognitive impairment that occurs in about 30%–70% of cancer victims treated with various chemotherapeutics and lasts long after the discontinuation of the chemotherapies. The mechanism by which a chemotherapeutic such as doxorubicin (Dox; also known as adriamycin) induces CICD is unclear, as Dox is prevented from crossing the BBB by Pgp (Darvari and Boroujerdi, 2005). One mechanism (Fig. 7) by which Dox could induce CICD directly involves the BBB’s ability to transport TNF (Gutierrez et al., 1993). In brief, Dox produces superoxide free radicals, oxidizing ApoA1 in blood, which then induces monocytes and macrophages to release TNF into the bloodstream (Tangpong et al., 2006, 2007). TNF is transported across the BBB by a saturable transport system (Gutierrez et al., 1993) into the CNS. TNF transported across the BBB induces oxidative and nitrosative stress within the brain and acts directly on glial cells to induce them to release TNF (Joshi et al., 2010). Mn superoxide dismutase, which scavenges superoxide in mitochondria, is nitrated and dysfunctional, leading to mitochondrial damage and apoptosis (Tangpong et al., 2006, 2007). TNF, both that derived from the blood and from the CNS, induces neuronal toxicity and apoptosis, resulting in the behavioral changes of CICD (Aluise et al., 2011; Keeney et al., 2013; Butterfield, 2014).

Chemobrain or CICD. The BBB is involved in mediation of chemobrain in two known ways. The efflux transporter Pgp prevents doxorubicin from entering the brain. TNF is transported across the BBB, where it acts directly to induce apoptosis as well as to induce release of additional TNF from glial cells.
Notably, the oxidative stress and inflammation known to be caused by some chemotherapeutics may also affect their disposition in the brain, as many chemotherapeutic drugs are also ABC transporter substrates (Deeken and Loscher, 2007), and ABC transporters are regulated by neuroinflammation (see section I). For example, Dox was shown to increase MDR-1 expression in whole brains of rodents (Joshi et al., 2010). The relationship of chemotherapy-associated systemic and neuroinflammation and subsequent effects on the regulation of ABC transporters at the BBB is an important, yet understudied topic of relevance to brain cancer therapy.
I. Inflammation, ATP-Binding Cassette Transporters, and Central Nervous System Drug Delivery
The presence of multidrug efflux transporters at the BBB limits the distribution of many classes of drugs to the CNS, including those for cancer, viral and bacterial infections, epilepsy, and pain (Loscher and Potschka, 2005). In many of these conditions, systemic and/or neuroinflammation occurs, which can alter efflux transporter expression at the BBB. For example, Pgp expression and function are modulated by LPS and proinflammatory cytokines. However, the effects of inflammation on Pgp vary depending on the inflammatory model. Pgp activity at the BBB is downregulated in rodents when inflammation is initiated with LPS in the peritoneum, skin, or the lateral ventricle (Goralski et al., 2003; Salkeni et al., 2009; Jin et al., 2011; Erickson et al., 2012a). In contrast, inflammatory pain in rodents upregulates Pgp protein expression and function, limiting the uptake of morphine into the CNS (Seelbach et al., 2007). Isolated rat brain microvessels treated with low levels of LPS or TNF-α cause a rapid, reversible loss of Pgp activity with no change in protein expression (Hartz et al., 2006). However, longer exposure to TNF causes an upregulation of Pgp expression and activity (Bauer et al., 2007; Yu et al., 2007). TGF has also been shown to upregulate Pgp function (Dohgu et al., 2004). TNF also decreases expression of Mrp2 and Mrp4, and does not affect protein expression of Mrp1 and BCRP in isolated rat brain microvessels (Bauer et al., 2007). BCRP expression and activity are reduced by IL-1β, IL-6, and TNF-α in a human BEC line (Poller et al., 2010).
Neuroinflammation may alter the efficacy of CNS therapeutics by altering the expression of efflux transporters, as was discussed for ABC transporter expression in HIV infection in a prior section. There are multiple classes of drugs that are prescribed, typically in combination, for the treatment of HIV-1 infection that target various stages of the HIV replication cycle. These include fusion inhibitors, nucleoside reverse transcriptase inhibitors (NRTIs), non-NRTIs (NNRTIs), protease inhibitors, and integrase strand transfer inhibitors. The current HAART regimens recommended by the World Health Organization as first-line treatment of HIV-1 infection in adults consist of two NRTIs plus a NNRTI or an integrase strand transfer inhibitor. Currently, the preferred drug regimen to initiate ART in HIV-1–infected adults includes a fixed-dose combination of tenofovir (NRTI), lamivudine or emtricitabine (NRTI), plus efavirenz (NNRTI). Recommendations for alternative treatment regimens and second-line treatments in individuals with viral failure have also recently been updated by WHO (2016). HIV protease inhibitors, integrase inhibitors, and the nucleoside analog reverse-transcriptase inhibitor zidovudine are ABC transporter substrates. In human BECs, Pgp and MRPs limit the uptake of saquinavir and zidovudine (Eilers et al., 2008). Indinavir, nelfinavir, and atazanavir are also substrates of Pgp (Kim et al., 1998; Robillard et al., 2014a,b). Furthermore, saquinavir can upregulate the expression and function of Pgp on BECs (Roy et al., 2013), whereas ritonavir inhibits MRP-1 (Olson et al., 2002). Altered expression of brain efflux transporters was also observed in a transgenic rat model of HIV-1 infection (Robillard et al., 2014b).
Pgp upregulation has been reported in rodent stroke models with or without reperfusion (Spudich et al., 2006; Cen et al., 2013; DeMars et al., 2017). Furthermore, upregulation of Pgp expression in these models is concurrent with reduced CNS uptake of Pgp substrates, despite evidence of BBB disruption (Spudich et al., 2006; Cen et al., 2013). Hypoxia and low glucose mediate Pgp upregulation in brain endothelial cells in vitro, which may, in part, be due to the upregulation of hypoxia-inducible factor-1 (Comerford et al., 2002; DeMars et al., 2017). Notably, Pgp inhibition improved the neuroprotective effects of FK506 and rifampicin in a rodent stroke model (Spudich et al., 2006). Therefore, therapeutic efficacy of neuroprotective agents in stroke depends in part on whether they are Pgp substrates.
About 30% of patients are resistant to anti-epileptic drugs, and this may be because they are the group who is genetically determined to have an increased Pgp activity (Loscher and Potschka, 2002). Upregulation of Pgp from multiple seizures is thought to be neuroimmune mediated and may underlie why patients in status epilepticus are so resistant to treatment (Loscher and Potschka, 2002; Rizzi et al., 2002; Lazarowski et al., 2004; Kumar et al., 2014).
Pgp also plays a role in brain uptake, accumulation, and efflux of Aβ peptide, morphine, opiate peptides, and chemotherapeutics (Letrent et al., 1999; Thompson et al., 2000; Lam et al., 2001; Kastin et al., 2002; Darvari and Boroujerdi, 2005; Erickson et al., 2012b). As such, neuroimmune-related alterations in Pgp function at the BBB provide another mechanism by which inflammation can affect AD, analgesia, and response of CNS cancers to chemotherapeutics.
J. Emergence of Progressive Multifocal Leukoencephalopathy Associated with Therapies That Inhibit T-Cell Immune Surveillance
Progressive multifocal leukoencephalopathy is a devastating infection of the brain caused by lytic reactivation of the JC polyoma virus. About 30%–90% of the human population is seropositive for JC virus (Misbah, 2017), and early studies using polymerase chain reaction–based methods of detection indicated that about 50% of non-PML presenting cases had detectable levels of JC virus in their brains (White et al., 1992). However, JC virus persists as a latent, asymptomatic infection in immunocompetent individuals. PML was first reported as a rare side effect of chemotherapy in patients with leukemia and lymphoma (Astrom et al., 1958), and subsequent PML cases prior to the HIV epidemic were associated with immunosuppressive therapies or rare primary immunodeficiencies (Bag et al., 2010). An increase in PML cases emerged during the early AIDS epidemic, and about 5% of AIDS patients developed PML prior to the widespread use of HAART (Berger et al., 1998). The emergence of PML in AIDS patients led to the realization that severe deficiency in T-cell immunity is necessary for JC virus activation and neurotropism (Misbah, 2017).
In the post-HAART era, there has been a dramatic decrease in the incidence of PML associated with HIV-1 infection (Bag et al., 2010). However, new cases of PML have emerged as side effects of new immunosuppressive and immunomodulatory therapies. One example is natalizumab, which is a monoclonal antibody that binds α4β1 and α4β7 integrins, and interferes with α4 integrin interactions with VCAM-1 that mediates T-cell trafficking to the CNS. Soon after its approval, two participants from the phase III trial developed PML. Notably, PML had not previously been associated with MS, and so natalizumab was temporarily suspended from the market (Misbah, 2017). After a detailed assessment of all patients receiving natalizumab, the risk for PML was estimated to be approximately 1/1000, and thus determined to be sufficiently low to continue marketing. However, it is now appreciated that the PML risk for natalizumab may be as high as 1/44, depending on the length of treatment, JC virus seropositivity, and prior treatment with immunosuppressive drugs (Misbah, 2017). Notably, another α4 integrin inhibitor, vedolizumab, which is specific for α4β7 and indicated for Crohn’s disease, has not been associated with PML (Dallasta et al., 1999). This indicates that it is specifically the impairment of lymphocyte trafficking to the brain that increases PML risk, and that T-cells are important contributors to CNS immunity.
K. IL-1ra, Febrile Infection-Related Epilepsy Syndrome, and Neonatal-Onset Multisystem Inflammatory Disease
The treatment of two childhood diseases apparently depends on the ability of IL-1ra to cross the BBB. Neonatal-onset multisystem inflammatory disease, also known as chronic infantile neurologic cutaneous articular syndrome, presents as a rash before age 6 weeks and has several CNS manifestations, including chronic aseptic meningitis, cerebral atrophy with enlarged ventricles, increased intracranial pressures, papilledema with optic nerve atrophy, mental retardation, seizures, and sensorineural hearing loss. About 60% of cases have a mutation in the CIAS1 gene that encodes for cryopyrin, a protein that forms part of a macromolecular inflammasome, activating caspase 1, which, in turn, cleaves pro–IL-1β.
Inflammation can be causal in certain types of epilepsies, and febrile infection-related epilepsy syndrome (FIRES) is one example (Nabbout, 2012). FIRES involves acute onset of refractory seizures following a febrile infection. However, CSF reveals no pathogens and brain biopsies show gliosis but no inflammation. Treatment with anakinra, an analog of IL-1ra, is very effective at controlling CNS symptoms, including seizures, in both these syndromes. This is consistent with IL-1ra being able to control status epilepticus in a rodent model (Marchi et al., 2009). Anakinra was more effective than canakinumab, an antibody directed against IL-1β, in lowering CSF cytokines (Rodriguez-Smith et al., 2017). This difference between anakinra and canakinumab may be that IL-1α, which is blocked by anakinra but not by canakinumab, plays a role in seizure activity, as indicated by genetic data (Saghazadeh et al., 2014). Alternatively, anakinra may, like IL-1ra, be transported across the BBB to a much greater extent than antibodies (Gutierrez et al., 1994). CSF and serum levels of anakinra increase arithmetically with increasing doses of i.v. anakinra (Fox et al., 2010). CSF/Serum ratios calculated as individual time points or as areas under the curve give values of about 0.28%.
L. Immunomodulatory Therapies and Stroke
Ischemic stroke results from blockage of a cerebral artery, either due to the local formation of a thrombus or a distal embolism. Current treatments for ischemic stroke rely on rapid clearance of the blockage by proteolytic or mechanical approaches (Emberson et al., 2014; Campbell et al., 2015), and tissue plasminogen activator remains the only Food and Drug Administration–approved treatment of ischemic stroke. However, the prospects of immunomodulatory therapies in stroke have been and continue to be tested in clinical trails (Veltkamp and Gill, 2016). One approach to modulate neuroinflammation in stroke is via the inhibition of leukocyte trafficking to the brain by targeting specific cell adhesion molecules that are expressed on leukocytes or brain endothelial/epithelial cells. Such treatments have had varied success in clinical trials (Veltkamp and Gill, 2016). Recent findings that leukocytes can be protective under certain conditions of CNS injury (Shechter et al., 2013) and that novel pathways of leukocyte trafficking to peri-infarct areas have been identified that cirvumvent conventional routes of BBB transport (Llovera et al., 2017) may provide important insight for future directions in this therapeutic approach.
IL-1 is also an important mediator in stroke, and blockade of IL-1 receptor signaling is another therapeutic strategy for stroke under active investigation (Sobowale et al., 2016; Veltkamp and Gill, 2016). An IL-1ra analog modified by attaching a 21-amino-acid cell-penetrating peptide to the C terminus showed a twofold to fourfold increase in brain uptake. This increase in uptake was not accountable by improved pharmacokinetics. In a stroke model, both native IL-1ra and the analog showed improvements in infarct size, neurologic scores, brain edema, oxidative stress measures, and motor performance, but the analog was superior to native IL-1ra (Zhang et al., 2017a). That IL-1ra exerts its improvement on stroke through a central mechanism was further shown by positive effects after delivery to the brain using the intranasal route (Frey, 2002; Lee et al., 2017).
M. Methamphetamine and Blood–Brain Barrier Alterations
Methamphetamine is prescribed in the United States as a treatment of obesity, attention deficit hyperactivity disorder, narcolepsy, and idiopathic hypersomnia, and is over-the-counter in its levo-form as a nasal decongestant. It is best known as a recreational drug with high addiction potential and a high degree of neurotoxicity, especially to dopamine neurons. Methamphetamine alters BBB, BEC, and NVU function. In BECs, methamphetamine induces oxidative stress, inducing changes in myosin light chain kinases and actin-related protein 2/3, resulting in decreased TJ proteins and increased monocyte migration (Ramirez et al., 2009; Zhang et al., 2009; Park et al., 2013). HIV-1 and methamphetamine have a complex relationship, including each enhancing the oxidative stress and BBB changes induced by the other (Banerjee et al., 2010). Similar events occur in the blood–spinal cord barrier (Kiyatkin and Sharma, 2015). Autopsy material from methamphetamine users show increased expression of aquaporin-4 and matrix metalloproteases and decreased TJ protein expression (Wang et al., 2014a). Pericyte migration and alterations in astrocytic aquaporin 4 levels, the latter resulting in vasogenic edema, also occur after methamphetamine use (Leitão et al., 2017; Zhang et al., 2017b). These changes likely result from neuroinflammation, as evidenced by glial activation and cytokine release from astrocytes and BECs (Loftis and Janowsky, 2014), including TNF-α and IL-1β (Goncalves et al., 2017). TNF release from astrocytes plays a role in methamphetamine-induced BBB disruption, first resulting in an opening of the transcytotic pathways and later in the paracellular pathways (Coelho-Santos et al., 2015). Endothelin-1 from BECs activates endothelial nitric oxide synthase and induces vasoconstriction (Seo et al., 2016). Interestingly, exercise in mice protects against the BBB changes of methamphetamine, possibly mediated by the ability of exercise to lower levels of inflammatory cytokines ((Allen et al., 2015; Park et al., 2016).
N. Cerebral Cavernous Malformations
Cerebral cavernous malformations are hemangiomas in the brain and can result in seizures, focal neurologic deficits, and hemorrhagic strokes. Three genetic mutations resulting in loss of function have been identified that affect signaling through Notch, mitogen-activated protein kinase ERK1/2, WNT/β-catenin, and TFG-β pathways (Schulz et al., 2015; Stamatovic et al., 2015; Kim, 2016), leading to perictye loss and BBB leakiness. Activation of Toll-like receptor 4 on BECs has been associated with enhanced cerebral cavernous malformation formation, and the source of activation appears to be the gut microbiome (Tang et al., 2017).
O. Antibody-Associated Autoimmune Encephalitis Syndromes
A series of conditions that involve autoantibodies directed against CNS receptors has been described in recent years. The CNS antibodies presumably come in part from the periphery and so likely cross the BBB by way of extracellular pathways, although BBB disruption of antibodies secreted from immune cells trafficking into the CNS is also a possibility. Antibodies are usually IgG and can be targeted to a number of CNS receptors; Oldham (2017) lists over two dozen conditions, and Crisp et al. (2016) focus on 12. The most common receptors known to be targeted are leucine-rich glioma-inactivated protein 1, the N-methyl-D-asparate receptor, and the glycine receptor. Leucine-rich glioma-inactivated protein 1 is high in the hippocampus and may be a modulator of the AMPA receptor, and antibodies directed against it induce limbic encephalitis, characterized by progressive cognitive impairment and seizures, often with hyponatremia. Other symptoms presented by these syndromes include narcolepsy, psychoses, migraines, hallucinations, anxiety, weight loss, and ataxia (Crisp et al., 2016). Onset of symptoms is sometimes related to infections or linked to malignancies, tumors, or autoimmune diseases, but a few cases have been associated with immune therapy (Brown et al., 2017). A few case reports place onset after BBB-disrupting diseases, including stroke and methamphetamine use (Simal et al., 2012; Iriondo et al., 2017). Treatments are directed against the immune response in some way and include steroids, cyclophosphamide, i.v. immunoglobulins, monoclonal antibodies, and immunoadsorption (Hansen et al., 2016).
V. Conclusions and Future Directions
The BBBs endow the CNS with immune privilege by preventing the unregulated exchange of immune-related cells and substances between the CNS and blood. The barriers, however, do allow regulated communication between the CNS and the peripheral tissues by a number of integrated mechanisms that can be divided with current knowledge into five neuroimmune axes. These axes depend on specialized properties of the barrier cells, such as their abilities to secrete and respond to immune-related substances, to recruit immune cells and facilitate their passage across the BBB, and to transport immune substances such as cytokines from blood to brain. These axes allow the barrier cells to engage in a two-way communication with circulating immune cells and peripheral tissues via secretions into the bloodstream and most of the known cell types within the CNS; this concept of two-way communication is embodied in the term NVU. Transport of substances across the BBB and polarized secretions allows the BBB to act as a conduit of information between the CNS and peripheral tissues. As a result, the BBB is able to adapt to the needs of the CNS and allows communication between the peripheral tissues and the CNS.
These axes are important to the maintenance of health, and their breakdown or failure can result in disease. A number of conditions are recognized that involve barrier cell/neuroimmune interactions that are adaptive, such as sickness behavior and euthyroid sick syndrome, or are clearly pathologic, such as multiple sclerosis and chemobrain. These axes are relevant for treatment of CNS diseases as well. In some cases, the neuroimmune reactions run counter to therapeutic intervention, as illustrated by upregulation of Pgp activity with seizures and stroke, thus fostering drug resistance. In other cases, the neuroimmune axes participate in the therapeutic intervention, such as the transport of IL-1ra in the syndromes of FIRES and neonatal-onset multisystem inflammatory disease.
Future directions are likely to discover more functions, properties, and interactions among the cells of the NVU, and even new cell types in the periphery or CNS may be discovered. Recent studies have emphasized the importance of pericytes at the NVU, and there is renewed interest in the mast cell. New functions and even axes are likely to be discovered for the barrier cells: the vascular BBB is an area that is greatly understudied, but the choroid plexus and tanycytic barriers have even less work done on them. The literature of the neuroimmunology of nerve blood barriers, the blood–retinal barrier, and other specialized barriers is very small. The number of disease states in which the brain plays a role and that have an inflammatory or immune aspect is growing tremendously. This review will hopefully serve as a guide as those conditions and diseases are investigated from a neuroimmune point of view, but will likely quickly become antiquated as new types of interactions are discovered.
Perhaps the most exciting and promising area is that of pharmacology and the treatment of conditions with barrier/interface-neuroimmune components. Drugs that can cross the BBB to access the CNS are critically needed. But the various axes provide ways that neuroimmune-modifying drugs can have targets on the blood side of the barrier and still affect CNS function. For example, decreasing Pgp activity in status epilepticus could restore responsiveness. As Pgp is on the luminal side of the BBB, allosteric regulators or competitive blockers do not have to cross the BBB to access the transporter. The ability of an immune substance to act at the luminal surface of the BBB and modulate secretions from the BBB abluminal surface is another strategy that does not require the xenobiotic to enter the CNS. This strategy also has the advantage that the therapeutic activity in the brain is produced by the patient’s own body. Modulation of barrier cell activities is also a strategy that only requires action at the barrier cell itself, not penetration into brain. Reversal of euthyroid sick syndrome by targeting tanycyte type 2 deiodinase or inhibiting fever by targeting barrier cell cyclooxygenase in sickness behavior are additional examples. Disruption of the BBB to enhance drug delivery, although often tried, has yet to prove a viable strategy because of the neurotoxicity that results from a leaky BBB. Blockade of substances entering the brain is another approach; for example, blocking circulating antibodies from entering the brain could be effective in treating NMO and autoimmune encephalitis syndromes. The development of these and other strategies for the treatment of CNS diseases will be dependent on our understanding of the interplay of the neuroimmune system with the BBBs and blood–brain interfaces.
Acknowledgments
We thank Emily Wing for outstanding contribution to the figures.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Banks, Erickson.
Footnotes
This work was supported by the Department of Veterans Affairs and the National Institutes of Health National Institute on Aging [Grant R01 AG046619].
Abbreviations
- Aβ
- amyloid β
- ABC
- ATP-binding cassette
- AD
- Alzheimer disease
- AJ
- adherens junction
- ART
- anti-retroviral therapy
- BALT
- bronchus-associated lymphoid tissue
- BBB
- blood–brain barrier
- BCRP
- breast cancer resistance protein
- BCSFB
- blood–CSF barrier
- BEC
- brain endothelial cell
- BUI
- brain uptake index
- CCL
- C-C motif chemokine ligand
- CICD
- chemotherapy-induced cognitive dysfunction
- CMB
- cerebral microbleed
- CNS
- central nervous system
- COX
- cyclooxygenase
- CP
- choroid plexus
- CPE
- choroid plexus epithelium
- CSF
- cerebrospinal fluid
- CVO
- circumventricular organ
- DHA
- docosahexaenoic acid
- DOX
- doxorubicin
- EAE
- experimental autoimmune encephalomyelitis
- ECM
- extracellular matrix
- FIRES
- febrile infection-related epilepsy syndrome
- GRP78
- glucose-regulated protein 78
- HAART
- highly active antiretroviral therapy
- HAND
- HIV-associated neurologic disorder
- HIV
- human immunodeficiency virus
- HIVE
- HIV encephalitis
- IDT
- indicator diffusion technique
- IFN
- interferon
- IL
- interleukin
- IL-1R1
- type 1 IL-1 receptor
- ISF
- interstitial fluid
- JAM
- junctional adhesion molecule
- LPC
- lysophosphatidylcholine
- LPS
- lipopolysaccharide
- LRP
- low-density lipoprotein receptor-related protein
- LTP
- long-term potentiation
- ME
- median eminence
- MFSD2A
- major facilitator superfamily domain-containing 2A
- MMP
- matrix metalloproteinase
- MRP
- multidrug resistance protein
- MS
- multiple sclerosis
- NF-κB
- nuclear factor κB
- NLR
- nucleotide-binding oligomerization domain-like receptor
- NMO
- neuromyelitis optica
- NNRTI
- non-NRTI
- NOD
- nucleotide-binding oligomerization domain
- NRTI
- nucleoside reverse transcriptase inhibitor
- NVU
- neurovascular unit
- OVLT
- organum vasculosum of the lamina terminalis
- PDGF-B
- platelet-derived growth factor subunit B
- PDGFRβ
- platelet-derived growth factor receptor β
- Pgp
- P-glycoprotein
- Plvap
- plasmalemma vesicle-associated protein
- PML
- progressive multifocal leukoencephalopathy
- RAGE
- receptor for advanced glycation endproducts
- RMT
- receptor-mediated transcytosis
- T3
- triiodothyronine
- TEER
- transendothelial electrical resistance
- TGF
- transforming growth factor
- TJ
- tight junction
- TNF
- tumor necrosis factor
- TSH
- thyroid-stimulating hormone
- VCAM
- vascular cell adhesion molecule
- VEGF
- vascular endothelial growth factor
- ZO
- zonula occulins
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
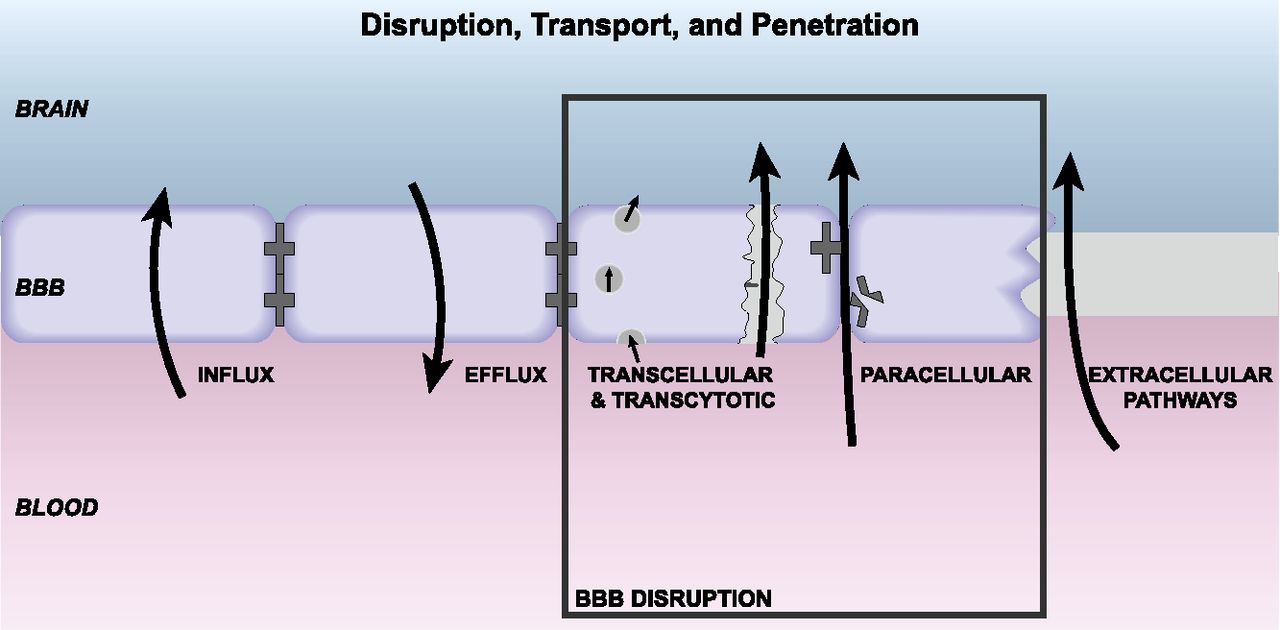{kind=link}
{kind=link}
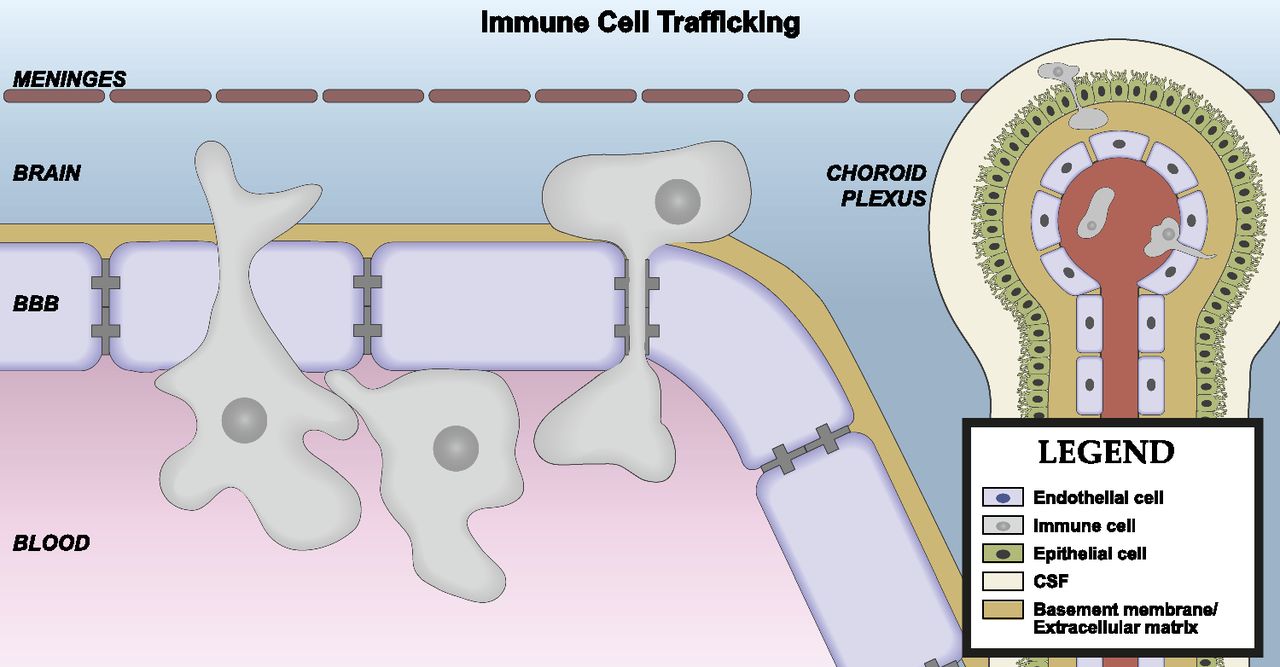{kind=link}
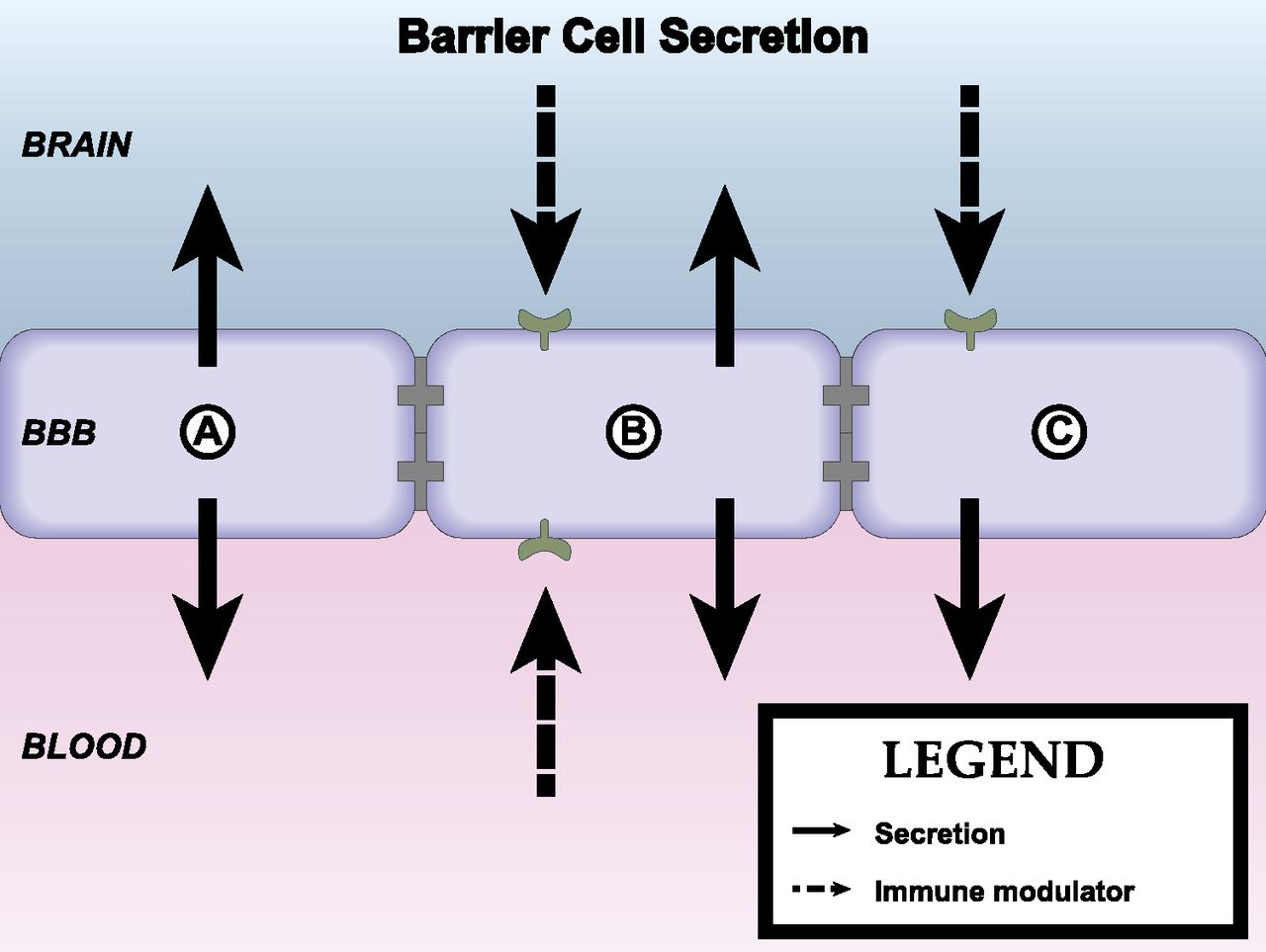{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Features and Functions of the Blood–Brain Barrier and Blood–Brain Interface
- III. The Neuroimmune Axes
- IV. Physiologic Conditions, Disease States, and Pharmacologic Agents
- V. Conclusions and Future Directions
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters