


Abstract
The cardiovascular and other actions of angiotensin II (Ang II) are mediated by AT1 and AT2 receptors, which are seven transmembrane glycoproteins with 30% sequence similarity. Most species express a single autosomal AT1 gene, but two related AT1A and AT1B receptor genes are expressed in rodents. AT1 receptors are predominantly coupled to Gq/11, and signal through phospholipases A, C, D, inositol phosphates, calcium channels, and a variety of serine/threonine and tyrosine kinases. Many AT1-induced growth responses are mediated by transactivation of growth factor receptors. The receptor binding sites for agonist and nonpeptide antagonist ligands have been defined. The latter compounds are as effective as angiotensin converting enzyme inhibitors in cardiovascular diseases but are better tolerated. The AT2receptor is expressed at high density during fetal development. It is much less abundant in adult tissues and is up-regulated in pathological conditions. Its signaling pathways include serine and tyrosine phosphatases, phospholipase A2, nitric oxide, and cyclic guanosine monophosphate. The AT2 receptor counteracts several of the growth responses initiated by the AT1 and growth factor receptors. The AT4 receptor specifically binds Ang IV (Ang 3–8), and is located in brain and kidney. Its signaling mechanisms are unknown, but it influences local blood flow and is associated with cognitive processes and sensory and motor functions. Although AT1 receptors mediate most of the known actions of Ang II, the AT2 receptor contributes to the regulation of blood pressure and renal function. The development of specific nonpeptide receptor antagonists has led to major advances in the physiology, pharmacology, and therapy of the renin-angiotensin system.
I. Introduction
A. Historical Background
Blood pressure was measured for the first time in 1733 by Stephen Hales, in a dramatic experiment on a horse, by inserting a brass pipe into the carotid artery. The technique of modern blood pressure measurement was introduced in 1905 by Nicolai Korotkov using the stethoscope invented by Laennec in 1815 and the relatively recently devised wraparound inflatable rubber cuff. The latter was first described by Riva-Rocci in 1896 and was improved by von Recklinghausen in 1901 (Freis, 1995).
The first insight into the regulation of blood pressure came from the discovery of a pressor principle by Tigerstedt and Bergman in 1897. They called this factor “renin” because it was extracted from the kidney. This pioneering work led to the description of reno-vascular hypertension in animals and in humans (Goldblatt et al., 1934). However, it was not until 1940 (Braun-Menendez et al., 1940) that a vasoconstrictor substance was isolated from renal venous blood from the ischemic kidney of a Goldblatt hypertensive dog. A similar finding was made simultaneously and independently by Page and Helmer (1940) after the injection of renin into an intact animal. This group also isolated a so-called “renin activator” that later proved to be angiotensinogen. The pressor substance was named “hypertensin” in Argentina and “angiotonin” in the United States and was later isolated and shown to be an octapeptide (Skeggs et al., 1956; Bumpus et al., 1957; Elliott and Peart, 1957). There were differences between laboratories concerning interpretations and nomenclature but in fact hypertensin and angiotonin were the same substance. In 1958, Braun-Menéndez and Page agreed on the hybrid term angiotensin for the highly potent pressor octapeptide. This proved to be an appropriate choice, given the later recognition of angiotensin's numerous actions in addition to its hypertensive effects. The sequence of angiotensin II is Asp-Arg-Val-Tyr-Ile-His-Pro-Phe in the human, horse, and pig. In bovine angiotensin II, the isoleucine residue in position 5 is replaced by valine.
Following this major discovery, the various components of the cascade leading to the formation of angiotensin II were characterized, including angiotensinogen, angiotensin converting enzyme (ACE),2 and angiotensins I, II, and III (Table 1). The synthesis of the peptide angiotensin II by Bumpus et al. (1957) and by Rittel et al. (1957) was followed by a continuing series of investigations into the structure-activity relationship of angiotensin analogs, mainly in the hope of finding a peptide antagonist.
Amino acid sequences of Ang II precursors and metabolites
In 1987, a committee of the International Society for Hypertension, The American Heart Association, and the World Health Organization proposed abbreviating angiotensin to Ang using the decapeptide angiotensin I as the reference for numbering the amino acids of all angiotensin peptides (Dzau et al., 1987).
Angiotensin II plays a key role in the regulation of cardiovascular homeostasis. Acting on both the “content” and the “container”, Ang II regulates blood volume and vascular resistance. The wide spectrum of Ang II target tissues includes the adrenals, kidney, brain, pituitary gland, vascular smooth muscle, and the sympathetic nervous system. Angiotensin is not only a blood-borne hormone that is produced and acts in the circulation but is also formed in many tissues such as brain, kidney, heart, and blood vessels. This has led to the suggestion that Ang II may also function as a paracrine and autocrine hormone, which induces cell growth and proliferation and controls extracellular matrix formation (Dzau and Gibbons, 1987; Griffin et al., 1991; Weber et al., 1995a,b). Other angiotensin-derived metabolites such as angiotensin 2–8 (Ang III), angiotensin 1–7, or angiotensin 3–8 (Ang IV) have all been shown to have biological activities (Table 1) (Peach, 1977; Schiavone et al., 1990; Ferrario et al., 1991; Ferrario and Iyer, 1998; Wright et al., 1995).
As for other peptide hormones, Ang II was postulated to act on a receptor located on the plasma membrane of its target cells. This receptor should possess the dual functions of specific recognition of the ligand and stimulation of the characteristic cellular response. Comparison of changes in steroidogenesis in the adrenal cortex, adrenal catecholamine release, and developed tension in aortic strips in response to Ang I, Ang II, and Ang III clearly indicated different affinities of these target organs for the three peptides (Peach, 1977;Devynck and Meyer, 1978). These pharmacological experiments showed that effector organs responded to Ang I, II, and III with 2 to 3 log differences in potency from tissue to tissue. Based on these studies, Ang II receptor selectivity for the agonists was proposed to be structure-activity related. Comparison of Ang II and a large number of synthetic agonists and antagonists formed by substituting various amino acids of Ang II indicated marked dissimilarities between the analogs in each of the preparations, suggesting differences in the structure of the receptor sites (Khosla et al., 1974; Papadimitriou and Worcel, 1974; Peach and Levens, 1980).
Early binding studies detected sites with binding characteristics that differed between the various target tissues (Peach and Levens, 1980). Also, receptor density was up- or down-regulated in different tissues following either Ang II infusion or Na+restriction (Aguilera and Catt, 1978). The characterization of receptor types in rat liver and kidney cortex (Gunther, 1984; Douglas, 1987; Bouscarel et al., 1988b) suggested further Ang II receptor heterogeneity. An early classification proposed for Ang II receptor types was based on studies in only a few tissues or species (Levens et al., 1980; Peach and Levens, 1980; Ferrario et al., 1991). It was not until the end of the 1980s that tools became available to demonstrate the existence of at least two receptor types in many tissues for which the conventional peptide analogs such as saralasin have high affinity but little or no selectivity. These included the nonpeptide antagonists losartan (or Ex89 or DuP 753) and PD123177, and a new generation of peptide ligands such as CGP42112 and p-aminophenylalanine Ang II (Chiu et al., 1989a; Whitebread et al., 1989; Speth and Kim, 1990). This new development was made simultaneously and independently in three different laboratories, and the initial nomenclature was confusing: the receptor sensitive to losartan was called 1, B, or α, and that with no affinity for losartan was termed 2, A, or β. The High Blood Pressure Research Council in 1990 and the International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC-IUPHAR) in 1992 therefore appointed a subcommittee3 to address the problem, and a classification was proposed in 1991 and updated in 1995 (Bumpus et al., 1991; de Gasparo et al., 1995).
B. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification Criteria for Classification
To obtain a “fingerprint” capable of identifying distinct receptors, three main criteria have been proposed: operational, transductional, and structural (Humphrey et al., 1994). The operational criteria include the drug-related characteristics of the receptor, such as ligand binding affinities, and selective agonists and antagonists. The receptor-effector coupling events constitute the transductional criteria, and the receptor sequence and gene cloning represent the structural criteria. It is clear that all of these criteria are not necessarily achieved simultaneously and at an early stage. The coupling mechanism may not have a major influence on receptor pharmacology but it helps in differentiating receptor types. Also, receptors with diverse structures may respond to the same endogenous ligands. Finally, receptors may be cloned without having a known pharmacology. The combination of the three criteria should clearly help in defining true receptor types.
Any such classification will essentially evolve as our knowledge increases. Nevertheless, there is a need for an official scheme that will help to avoid confusion among investigators. Two Ang II receptor types fulfill the three classification criteria, and are termed AT1 and AT2 receptors. According to the NC-IUPHAR recommendation, the AT1 and AT2 receptors have an IUPHAR Receptor Code of 2.1.Ang.01.000.00.00 and 2.1 Ang.02.000.00.00 (Humphrey and Barnard, 1998). The first two numbers indicated the structural class: they are seven transmembrane domain, G protein-coupled receptor (GPCR) member of the rhodopsin subclass (2.1). The receptor family is abbreviated Ang. The types indicated as 01 and 02 for AT1 and AT2. The following series of null are reserved for splice variants chronologically numbered according to identification within species.
Two other receptors (AT3 and AT4) have been proposed, based on operational criteria, but their transduction mechanisms are unknown and they have not yet been cloned. The name AT3 was initially given to a binding site described in the Neuro-2a mouse neuroblastoma cell line that was not blocked by either AT1-specific losartan, or AT2-specific PD123319 and was not affected by GTP analogs (Chaki and Inagami, 1992). This AT3binding site, which has a low affinity for Ang III, should be called a non-AT1-non-AT2 site until more information about its nature has been obtained. The endogenous ligand for the AT4 receptor is Ang 3–8 or Ang IV. Its binding properties and physiological characteristics, described in more detail in another section, are sufficiently different from those of the AT1 and AT2receptor to warrant keeping the name AT4 for this putative Ang IV-selective receptor until the binding protein is cloned and further characterized.
C. Current Nomenclature
The present angiotensin receptor identification is based on six principles. 1) The receptor is abbreviated to AT followed by a numerical subscript. 2) Further subdivisions are indicated by subscript letters that are in upper case for pharmacologically defined receptor subtypes (e.g., AT1B). 3) The species is identified by a lowercase prefix preceding AT (e.g., r AT1, h AT2). There is a space between the species and the receptor name. 4) Mutant receptors should be designated with specification of the position of the amino acid substitution in bracket (e.g., [L112P]AT1Awhen leucine at position 112 has been changed to proline. 5) The human gene is written in upper case and preferably but not essentially in italics (e.g., AGTR1 and AGTR2). In mouse and rats, it would be Agtr1a, Agtr1b and Agtr2 in lowercase.
D. Structural Analysis
The strategy of expression cloning was successfully applied to the AT1 receptors of rat smooth muscle and bovine adrenal gland, and subsequently the corresponding receptors of mouse, rabbit, human, pig, dog, turkey, and frog angiotensin receptors were cloned and sequenced. The nonmammalian receptors have 60% identity with the mammalian receptor and are pharmacologically distinct in their ligand binding properties. Hydropathy analysis indicated that both AT1 and AT2 receptors contain seven hydrophobic transmembrane segments forming α helices in the lipid bilayer of the cell membrane. The structural information for the AT1 receptor is coded as follows: h 359 aa, P30556, chr.3. This indicates that the human AT1receptor contains 359 amino acids, with the sequence reported in the SwissProt file under the number 30556 and the gene coding for the receptor (abbreviated AGTR1) is located on chromosome 3 q. Similarly, the structural coding for rat and mouse AT1 receptor is r 359 aa, P29089, P25095 and m 359 aa, P29754, P29755 as there are two subtypes A and B in rat and mouse located on chromosomes 17 and 2 and 13 and 3, respectively. The AT2 receptor is only 34% identical with its AT1 counterpart (Fig.1). The structural information is coded h 363 aa, P50052, chr.X q22-q23 as the gene AGTR2 is located on human chromosome X with the cytogenetic location q23-q24. For rat and mouse, the respective information is r 363 aa, P35351 and m 363 aa, P35374. As in human, the AT2 receptor in rodents is also located on chromosome X.

Secondary structure and consensus sequence of the mammalian angiotensin AT1 receptor. The amino acid sequence shown is based on the derived sequences of five individual cloned mammalian AT1 receptors. The amino acid residues that are highly conserved among G protein-coupled receptors are indicated by bold letters. The positions of the three extracellular carbohydrate chains, and of the two extracellular disulfide bonds, are also indicated.
An evolutionary analysis based on the alignment of cloned AT1 receptor sequences, using the CLUSTAL algorithm of PC/gene, has suggested that rat and mouse AT1 receptors coevolved. (Sandberg, 1994). Amphibian and avian receptors diverged early during evolution. So far, gene duplication has been observed only in rats and mice (see following section). Two isoforms of the AT1 receptor derived by alternative splicing of the same gene have been reported in man (Curnow et al., 1995). They have similar binding and functional properties. A receptor with as much as 97% identity to the AT1 receptor has been cloned from human placenta (Konishi et al., 1994). It differs in its C-terminal amino acid sequence, tissue distribution, and pharmacological properties. The gene has not been cloned and it may well be a splice variant of the AT1 receptor.
II. The Type 1 (AT1) Angiotensin Receptor
The angiotensin AT1 receptor mediates virtually all of the known physiological actions of angiotensin II (Ang II) in cardiovascular, renal, neuronal, endocrine, hepatic, and other target cells. These actions include the regulation of arterial blood pressure, electrolyte and water balance, thirst, hormone secretion, and renal function. The AT1 receptor belongs to the G protein-coupled receptor (GPCR) superfamily and is primarily coupled through pertussis toxin-insensitive G proteins to the activation of phospholipase C and calcium signaling. The AT1receptors of several species have been cloned and their amino acid sequences determined from the respective cDNAs. Ang II binding to the AT1 receptor induces a conformational change in the receptor molecule that promotes its interaction with the G protein(s), which in turn mediate signal transduction via several plasma membrane effector systems. These include enzymes, such as phospholipase C, phospholipase D, phospholipase A2, and adenylyl cyclase, and ion channels, such as L-type and T-type voltage-sensitive calcium channels. In addition to activating several intracellular signaling pathways that mediate agonist-induced phenotypic responses in a wide variety of Ang II target cells, the agonist-occupied AT1 receptor undergoes desensitization and internalization in the same manner as many other GPCRs.
The cellular responses to AT1 receptor signaling include smooth muscle contraction, adrenal steroidogenesis and aldosterone secretion, neuronal activation, neurosecretion, ion transport, and cell growth and proliferation. The AT1 receptor is coupled not only to the well recognized Gq-mediated calcium and protein kinase C signaling pathways, but also to intracellular signaling cascades that extend into the nucleus. These pathways regulate gene transcription and the expression of proteins that control growth responses and cell proliferation in several Ang II target tissues. Some of the latter consequences of AT1 receptor activation are counteracted by the structurally dissimilar AT2receptor, which antagonizes the effects of AT1-mediated growth responses in several cell types, in particular endothelial cells, cardiomyocytes, and ovarian granulosa cells. These actions of the AT2receptor are described in more detail below. This account of the AT1 receptor will address its gene expression, ligand binding, activation and signal transduction pathways, and physiological roles in the regulation of the activity and growth of its major target cells in cardiovascular, neuronal, and endocrine tissues.
A. Angiotensin II Receptors: Early Studies
The angiotensin receptor was identified as a functional entity byLin and Goodfriend (1970), who first described the binding of radioiodinated Ang II to its receptor sites in the adrenal gland. These sites were subsequently shown to be located in the plasma membrane (Glossmann et al., 1974a), and the binding reaction was found to be influenced by the ambient Na+ concentration and guanyl nucleotides (Glossmann et al., 1974b,c). The G proteins had not been discovered at that time, but this finding indicated that the binding activity of a noncyclic AMP-coupled receptor is regulated by guanine nucleotides. Subsequent studies showed that the AT1 receptor is coupled to both Gq and Gi proteins in the adrenal glomerulosa zone and several other tissues in the rat.
Many of the properties of the angiotensin II receptor were first identified in studies on the adrenal gland and liver, both of which are abundant sources of receptors that are coupled to well defined physiological responses (Saltman et al., 1975; Campanile et al., 1982). As in the rat adrenal gland, guanine nucleotides reduced agonist binding of 125I-Ang II to hepatic receptors, largely by increasing its dissociation rate constant. Guanine nucleotides also decreased the number of high-affinity binding sites for Ang II, but not those for the peptide antagonist, [Sar1,Ala8]Ang II. These changes were accompanied by inhibition of adenylyl cyclase activity in hepatic membranes, and of cyclic AMP production in intact hepatocytes (Crane et al., 1982). The high-affinity Ang II receptors in the liver were found to be inactivated by dithiothreitol, with a concomitant loss of Ang II-induced stimulation of glycogen phosphorylase in isolated hepatocytes (Gunther, 1984). These and related studies also presaged the existence of angiotensin II receptor types with distinct biochemical properties and intracellular mechanisms of action. Differential effects of guanine nucleotides on receptor binding of Ang II agonist and antagonist ligands were also observed in the bovine adrenal gland. This effect was evident for both membrane-bound and solubilized receptors. Concerning the latter, the association of the agonist-occupied receptor with a putative G protein was suggested by its larger size on steric exclusion HPLC (De Lean et al., 1984).
The ability of Ang II to inhibit glucagon-stimulated cyclic AMP production in hepatocytes, and adenylyl cyclase activity in hepatic membranes, was consistent with its coupling to an inhibitory G protein, now termed Gi. This was confirmed by the ability of pertussis toxin to prevent the inhibitory action of Ang II on adenylyl cyclase. The ability of GTPγS to further reduce receptor binding affinity when all Gi molecules were ADP-ribosylated by the toxin indicated that Ang II receptors are also coupled to other G protein(s) that could mediate actions of Ang II on additional signaling pathways (Pobiner et al., 1985). Subsequent studies on cultured hepatocytes revealed a single population of Ang II binding sites and demonstrated that agonist and antagonist analogs had parallel actions on cytosolic calcium and phosphorylase activity, as did treatment with dithiothreitol to inactivate the receptors (Bouscarel et al., 1988a). Reconstitution studies in hepatocyte membranes showed that Gi3 is the major form of Gi in these cells and is responsible for coupling the Ang II receptor to agonist-induced inhibition of adenylyl cyclase (Pobiner et al., 1991). One of the few physiological actions of Ang II that is mediated by Gi, rather than Gq/11, is the AT1receptor-dependent stimulation of angiotensinogen production in the rat liver (Klett et al., 1993).
B. Cloned AT1 Receptors
The relatively low abundance of the AT1receptor in most Ang II target tissues, and the instability of the solubilized receptor molecule, impeded efforts to isolate and sequence the receptor protein. For this reason, expression cloning from bovine adrenal and rat smooth muscle cells was necessary to isolate the cDNAs encoding the receptor proteins of these species (Sasaki et al., 1991;Murphy et al., 1991). Both AT1 receptors were found to be typical seven transmembrane domain proteins, composed of 359 amino acids and with a molecular mass of about 41 kDa. The extracellular regions, composed of the N terminus and the three extracellular loops, contain three N-glycosylation sites and four cysteine residues (Fig. 1). Each of the consensus sites is glycosylated in the native AT1 receptor (Jayadev et al., 1999), which has a molecular mass of about 65 kDa. In addition to the two conserved cysteines that form a disulfide bond between the first and second extracellular loops of all GPCRs, the AT1 receptor contains an additional pair of extracellular cysteine residues. These are located in the N-terminal region and the third extracellular loop, and form a second disulfide bond that maintains the conformation of the AT1receptor protein (Ohyama et al., 1995). The latter disulfide bond, which is not present in the AT2 receptor, renders the AT1 receptor susceptible to inactivation by dithiothreitol and other reducing agents. The cytoplasmic region of the receptor, which is composed of the three intracellular loops and the C-terminal cytoplasmic tail, contains consensus sites for phosphorylation by several serine/threonine kinases, including protein kinase C (PKC) and GPCR kinases. Several of the specific residues that are phosphorylated during AT1receptor activation have been identified, but there are no confirmed reports of agonist-induced tyrosine phosphorylation of the AT1 receptor or other GPCRs.
Similar structural features are present in several other cloned mammalian and nonmammalian AT receptors. The rat and mouse AT1 receptors exist as two distinct subtypes, termed AT1A and AT1B, that are 95% identical in their amino acid sequences. The two subtypes are also similar in terms of their ligand binding and activation properties but differ in their tissue distribution, chromosomal localization, genomic structure, and transcriptional regulation. None of the other cloned mammalian AT1 receptors, including those from cow (Sasaki et al., 1991), human (Bergsma et al., 1992; Curnow et al., 1992), pig (Itazaki et al., 1993), rabbit (Burns et al., 1993), and dog (Burns et al., 1994) appear to have subtypes. The two AT1 subtypes in the rodent genome may be the consequence of a gene duplication event that occurred during evolution after the branching of rodents from the mammalian phylogenetic tree (Aiyar et al., 1994b).
C. Genomic Organization of Rat AT1A and AT1B Receptor Genes
The rat AT1A receptor gene is 84 kb in length and contains three introns and four exons, the third of which (∼2 kb) includes the entire 1077-base pair (bp) coding sequence of the receptor protein as well as 5′ and 3′ untranslated sequences (Langford et al., 1992; Murasawa et al., 1993; Takeuchi et al., 1993). The first two small exons encode alternatively spliced 5′ untranslated sequences, and the fourth exon (1 kb) encodes an additional 3′ untranslated sequence. A 2.3-kb transcript is found in all AT1A-expressing tissues and contains exons 2 and 3. An additional 3.3-kb transcript containing exons 2, 3, and 4 is present in vascular smooth muscle cells and several other tissues but is not found in the brain. The transcription start site of the AT1A receptor gene is located about 70 kb upstream from the exon that encodes the receptor protein. The rat AT1B receptor gene is about 15 kb in length and contains two introns and three exons, the first two of which encode 5′ untranslated sequences. The third exon contains the entire coding region of the receptor and the 3′ untranslated sequence. The AT1B receptor has 92 and 95% homology with the AT1A at the nucleotide level and amino acid levels, respectively (Guo and Inagami, 1994) and is expressed in relatively few tissues as a 2.4-kb transcript. The rat AT1A and AT1B receptor genes are located on chromosomes 17q12 and 2q24, respectively (Tissir et al., 1995).
D. Expression and Regulation of Rat AT1A and AT1B Receptor
AT1A and AT1Breceptors exhibit similar ligand binding and signal transduction properties but differ in their tissue distribution and transcriptional regulation. In the rat, AT1A and AT1B receptor mRNAs are expressed in numerous tissues, including adrenal, kidney, heart, aorta, lung, liver, testis, pituitary gland, and brain. AT1A transcripts are predominantly expressed in all tissues except the adrenal and pituitary glands, where the AT1B message is the major subtype. AT1A receptors are abundantly expressed in vascular smooth muscle cells, in which their properties and regulation have been extensively investigated. In the adult mouse, AT1A receptors are expressed in the kidney, liver, adrenal gland, ovary, brain, testis, lung, heart, and adipose tissue. In contrast, AT1B receptors are confined to the adrenal gland, brain, and testis (Burson et al., 1994).
Studies on the tissue-specific expression of AT1receptor by in situ hybridization revealed that liver, heart, and lung contain solely AT1A receptors, whereas the anterior pituitary gland contains only AT1Breceptors (Gasc et al., 1994). In the adrenal gland, the zona glomerulosa contains both AT1A and AT1B transcripts, the zona fasciculata contains little of either subtype, and only AT1A mRNA is present in the medulla. In the kidney, AT1A mRNA is present in mesangial and juxtaglomerular cells, proximal tubules, vasa recta, and interstitial cells, whereas AT1BmRNA is found only in mesangial and juxtaglomerular cells, and in the renal pelvis. In male rats, quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) showed that the relative abundance of AT1A transcripts is 100% in liver, 85% in lung, 73% in kidney, 48% in adrenals, and 15% in the pituitary gland (Llorens-Cortes et al., 1994). In contrast to the adult animal, only AT1A receptors are expressed in the pituitary gland during fetal and postnatal life.
The expression of the AT1A receptor is stimulated by glucocorticoids, which act via one of three putative glucocorticoid responsive elements located in its promoter region (Guo et al., 1995). In the rat heart, where the AT1A receptor is expressed in 10-fold excess over the AT1Breceptor, treatment with dexamethasone increased AT1A and AT1B mRNA levels by 100 and 300%, respectively (Della Bruna et al., 1995). Conversely, deoxycorticosterone acetate suppressed AT1A mRNA levels by 70%, indicating that glucocorticoids and mineralocorticoids exert reciprocal actions on AT1A receptor levels in the heart. In the heart and aorta, transcripts for both AT1 subtypes were reduced by treatment with an AT1 receptor antagonist. However, the AT1B subtype was preferentially reduced, suggesting that the expression of AT1B receptors in the adrenal is dependent on the activity of the renin-angiotensin system (Kitami et al., 1992).
Estrogens also influence the expression of AT1receptors, and exert divergent actions on subtype abundance in the pituitary gland and vascular smooth muscle. Estrogen treatment suppresses the expression of AT1B but not AT1A mRNA in the pituitary gland (Kakar et al., 1992). On the other hand, AT1A receptor expression in vascular smooth muscle is elevated in ovariectomized rats and restored to normal by estrogen replacement (Nickenig et al., 1996). In cultured vascular smooth muscle cells, a high concentration of estradiol (1 μM) reduced AT1A mRNA by about 30%. Whether estrogen deficiency leads to increased vascular AT1 receptor expression in the human has yet to be determined.
Other forms of hormonal regulation of AT1receptor expression include the insulin-induced up-regulation of vascular AT1 receptor expression, which has been attributed to a post-translational mechanism (Nickenig et al., 1998). In cultured vascular smooth muscle cells, insulin caused a doubling of AT1 receptor density and a concomitant increase in the Ang II-induced intracellular Ca2+response. This increase in receptor content, which was dependent on tyrosine phosphorylation and the intracellular Ca2+ response, was due to an increase in receptor mRNA stability rather than increased gene transcription. In rat astrocytes, growth hormone but not insulin-like growth factor 1 (IGF-1) also increased AT1A receptor expression. This was associated with an increase in gene transcription and elevated mRNA levels. AT1B receptors, which were much less abundant than the AT1A subtype, were not affected by growth hormone treatment (Wyse and Sernia, 1997). On the other hand, nitric oxide (NO) caused a marked decrease in AT1A gene expression in vascular smooth muscle cell (VSMC) that was independent of changes in cyclic GMP. This was accompanied by an inhibitory action of NO on the expression of a reporter gene containing 616 bp of the AT1receptor gene promoter, and reduced association with a DNA binding protein that interacts with this region (Ichiki et al., 1998).
E. The Human AT1 Receptor
The human AT1 receptor contains 359 amino acids, and its deduced amino acid sequence is 95% identical with those of the rat and bovine AT1 receptors (Curnow et al., 1992; Bergsma et al., 1992; Furuta et al., 1992). The receptor is derived from a single large gene that contains five exons ranging in size from 59 to 2014 bp (Guo et al., 1994). The open reading frame of the AT1 receptor is located on exon 5. The other four exons participate to varying degrees in alternative splicing to produce mature RNAs that encode two receptor isoforms that are translated with different efficiencies (Curnow et al., 1995). The inclusion of exon 2 occurs in up to 50% of AT1mRNAs and inhibits the translation of the downstream AT1 receptor sequence. In about one-third of AT1 transcripts, the splicing of exon 3 to exon 5 yields a receptor with a 32 amino acid N-terminal extension. The ligand binding and signaling properties of this receptor are similar to those of the predominant shorter isoform of the AT1receptor.
The human AT1 receptor gene is located on the q22 band of chromosome 3 (MEM number 106165) (Curnow et al., 1992;Davies et al., 1994). An additional human AT1 receptor gene was suggested by the report of a human cDNA clone that differed from the known sequence in 10 of its 359 residues (Konishi et al., 1994), but subsequent studies have not confirmed the existence of a second gene (Curnow, 1996; Su et al., 1996). However, most human Ang II target tissues also express the slightly longer and functionally similar AT1receptor that results from alternative splicing of exons 3/5 as noted above. The longer isoform appears to be better expressed at the plasma membrane in cell transfection studies, but there is no evidence to suggest that it has a significant physiological role in AT1 receptor function (Curnow, 1995).
Expression of the human AT1 receptor is enhanced by epidermal growth factor in transfected COS-7 cells (Guo and Inagami, 1994b). Relatively little is known about the control of expression of the AT1 receptor in most Ang II target tissues in the human. In the reproductive system, both Ang II and its AT1 and AT2 receptor types are present in the endometrium and exhibit cyclic changes during the menstrual cycle with a maximum in the early secretory phase (Ahmed et al., 1995). AT1 receptors are expressed in the glands and the endometrial blood vessels and may participate in uterine vascular regulation and regeneration of the endometrium after menstruation. The human placenta expresses the AT1 receptor and all other components of the renin-angiotensin system. The receptors are present throughout gestation in the syncytiotrophoblast and cytotrophoblast, and in the fetal vascular endothelial cells (Cooper et al., 1999). AT1 receptor mRNA transcripts (2.4 kb) and receptor protein (83 kDa) increase progressively during pregnancy and reach their maximal level in the term placenta (Petit et al., 1996).
A local renin-angiotensin system is also present in human adipose tissue, with expression of angiotensinogen, ACE, and AT1 receptor genes in omental and s.c. fat and cultured adipocytes (Engeli et al., 1999). The extent to which these components are related to the development of hypertension and obesity-related disorders has yet to be established. In the human kidney, AT1 receptors are expressed in the renal vasculature, glomeruli, and the vasa recta bundles in the inner stripe of the outer medulla (Goldfarb et al., 1994). AT1 receptors are diminished in the glomeruli of patients with chronic renal disease (Wagner et al., 1999). The AT1 receptors expressed in cultured human mesangial cells mediate Ang II-induced hypertrophy and proliferative responses, implying that Ang II may be involved in the pathogenesis of glomerulosclerosis (Orth et al., 1995).
Similar effects of Ang II are mediated by AT1receptors in human pulmonary artery smooth muscle cells, in which Ang II stimulates DNA and protein synthesis. This response was associated with activation of mitogen-activated protein kinase (MAPK) and was prevented by losartan and by the MAPK inhibitor, PD-98059. These findings suggest that Ang II-induced activation of the AT1 receptor initiates signaling pathways that participate in growth and remodeling of the human vascular system (Morrell et al., 1999).
In erythroid progenitor cells, which express both AT1 and erythropoietin (EPO) receptors, Ang II enhances EPO-stimulated erythroid proliferation in vitro (Mrug et al., 1997). In vivo, the β2-adrenergic receptor-induced production of EPO in normal subjects was inhibited by losartan treatment, implying that Ang II is a physiological regulator of EPO production in the human (Freudenthaler et al., 1999).
1. AT1 Receptor Gene Polymorphisms and Cardiovascular Disease.
The discovery of several polymorphisms in the human AT1 receptor gene, one of which (A1166C) was more frequent in hypertensive subjects (Bonnardeaux et al., 1994), initiated a series of studies on the role of such mutations in the genesis of hypertension and other cardiovascular disorders. Subsequently, this polymorphism was reported to act synergistically with the angiotensin converting enzyme DD genotype on the risk of myocardial infarction (Tiret et al., 1994). However, the results of subsequent reports on this topic have not been consistent. In some studies, the A1166C polymorphism had no effect on ambulatory blood pressure, left ventricular mass, or carotid arterial wall thickness (Castellano et al., 1996; Schmidt et al., 1997). In other reports, the same AT1 receptor gene polymorphism was associated with increased coronary arterial vasoconstriction in response to methylergonovine maleate (Amant et al., 1997), essential hypertension (Szombathy et al., 1998; Kainulamen et al., 1999), and increased left ventricular mass but not hypertension (Takami et al., 1998). An analysis of the role of this polymorphism in rats overexpressing the mutant human AT1 receptor in the myocardium suggested that it is associated with increased responsiveness to Ang II. This may lead to cardiac hypertrophy under high-renin conditions or during pressure and volume overload (Van Geel et al., 1998).
F. The Amphibian AT1 Receptor
In the Xenopus laevis oocyte, endogenous Ang II receptors were detected in the ovarian follicular cells that surround the oocyte. These receptors mediate Ang II-induced elevations of cytoplasmic Ca2+ in the oocyte via gap junctions between follicular cells and oocyte (Sandberg et al., 1990, 1992b) and are thus functionally identifiable as AT1receptors. However, the amphibian (xAT) receptor for Ang II did not recognize the nonpeptide antagonist, DuP753, that inhibits the binding and actions of Ang II at the mammalian AT1receptor (Sandberg et al., 1991). The xAT receptor cDNA was cloned from a Xenopus myocardial cDNA library to investigate the structural basis of this functional distinction in ligand binding. The xAT receptor is a 41-kDa protein containing 362 amino acids that has 60% amino acid identity and 65% nucleotide homology with the coding regions of known mammalian AT1 receptors (Ji et al., 1993; Aiyar et al., 1994a). When expressed in Xenopusoocytes, xAT receptors mediate Ang II-induced Ca2+ mobilization and are pharmacologically distinct from mammalian AT1 receptors. Receptor transcripts are present in Xenopus lung, liver, kidney, spleen, and heart, but not in adrenal, intestine, and smooth muscle. Mutational analyses of xAT and rat AT1 receptors have largely elucidated the structural basis of their individual ligand binding properties, as described below.
G. The AT1 Receptor Null Mouse
Gene targeting experiments have provided several important insights into the physiological role of the renin-angiotensin system in cardiovascular regulation, fluid and electrolyte balance, and development. Deletion of the genes encoding angiotensinogen (Tanimoto et al., 1994; Kim et al., 1995) and ACE (Krege et al., 1995; Esther et al., 1996) revealed that the lack of Ang II in Agt−/− orAce−/− mice was associated with hypotension, reduced survival, and marked abnormalities in renal development. Most of the Agt null animals died before weaning and most of the ACE null mice died within 12 months. The Agt and ACE null animals that survived until adult life had severe renal lesions. In both cases, the kidneys showed focal areas of cortical inflammation, thickened arterial walls, and medullary hypoplasia with a consequent deficit in urinary concentration. An additional feature of interest in theAce−/− animals was impaired fertility in the male animals, although histologically the sperm appeared to be normal and had normal motility (Krege et al., 1995; Esther et al., 1997). This was dependent on the loss of a testis-specific ACE isozyme that is expressed in round and elongating spermatids and was associated with defects in sperm transport in the oviduct and binding to the zona pellucida of the oocyte (Hagaman et al., 1998). In mice lacking both ACE isozymes, male fertility was selectively restored by sperm-specific expression of the testicular isoenzyme (Ramaraj et al., 1998).
The effects of disruption of the mouse gene encoding the AT1 receptor types were in part predictable from the results of deleting the genes encoding angiotensinogen and ACE. Mice lacking a functional AT1A receptor had a significant reduction of resting blood pressure (ca. 20 mm Hg) and lacked the pressor/depressor responses to infused Ang II that occur in normal animals. However, the Agtr1a(−/−) animals showed no marked impairment of development and survival and had no major abnormalities in the heart, vascular system, and kidney (Ito et al., 1995; Sugaya et al., 1995a,b; Chen et al., 1997). Closer examination of the Agtr1a(−/−) animals revealed a slight decrease in survival, marked hypertrophy of the renal juxtaglomerular cells, and a moderate degree of mesangial expansion (Oliviero et al., 1997). Also, the tubuloglomerular feedback loop that regulates sodium delivery to the distal tubule was not detectable in AT1A knockout mice, indicating a specific role of AT1 receptor in the operation of this homeostatic mechanism (Schnermann et al., 1997). The absence of the severe renal lesions observed in animals lacking angiotensinogen or ACE at first suggested that the AT1A receptor is not a critical determinant of normal renal development and structure. However, the more severe effects of angiotensinogen and ACE deficiency, and the prominent inhibitory action of losartan treatment on renal structure and function in neonatal mice, indicated that Ang II action through the AT1 receptor is essential for normal renal growth and development (Tufro-McReddie et al., 1995).
Subsequent studies revealed that Ang II infusions in Agtr1a(−/−) mice treated with enalapril to reduce endogenous Ang II production cause dose-related elevations in blood pressure that were prevented by AT1 receptor antagonists. These pressor responses were much smaller than those observed in wild-type mice, but nevertheless demonstrated that AT1B receptors participate in blood pressure regulation in the absence of the AT1A receptor (Oliverio et al., 1997). This was confirmed by the finding that Ang II-induced calcium mobilization was similar in vascular smooth muscle cells from AT1A-deficient and wild-type mice, and was blocked by losartan (Zhu et al., 1998b). Thus, the AT1B receptor contributes substantially to Ang II action in the cardiovascular system in the absence of the AT1A receptor, and presumably is subsidiary to the major AT1A subtype in normal animals. This is supported by data obtained in mice with a double knockout of the AT1A and AT1B receptors, which have a more severe phenotype and lower blood pressure than mice lacking only the AT1A receptor (Oliviero et al., 1998a; Tsuchida et al., 1998).
These conclusions were confirmed by the finding that mice lacking both AT1A and AT1B receptors showed impaired growth and marked abnormalities in renal structure and function. The renal abnormalities in the double knockout animals were similar to those seen in Agtr−/− and Ace−/− mice and were accompanied by comparable decreases in blood pressure and the complete absence of pressor responses to Ang II. In these animals, inhibition of converting enzyme by enalapril caused a paradoxical increase in blood pressure that could result from impairment of AT2 receptor signaling and possibly an inhibitory effect on renal sodium excretion (Oliviero et al., 1998). These observations demonstrated that although the AT1Breceptor has a relatively minor role in normal animals, its contributions to growth, renal development, and cardiovascular regulation can compensate for much of the loss of the major regulatory actions of the AT1A receptors in Agtr1a−/− animals.
Studies on the role of the AT1 receptors in sodium homeostasis revealed that Agtr1a−/− mice have a further fall in blood pressure during sodium restriction and, unlike wild-type mice, develop negative sodium balance. However, these animals showed normal increases in plasma aldosterone levels during sodium depletion, consistent with the abundance of AT1B receptors in the adrenal zona glomerulosa. These findings suggest that the hypotension observed in Agrt1a−/− mice results from sodium deficiency and blood volume depletion and are consistent with the major role of AT1A receptors in renal sodium resorption. The mechanisms by which Ang II regulates water homeostasis through its actions in the kidney and the brain were also clarified by observations in AT1A receptor-deficient mice (Oliviero et al., 1998a,b). Agtr1a−/− mice have a significant defect in urinary sodium concentration and develop marked serum hypotonicity during water deprivation. This does not result from impairment of vasopressin action on water permeability in the distal tubule but from the inability to maintain a maximal sodium gradient in the kidney. This change is not increased by losartan treatment and appears to be solely dependent on AT1A receptor function. In contrast, the central action of Ang II on drinking responses appears to be mediated by AT1B receptors, since it is largely retained in Agtr1a−/− mice but is almost abolished in the mice lacking AT1B receptor (Davisson et al., 1998).
In contrast to the hypertension and impaired vascular responses observed in AT1-deficient mice, knockout of the AT2 receptor leads to elevation of blood pressure and increased vascular sensitivity to Ang II (Hein et al., 1995a;Ichiki et al., 1995b). This has suggested that the AT2 receptor may exert a protective action in blood pressure regulation by counteracting AT1receptor function. Such an action could be exerted in part by reduced expression of the AT1 receptor, which is increased in the vascular smooth muscle of AT2-deficient mice (Tanaka et al., 1999). However, the sustained hypersensitivity to Ang II in such animals is also attributable to loss of the counter-regulatory action of renal bradykinin and cyclic GMP formation (an index of NO production) (Siragy et al., 1999). The relative contributions of these two factors to AT2-dependent vascular regulation, and the extent to which AT2 receptor deficiency could contribute to sustained blood pressure elevation, as in human hypertension, have yet to be determined (see Section III, D).
H. Structural Basis of Ligand Binding to the AT1Receptor
The cloning of Ang II receptors from several species was followed by extensive studies on the structural features that are responsible for many of the specific functional properties of the AT1 receptor. Mutational analyses of AT1 receptors have identified many of the amino acid sequences and residues that are involved in the processes of ligand binding, agonist activation, G protein coupling, and internalization of agonist-receptor complexes.
1. Determinants of Ang II Bioactivity.
The major features of the Ang II octapeptide (Asp 1 -Arg-Val-Tyr-Ile/Val-His-Pro-Phe 8) that determine its biological activity were identified in early studies on the in vivo and in vitro actions of structurally modified Ang II peptides (Khosla et al., 1974). All of the biological responses that were analyzed in these early reports were mediated by what is now defined as the AT1 receptor. These findings were extended by the development of radioligand-receptor binding assays that used radioiodinated Ang II or its peptide analogs for in vitro analysis of the hormone-receptor interaction. More recent studies on the properties of cloned and mutant AT1 receptors have led to the development of ligand-receptor models that incorporate the major features currently believed to be important in agonist binding to the AT1 receptor. They have also provided further insights into the nature of the peptide binding site and the structural features that determine receptor activation, G protein coupling, and agonist-induced desensitization and internalization of the receptor.
The biological activity of Ang II is highly dependent on the aromaticity of its Phe 8 C-terminal residue. The aromatic side groups of Tyr 4 andHis 6, the guanidine group ofArg 2 , and the charged carboxyl terminus, are also essential for receptor activation (Khosla et al., 1974). In contrast, the N-terminal residues are important for receptor binding and the duration of action of Ang II agonists but are not specifically required for biological activity. The Ang II (2–8) heptapeptide (Ang III) formed by deletion of theAsp 1 residue is almost as potent as the native octapeptide. The Ang II (3–8) hexapeptide (Ang IV) and the Ang II (4–8) pentapeptide also retain full biological efficacy but are weak agonists due to their low-binding affinity for the AT1 receptor. The des-Phe 8 Ang II (1–7) heptapeptide also binds to the receptor with low affinity, but has no agonist activity (Capponi and Catt, 1979) in most Ang II target cells, and is accordingly a weak Ang II antagonist in vitro (Mahon et al., 1994).
NMR analyses of Ang II and its peptide analogs yielded models for the spatial arrangement of the four pharmacophore groups, Tyr4, His6, Phe8, and the C-terminal carboxyl group, that determine the biologically active conformation of the peptide (Nikiforovich and Marshall, 1993; Matsoukas et al., 1994; Nikiforovich et al., 1994). These studies indicated that the three aromatic rings cluster together and suggested that a bend in the Tyr-Ile-His region of the molecule is a prominent feature of its agonistic conformation. The charged amino- and carboxyl-terminal regions of the Ang II molecule are believed to form an ion pair that maintains the hairpin shape of the Ang II molecule and also stabilizes the binding of Ang II to the receptor. Structural comparisons of Ang II and its nonpeptide antagonist analogs by overlapping the imidazole, phenyl ring, and acidic moieties confirmed the model of Ang II as a twisted hairpin shape with about 9 Å between the His6 imidazole and terminal carboxylate groups (Pierson and Freer, 1992). Also, significant interactions between the Asp 1and Arg 2 side chains of Ang II were suggested by proton NMR studies (Zhou et al., 1991).
Subsequent studies confirmed the compact folded shape of the Ang II peptide backbone, and the relative positions of the four pharmacophore groups. This shape resulted from electrostatic interactions between the N- and C-terminal groups and was similar to that for antibody-bound Ang II as determined by crystallography (Joseph et al., 1995a,b). The conformation of Ang II in phospholipid micelles also confirmed the hairpin structure of the molecule and suggested the presence of a stable hydrogen bond between thePhe 8 NH andHis 6 carbonyl group, with an inverse gamma turn centered on Pro 7(Carpenter et al., 1998).
The Phe 8 residue of Ang II has long been recognized to be crucial for its biological activity. This residue is important for both the binding activity and the intrinsic activity of Ang II, and even minor charges in its structure have marked effects on biological activity. Its aromaticity and steric influence determine the biologically active conformation of the C terminus of Ang II, which requires an appropriate orientation of the position 8 carboxyl group relative to the aromatic group of the phenylalanine residue. Replacement of Phe 8 by nonaromatic residues endows antagonist properties that result from distortion of the orientation of the C-terminal carboxyl group (Aumelas et al., 1985). Consistent with its central role in receptor activation, replacement ofPhe 8 byd -Phe 8 also caused antagonist activity, and replacement by Phe(Br5)8 causes prolonged pressor responses attributed to slow dissociation of the more hydrophobic peptide from the Ang II receptor (Bosse et al., 1990).
2. Agonist Binding Site of the AT1Receptor.
Amino acids in the AT1 receptor that are essential for Ang II binding include the four cysteine residues that form the two external disulfide bonds and several other residues located in the exposed surface regions of the receptor (Fig.2). In addition, polar or charged residues located within the hydrophobic transmembrane domains, including Lys102 at the top of TM helix III and Lys199 near the top of TM helix V, participate in agonist binding. Some of the extracellular residues contribute to ligand interaction and stabilization of Ang II binding and others to the conformational change that causes receptor activation. The additional disulfide bridge between the amino terminal region and the third extracellular loop of the AT1 receptor appears to stabilize the receptor and may be necessary to maintain the proximity of the extracellular amino acids that are involved in peptide binding. Cleavage of this disulfide bond probably accounts for the impairment of AT1 receptor binding by reducing agents (Ohyama et al., 1995).

Amino acids that contribute to the agonist binding site of the AT1 receptor. Residues implicated in Ang II binding are shown as blue letters on pink background. The positions of the glycosylation sites, and of the conserved residues among GPCRs, are also shown.
Ang II binds primarily to the extracellular region of the AT1 receptor (Hjorth et al., 1994) by interacting with residues in its N terminus and its first and third extracellular loops. However the transmembrane helices also participate in Ang II binding, since its C-terminal carboxyl group interacts with Lys199 in the upper part of helix 5 of the receptor (Underwood et al., 1994; Noda et al., 1995a; Yamano et al., 1995). This could involve the formation of a salt-linked triad between Lys199 of the receptor and the carboxyl groups of Asp 1 andPhe 8 of the Ang II peptide (Joseph et al., 1995a,b). The Trp253 residue has been proposed to stabilize the ionic bridge formed between Lys199and the carboxyl-terminal group of the Phe 8residue. In addition, Phe259 and Asp263 in transmembrane helix VI could provide the docking site for His 6 of the ligand (Yamano et al., 1995). Two other residues (Lys102and Ser105) in the outer region of transmembrane helix III of the receptor have also been implicated in Ang II binding (Groblewski et al., 1995; Noda et al., 1995a). This region may participate in the formation of the intramembrane binding pocket and possibly in stabilization of the receptor's conformation.
The Asp281 residue, located at the C-terminal end of the third extracellular loop of the AT1receptor, serves as a major docking point for Ang II through its charge interaction with Arg 2 of the ligand (Feng et al., 1995). The Asp278 residue could also be important in this regard, since its mutation causes an even greater loss of receptor binding affinity (Hjorth et al., 1994). The N-terminalAsp 1 residue of Ang II has been proposed to interact with His123 in the second extracellular loop of the AT1 receptor (Yamano et al., 1995). These findings support the view that Ang II attaches primarily via its charged amino-terminal end to the extracellular binding region of the receptor. The major docking points for the amino- and carboxyl-terminal ends of Ang II, Asp281 and Lys199, respectively, are located at the outer ends of helices 7 and 5, respectively. The intramembrane binding pocket lies between these proposed contact points, and is adjacent to the cleft that contains the binding sites of receptors for smaller ligands, as well as the nonpeptide binding site of the AT1receptor. This region contains docking sites for the apolar/aromatic mid-portion of the Ang II molecule and for the carboxyl-terminal phenyl group that elicits the conformational change(s) leading to receptor activation.
The apolar nature of the essential aromatic ring of thePhe 8 residue of Ang II binds to the AT1 receptor suggests that the phenyl group could interact with residues located within the membrane-spanning helices. This is consistent with NMR-based predictions of the receptor-bound conformation of Ang II that position the phenyl group in an appropriate location for such interactions (Nikiforovich et al., 1993, 1994). The phenyl group could interact with aromatic residues in the helices, some of which have been suggested to form an aromatic floor for charge interactions between ligand and receptor (Trumpp-Kallmeyer et al., 1992; Findlay et al., 1993). Modeling studies on the AT1 receptor have also suggested that conserved aromatic residues in helices IV and VI could form the base of the ligand binding site, as in other G protein-coupled receptors (Underwood et al., 1995). Mutations of two of these residues, Phe259 and Trp253, reduces Ang II binding to the receptor (Yamano et al., 1995). Whether the aromatic group of Phe 8 of the ligand interacts with the binding pocket of the receptor via these amino acids, or with aromatic residues in other segments of the membrane domains, has yet to be determined. The Phe 8residue could also form a polar/aromatic interaction with the hydroxyl group of Ser105 in the third transmembrane domain of the receptor (Joseph et al., 1995a,b).
The Tyr 4 residue of Ang II is an important determinant of its binding and biological activities (Bumpus et al., 1977; Capponi and Catt, 1979; Nikiforivich et al., 1993, 1994). In fact, reversing the Tyr 4 andPhe 8 residues of the Ang II molecule (Marshall et al., 1970) formed the first Ang II antagonist. TheTyr 4 residue has been proposed to interact with Arg167 at the top of the fourth transmembrane helix (Yamano et al., 1995), and could also disrupt the hydrogen bonding between Asn111 and Tyr292 in transmembrane domains 3 and 7 of the unoccupied receptor by competing with Tyr292. This would permit the latter to interact with Asp74 in the second transmembrane domain during receptor activation. In this mechanism, the loss of a proton from theTyr 4 phenolic hydroxyl group to the carboxyl group of Glu91 could be part of a relay system initiated by the interaction of His 6with Thr88 and Glu91 of the receptor (Joseph et al., 1995a,b).
3. Antagonist Binding of the AT1 Receptor.
Since Ang II is a major regulator of blood pressure, aldosterone secretion, and fluid homeostasis, and is also an important etiological factor in hypertension and other cardiovascular disorders, blockade of Ang II formation or action by ACE inhibitors or receptor antagonists is of major therapeutic importance. Early attempt to develop therapeutic agents able to block the Ang II receptor impeded by the peptidic nature of antagonists such as saralasin, which lacked oral activity and showed agonistic properties (Pals et al., 1979). More recently, based on imidazole derivatives first described by Furukawa et al. (1982), it became possible to develop specific nonpeptide Ang II receptor antagonists that specifically and selectively block the angiotensin AT1 receptor (Timmermans et al., 1993; Goodfriend et al., 1996). The first of this series to reach the clinic, losartan, was followed by a large number of orally active AT1 antagonists (Table2).These can be classified in two groups depending on the presence of a biphenyltetrazole moiety, as in the prototype drug, losartan, in their structure. Receptor binding of nonpeptide Ang II antagonists is saturable and usually reversible and is independent of the pathway responsible for the synthesis of Ang II. This could be relevant to comparisons with ACE inhibitors, given the possible role of alternative Ang II-generating enzymes such as chymase, in human tissues (Urata et al., 1996).
Structure of AT1 and AT2 receptor nonpeptidic antagonists
In pharmacological studies on the properties of angiotensin and its synthetic analogs, certain AT1 receptor antagonists not only cause a rightward shift in the Ang II dose-response curve but also reduce the maximal response to agonist stimulation (Wienen et al., 1993; Morimoto and Ogihara, 1994; Criscione et al., 1995; Goa and Wagstaff, 1996; Gillis and Markham, 1997;McClellan and Balfour, 1998). The latter compounds (candesartan, EXP 3174, valsartan, irbesartan) are termed insurmountable antagonists, in contrast to surmountable antagonists such as losartan, eprosartan, and telmisartan, which do not impair the maximum response to Ang II. One explanation for this difference is that nonpeptide antagonists can act by interfering with receptor activation by occupying an intramembrane site that overlaps with the space occupied by the agonist (competitive or surmountable antagonists) or by inducing conformational changes that prevent agonist binding (noncompetitive or insurmountable antagonists). Another proposal is that surmountable antagonists such as losartan dissociate rapidly from the receptor, whereas insurmountable antagonists, exemplified by candesartan, bind tightly and dissociate so slowly as to cause functional loss of the occluded receptors. Recent studies on the properties of the human AT1receptor expressed in Chinese hamster ovary (CHO) cells have shown that the agonist-receptor complexes are divisible into a rapidly reversible, surmountable population, and a tightly binding, insurmountable population (Fierens et al., 1999).
Although losartan is a potent antagonist in its own right, about 10% of the dose is metabolized to EXP 3174, which has 10-fold higher affinity for the AT1 receptor and is responsible for the 24 h decrease in blood pressure. Candesartan cilexetil is an inactive ester prodrug and is completely cleaved during absorption in the gastrointestinal tract. Twenty-five percent of candesartan is eliminated by metabolism to an inactive metabolite. Among the other antagonists, irbesartan, valsartan, and eprosartan do not require metabolism to be active. Irbesartan is mainly eliminated by the liver (75%) and less than 2% is excreted unchanged in the urine. Sixty percent of eprosartan is cleared unchanged via the bile. Valsartan is essentially eliminated by biliary excretion and 10% of the dosage appears intact in urine.
As expected, significant increases in renin activity, Ang I and Ang II are observed after blockade of the AT1 receptor. It is conceivable that the increased circulating Ang II level could stimulate the AT2 receptor, which appears to counterbalance the effect of the AT1 receptor (see below). Blockade of the AT1 receptor not only inhibits smooth muscle contraction but also reduces the production of pressor agents including aldosterone, vasopressin, catecholamine, and endothelin. AT1 receptor antagonists have shown exceptionally good tolerability and their incidence of adverse effects is similar to that of placebos. The AT1receptor antagonists are approved for the treatment of hypertension, and in early clinical studies also appear to be of use in the treatment of congestive heart failure, postmyocardial infarction, and renal failure. Several large clinical trials are in progress such as Scope, Life and Value for hypertension, Regaal and Silver for left ventricular hypertrophy, ValHeft and Charm for congestive heart failure, Optimaal and Valiant for postmyocardial infarction, and Irma, IDNT, Renaal and ABCD 2V for diabetic nephropathy.
Identification of the losartan binding region of the mammalian AT1 receptor was facilitated by the finding that the amphibian Ang II receptor, which resembles the mammalian AT1 receptor in its signal transduction mechanisms, does not recognize nonpeptide antagonists such as losartan. This enabled the amino acid residues involved in the binding of losartan to the mammalian AT1 receptor to be determined by analysis of the ligand binding properties of mutant rat AT1 receptors in which nonconserved amino acids were replaced by the corresponding amphibian residues (Ji et al., 1994). Most of these mutant receptors showed only minor changes in binding affinity for Ang II and its peptide antagonist [Sar1,Ile8]Ang II, indicating that the overall conformation of the receptor was unaltered by such replacements. However, several residues located in the transmembrane domains (TMDs) of the receptor were found to be required for binding of the nonpeptide antagonist. These included Val108 in TMD III, Ala163in TMD IV, Pro192 and Thr198 in TMD V, Ser252 in TMD VI, and Leu300 and Phe301 in TMD VII.
These findings demonstrated that the nonpeptide AT1 antagonist binds to a site defined by amino acids located within the membrane-spanning regions of the receptor. Also, the nonpeptide binding site was largely distinct from the receptor domain that is involved in binding of Ang II and other peptide ligands. This conclusion is consistent with the presence of a primordial binding site for small ligands between the transmembrane helices of all GPCRs that can be used for the development of nonpeptide analogs for a wide variety of peptide hormones. Other amino acid residues in the rat AT1B receptor that influence losartan binding include Ala73 in TMD II; Ser104, Ala114, and Ser115 in TMD III; Lys199in TMD V; Phe248 in TMD VI; and Asn295 in TMD VII (Schambye et al., 1994; Ji et al., 1994; Noda et al., 1995). These and other observations have implicated TMD III in losartan binding to the mammalian AT1 receptor. The locations of the multiple amino acids that contribute to losartan binding in the receptor are shown in Fig. 3.

Amino acids that contribute to the nonpeptide antagonist binding site of the AT1 receptor. Residues implicated in losartan binding are indicated by green letters on orange background. The positions of the glycosylation sites, and of the conserved residues among GPCRs, are also shown.
A mutant amphibian receptor formed by exchanging these residues for the corresponding amino acids in the Xenopus AT receptor bound losartan with the same high affinity as the rat AT1 receptor (IC50 values: rat AT 2.2 ± 0.2 nM; xAT > 50 :M: mutant xAT 2.0 ± 0.1 nM) (Ji et al., 1993, 1995). This gain-of-function mutation, in which the residues known to participate in the formation of the ligand binding site in a mammalian hormone receptor were transferred to an unresponsive amphibian receptor, confirms the validity of the proposed losartan binding site. The identification of residues involved in receptor binding of nonpeptide Ang II analogs should aid in modeling studies on the structural basis of ligand recognition and activation of the Ang II receptor.
Although there is little overlap between the intramembrane residues that influence Ang II and losartan binding, both ligands appear to interact with the region located between helices III, V, VI, and VII of the AT1 receptor. This area also contains the binding sites of GPCRs for small ligands such as catecholamines and acetyl choline, as well as other nonpeptide antagonist analogs. In addition to sharing the binding pocket formed by the transmembrane helices of the receptor, Ang II and its nonpeptide analogs probably interact with common contact points at other locations in the receptor. In addition to the Lys102, Ser105, Arg167, and Lys199 residues, this could apply to Asn295, replacement of which by serine impairs the binding of Ang II as well as its nonpeptide agonist and peptide antagonist analogs (Hunyady et al., 1998). It is noteworthy that peptide agonist binding is also influenced by Phe301, an aromatic residue located near the end of helix VII (Hunyady et al., 1996a, 1999). Mutation of this residue to alanine impairs Ang II and losartan binding, as well as that of the peptide antagonist, [Sar1,Ile8]Ang II. These findings indicate that multiple residues contribute to the integrity of the binding pocket and are required for optimal agonist and antagonist binding.
Although the extracellular loops of the receptor contribute significantly to the binding of Ang II and its peptide antagonists, the intramembrane binding pocket has a major functional role in the AT1 and probably most other peptide hormone receptors. In receptors for large and/or bulky ligands, the intramembrane binding pocket per se may not permit efficient interaction without additional stabilization of the ligand through binding to residues in the extracellular surface of the receptor. Nevertheless, the binding pocket is essential for the initiation of receptor activation, and its interaction of agonists triggers the change in receptor conformation that leads to G protein coupling and second messenger generation.
The importance of the transmembrane domains in receptor activation also applies to glycoprotein hormone receptors, which bind extremely large ligands. These data suggest that a common mechanism for activation of GPCRs is based on critical interactions within the intramembrane binding pocket that evoke specific changes in receptor conformation. The extracellular domains of the receptor provide additional interactions with the ligand that are necessary for high-affinity binding. This is a prominent feature of receptors for larger agonists such as glycoprotein hormones, but also occurs in those for small peptide hormones such as angiotensin II, bradykinin, and vasopressin.
I. AT1 Receptor Signaling Mechanisms
1. AT1 Receptor Activation and Signal Transduction.
The AT1 receptor, like other GPCRs, has been proposed to undergo spontaneous isomerization between its inactive (R) and active (R*) states. The inactive R state is favored in the absence of agonist ligands and is in equilibrium with a small proportion of the active R* state in its unliganded form. In the presence of Ang II, the active R* form of the receptor is either selected or induced by agonist binding. The shift in equilibrium during agonist activation favors the R* state, in which the altered conformation of the receptor permits coupling to one or more G proteins that mediate intracellular signaling via phospholipase C and other pathways. Most receptors appear to be kept in their inactive state by structural restraints that are removed by agonist binding, leading to the formation of the activated R* state. Recent evidence suggests that the agonist-activated AT1 receptor, and possibly other GPCRs, undergoes transitions into multiple conformational states that are associated with the individual stages of receptor activation and regulation (Thomas et al., 2000).
The proportion of receptors in the activated state varies among individual GPCRs and is manifested by varying degrees of basal signaling activity when receptors are overexpressed in transfected cells. Significant degrees of agonist-independent or constitutive activity have been observed in wild-type thyroid-stimulating hormone and muscarinic receptors (Burstein et al., 1997; Wang and Gershengorn, 1999) but are not evident in the AT1 and most other GPCRs. Agonist-induced activation of the AT1 receptor appears to be triggered by an interaction between the Tyr 4 residue of Ang II and the Asn111 residue located in the third transmembrane domain of the receptor (Noda et al., 1996). This, and the interaction of Phe 8 of the angiotensin molecule with His256 in the sixth transmembrane domain of the receptor, appear to drive the AT1receptor into its fully active R* state (Noda et al., 1995a).
The importance of Asn111 as a switch residue in agonist-induced AT1 receptor activation is further indicated by the ability of its mutation to Gly111 or other small residues to induce constitutive activation of the receptor (Feng et al., 1998). This suggests that the ability of Asn111 to act as a conformational switch is related to its side chain size, rather than polarity or hydrogen binding. On the other hand, it is the aromaticity rather than the size of the Tyr 4 side chain that is important in receptor activation, which probably depends on amino-aromatic binding between Tyr 4 and Asn111 of the receptor (Miura et al., 1999a). Aromaticity is likewise important in the interaction between Phe 6 and His256, which is critical for full receptor activation by agonist ligands.
Like other constitutively activated GPCRs, the Asn111Gly and Asn111Ala mutant receptors show increased affinity and efficacy for agonist ligands, and exhibit marked increases in the sensitivity of their biological responses to agonist stimulation. They also discriminate between inverse agonists, which stabilize the inactive conformation of the receptor, and neutral antagonists. For these reasons, constitutively active AT1 receptors are highly sensitive to Ang II derivatives that are weak or partial agonists at the wild-type receptor (Miura et al., 1999b).
A proposed model of GPCR activation predicts that rigid body movement of the third, sixth, and seventh transmembrane domains induces conformational changes in the cytoplasmic loops that permit G protein interaction with the agonist-activated receptor (Gether and Kobilka, 1998). However, the manner in which the activating regions in the cytoplasmic domains of individual GPCRs interact with specific subsets of G proteins is not yet known. Although many amino acids in the transmembrane and cytoplasmic regions of a wide range of receptors have been implicated in G protein coupling, there are very few cases in which individual residues can be correlated with binding to specific types of G proteins (Hunyady et al., 1996b). The limited degree of selectivity of most such residues presumably reflects the precise conformational requirements for specific interaction of receptors with their cognate G proteins.
Although the AT1 receptor has been reported to interact with several G proteins, its major physiological functions are expressed through Gq-mediated activation of phospholipase C followed by phosphoinositide hydrolysis and Ca2+ signaling. Although all GPCRs possess the basic seven transmembrane structure, only a few amino acids are highly conserved among the superfamily of G protein-coupled receptors (Fig.1). One such conserved motif is the characteristic NPX2–3Y sequence that is located in the seventh transmembrane domain of most receptors, and in the rat AT1A receptor is Asn298-Pro299-Leu300-Phe301-Tyr302. A recent model of aminergic GPCRs suggests that the NPX2–3Y sequence is ideally placed to receive a signal from agonist-induced conformational changes in the ligand binding region (Donnelly et al., 1994). In the three-dimensional structure of the receptor, this sequence is in close proximity to the asparagine-aspartic acid pair located in transmembrane segments 1 and 2 and may form functionally important hydrogen bonding interactions with this region. Such models assume that the highly conserved proline residues, which disrupt the α-helical structure of the transmembrane domains, could serve as hinges during the conformational changes that occur in agonist-activated receptors. One such proline residue is located within the seventh transmembrane domain in the conserved NPX2–3Y sequence and has been found to be important for signal transduction by the m3 muscarinic receptor (Wess et al., 1993). However, this residue does not appear to be essential for signaling by Ang II and gonadotropin-releasing hormone (GnRH) receptors.
Another interesting feature of the NPX2–3Y sequence is its similarity to the NPXY internalization sequence that is present in the cytoplasmic segment of receptors for low-density lipoprotein and several growth factors. The tyrosine residue in this sequence has been reported to be essential for sequestration of the β-adrenergic receptor (Barak et al., 1994). However, this does not apply to the gastrin-releasing peptide receptor (Slice et al., 1994) and the AT1 receptor, both of which contain an additional aromatic amino acid (Phe321and Phe301, respectively) in their NPX2–3Y sequences. The presence of such residues might be important since phenylalanine can substitute for the tyrosine residue in the NPXY internalization sequence of nutrient receptors.
An analysis of the functional role of the Asn298-Pro299-Leu300-Phe301-Tyr302sequence (Hunyady et al., 1995b) revealed that the ability of the receptor to interact with G proteins and to stimulate inositol phosphate responses was markedly impaired by alanine replacement of Asn298 and was reduced by replacement of Pro299 or Tyr302. The Phe301Ala mutant receptor exhibited normal G protein coupling and inositol phosphate responses, and the binding of the peptide antagonist, [Sar1,Ile8]Ang II, was only slightly affected. However, its affinity for Ang II and the nonpeptide antagonist losartan was reduced by an order of magnitude, suggesting that Ang II and losartan share an intramembrane binding site, possibly through their aromatic moieties, in this region. None of the agonist-occupied mutant receptors, including Tyr302Ala and triple alanine replacement of Phe301, Tyr302, and Phe304 showed substantial changes in their internalization kinetics. These findings demonstrate that the NPLFY sequence of the AT1 receptor is not an important determinant of agonist-induced internalization. However, the Phe301 residue contributes significantly to agonist binding, and Asn298 is required for normal receptor activation and signal transduction.
Another highly conserved amino acid in the superfamily of G protein-coupled receptors is a tyrosine residue located in the fifth transmembrane helix, adjacent to the amino-terminal end of the third cytoplasmic loop. The location of this amino acid, which is Tyr215 in the rat AT1receptor, suggests that it could be involved in receptor activation. The amino-terminal region of the third cytoplasmic loop adjacent to this residue has been shown to be important in the signal generation and internalization of several G protein-coupled receptors. Furthermore, modeling of G protein-coupled receptors based on the crystal structure of bovine rhodopsin has suggested that this residue is in molecular proximity to regions that are important in receptor activation, including the conserved acidic-arginine-aromatic (DRY) triplet of the second intracellular loop (Baldwin et al., 1997). Although AT1 receptor internalization and signal transduction have different structural requirements, a 6-amino acid deletion of the amino-terminal end of the third cytoplasmic loop, which included Tyr215, was found to prevent both receptor internalization and signaling responses. The role of the highly conserved Tyr215 residue in the activation of GPCRs was analyzed in a mutant AT1 receptor created by replacing this amino acid with a phenylalanine residue (Hunyady et al., 1995b).
The resulting Tyr215Phe mutant receptor was normally expressed in transfected COS-7 cells, and its binding affinity for the peptide antagonist [Sar1, Ile8]Ang II was similar to that of the wild-type receptor. However, its affinity for Ang II was significantly reduced and its ability to mediate inositol phosphate responses to Ang II stimulation was abolished. These changes were associated with loss of coupling of the mutant receptor to G proteins, as indicated by the lack of effect of GTPγS on agonist binding to the receptor. The agonist-induced internalization of the mutant receptor was also impaired. The concomitant decreases in receptor internalization and G protein-mediated signaling of the Tyr215Phe mutant receptor indicate that this residue has a critical role in AT1 receptor activation. It is possible that the Tyr215 residue takes part in propagation of the agonist-induced conformational change to the intracellular loops that participate in G protein coupling. In view of its wide conservation among members of the seven transmembrane domain receptor superfamily, this residue is likely to be of general importance in signal transduction from G protein-coupled receptors. The AT1 receptor, like many other Ca2+-mobilizing GPCRs, signals primarily through coupling to a Gq/11 protein that activates phospholipase C-β (PLC-β), leading to polyphosphoinositide hydrolysis, stimulation of InsP3/calcium signaling, and activation of protein kinase C. However, in cultured rat VSMC it is predominantly PLC-γ rather than PLC-β that is activated during Ang II action. This response, which is dependent on tyrosine phosphorylation of the PLC-γ isozyme, is distinct from the usual process of activation of PLC-β1 and β2 by GPCRs through α and βγ subunits derived from the heterotrimeric G proteins, Gq/11and Gi/o, respectively. AT1receptors have also been reported to couple to the G12/13 family of pertussis-insensitive G proteins, which appear to mediate Ang II-induced L-channel activation in rat portal vein myocytes (Macrez et al., 1997). In the rat, the AT1 receptor is also coupled to pertussive-sensitive Gi/o protein(s) that inhibit adenylyl cyclase in several Ang II target tissues, including the adrenal glomerulosa zone, liver, kidney, and pituitary gland. Ang II has also been found to stimulate modest increases in cyclic AMP production in some of its target tissues. However, this may result from activation of Ca2+-sensitive adenylyl cyclases rather than coupling of the AT1 receptor to Gs. Interestingly, although the cloned rat AT1A and AT1B receptors expressed in COS-7 and Y-1 cells show the expected coupling to phosphoinositide hydrolysis and other Gq/11-mediated responses, they do not exhibit coupling to Gi-mediated responses such as inhibition of adenylyl cyclase (Tian et al., 1996).
2. AT1 Receptor and Tyrosine Phosphorylation.
In addition to its rapid actions on phosphoinositide/calcium signaling, Ang II elicits many of the intracellular signaling responses that are typically associated with activation of growth factors. These include tyrosine phosphorylation, most immediately of PLC-γ and subsequently of several other downstream proteins and effector enzymes. These include pp60c-src, pp120, pp125FAK, JAK2, STATs, paxillin, TYK2, and MAPK (Bernstein and Marrero, 1996). Because GPCRs do not have intrinsic tyrosine kinase activity, the ability of the AT1 receptor to induce tyrosine phosphorylation and activation of PLC-γ in rat VSMC, with consequent increases in InsP3 production and Ca2+signaling, must depend on other tyrosine kinases. Such ligand-induced activation of PLC-γ by tyrosine phosphorylation may occur during the actions of growth factors on their target cells, and accounts for the associated elevations in [Ca2+]i. This aspect of AT1 receptor function has been controversial, because the mechanisms by which the receptor couples to PLC in VSMC are poorly defined. In most cell types, calcium-mobilizing GPCRs are coupled through their cognate G proteins to activation of the β isozymes of PLC. The β1 and β2 enzymes are activated by Gα and Gβ γ subunits, respectively, the former derived from Gq/11 and the latter largely from Gi/o proteins. In contrast, AT1 receptor-mediated activation of PLC-γ is dependent on tyrosine phosphorylation and consequently on signaling from receptor or nonreceptor tyrosine kinases.
The mechanism by which Ang II stimulates tyrosine phosphorylation and activation of PLC-γ involves binding of the enzyme to the C-terminal cytoplasmic domain of the AT1 receptor. This interaction occurs between the C-terminal cytoplasmic domain of PLC-γ1 and a phosphorylated YIPP motif located within the intracellular tail of the receptor (Venema et al., 1998a). The same YIPP motif has been implicated in the binding of JAK2 to the AT1 receptor, probably as a complex with the phosphotyrosine phosphatase, SHP-2 (Ali et al., 1997a). The way by which AT1 receptor activation regulates ligand-dependent binding of two distinct intracellular signaling proteins to the same site in its cytoplasmic tail has yet to be determined. The relative roles of PLC-β and PLC-γ in Ang II-induced phosphoinositide hydrolysis in VSMC have been difficult to determine, and studies on the relative abundance of the PLC isoforms in VSMC have given variable results. Relatively little PLC-β1 was detected in rat and rabbit VSMC in some reports (Homma et al., 1993; Marrero et al., 1994), but others have found that all three isozymes (α, β, and γ) are abundant in rat VSMC (Ushio-Fukai et al., 1998). In the latter study, AT1 receptors were proposed to couple initially to PLC-β by heterotrimeric G proteins, and subsequently to PLC-γ. The G proteins responsible for the early activation of PLC-β, and the initial maximum InsP3 response at 15 to 20 s, included both Gq/11 and G12. The subsequent activation of PLC-γ and its contribution to the delayed InsP3 response are attributed to its tyrosine phosphorylation by a downstream kinase. This study also suggested that βγ subunits derived from G12 are involved in the activation of PLC in VSMC, and mediate Ang II-induced activation of PLD in these cells.
3. AT1 Receptor-Activated Growth Responses.
In addition to its contractile and secretory actions in smooth muscle and adrenal cells, Ang II stimulates growth and/or proliferative responses in these and others of its target cells. This effect was first documented in rat adrenal glomerulosa cells, which respond to sodium deficiency with hypertrophy and hyperplasia, and undergo regression in animals on high sodium intake (Gross, 1968). These changes are dependent on the circulating Ang II level, which is a major determinant of trophic changes in the adrenal glomerulosa zone (Aguilera et al., 1978). Ang II also has trophic actions on renal mesangial cells and vascular smooth muscle cells, leading to cellular hypertrophy and sometimes to cell division (Huckle and Earp, 1994). Cardiac myocytes and fibroblasts also exhibit hypertrophic responses to Ang II. However, cardiac fibroblasts undergo both hypertrophy and proliferation during stimulation by Ang II, an action that contributes to the development of ventricular hypertrophy. Treatment with AT1antagonists has been found to abolish the growth-promoting actions of Ang II in vitro and in vivo. As discussed below, many of the growth-related actions mediated by the AT1receptor are inhibited by concomitant binding of Ang II to the AT2 receptor.
The growth factor-like effects of Ang II include increases in tyrosine phosphorylation of numerous intracellular proteins, activation of MAPK and related pathways, and increased expression of several early response genes including c-fos, c-jun, and c-myc (Clark et al., 1992; Berk and Corson, 1997). These changes are associated with cell growth, increased thymidine incorporation, and cell proliferation (Tian et al., 1995). Such actions of Ang II are consistent with its well defined growth effects in adrenal glomerulosa and VSMC, and its role in the vascular and cardiac lesions associated with endothelial damage, atherosclerosis, hypertension, and cardiac failure. One of the earliest Ang II-induced events in VSMC is the rapid tyrosine phosphorylation and activation of PLC-γ, and the ensuing phosphoinositide/Ca2+signaling responses that initiate many of the actions of Ang II in these cells (Marrero et al., 1994). This is accompanied by the rapid activation of Ras, and its dependent phosphorylation cascades that extend through the cytoplasm and into the nucleus, with consequent effects on cell growth and differentiation. These include the MAPK and related pathways, in which sequential phosphorylations that are initiated at the plasma membrane lead to subsequent actions in the nucleus and other regions of the cell.
The consequences of Ang II-induced activation of Ras in cardiac myocytes resemble those elicited by agonist activation of growth factor receptors, in which the MAPK cascade is initiated by the binding of adaptor proteins, such as Grb2 and Shc, to the autophosphorylated receptors (Sadoshima et al., 1995). These adaptors interact with guanine nucleotide exchange factors that enhance the activities of small GTP binding proteins, including Ras, Rac, and Rho. The small G proteins of the Ras superfamily are activated by recruitment of guanine nucleotide exchange factors such as Sos, which promotes the exchange of GTP for Ras-bound GDP to form the active Ras-GTP complex. Ras-GTP then recruits and activates serine/threonine kinases such Raf-1 or Mos, which in turn activate dual-specificity MAPK kinases, such as MEKs, by serine phosphorylation. MEKs in turn phosphorylate the MAPKs, p44MAPK and p42MAPK (also known as extracellular signal-regulated kinases, or ERK1 and ERK2) on threonine and tyrosine residues that are located within a TEY phosphorylation motif in their activation loop. The activated MAPKs are translocated into the nucleus, where they phosphorylate other kinases and transcription factors that regulate the co-ordinated expression of genetic programs that control a wide variety of cellular functions. These include several early response genes and others such as c-myc, Elk1, Ets, RSK, and Mnk. MAPKs also exert regulatory actions on other signaling pathways in the cytoplasm and at the cell membrane. The multiple actions of activated MAPKs are terminated by a group of dual-specificity phosphatases that selectively dephosphorylate the individual MAPKs and related enzymes (Neel and Tonks, 1997).
Ang II is but one of several calcium-mobilizing GPCRs that mimic the effects of growth factors and other receptor kinases on the activation of MAPKs and early response genes. The way by which this response is linked to the well defined phosphoinositide/calcium signaling system that is used by many such receptors has only recently been established, and some aspects of this process still remain to be determined. In addition to the need for Ca2+ signaling (Sadoshima et al., 1995) a role of PKC was suggested by reports of phorbol ester-induced activation of Raf and Ras in certain cell types. In addition, the phorbol ester insensitive PKC isoform, PKC-ζ, was found to mediate Ang II-induced activation of ERK1/2 in VSMC and to associate with Ras during Ang II stimulation (Liao et al., 1997). Such findings indicated that additional upstream factors must be of primary importance in MAPK activation by calcium-mobilizing GPCRs. In rat VSMC, Ang II also activates Ras (Schieffer et al., 1996), an early and cardinal intermediate in stimulation of the MAPK pathway by growth factors. The Ang II-induced activation of Ras is more rapid and less prominent than that elicited by epidermal growth factor (EGF) and is substantially reduced by treatment with pertussis toxin. However, the concomitant increases in MAPK activity and c-fos expression are not impaired by pertussis toxin (Okuda et al., 1996), suggesting that these responses are not dependent on the Gi-mediated activation of Ras that occurs in Ang II-stimulated VSMC.
Ang II not only increases Ras-GTP levels but also stimulates the formation of Ras-Raf-1 complexes and tyrosine phosphorylation of GTPase-activating proteins (GAPs) such as p120 Ras-GAP and p190 Rho-GAP. These effects are prevented by the introduction of pp60c-src antibodies into the cultured cells, as is the tyrosine phosphorylation of PLC-γ1 and a substantial fraction of the Ang II-induced Ins(1,4,5)P3 response (Marrero et al., 1995; Schieffer et al., 1996). These and other findings have shown that Ang II partially mimics the action of growth factors by using the Ras pathway to activate MAPK. Furthermore, this appears to involve the nonreceptor tyrosine kinase, Src (pp60src), which probably acts through tyrosine phosphorylation and activation of a nucleotide exchange factor. Further evidence for the importance of Src in Ang II-stimulated activation of ERK has come from studies with tyrosine kinase inhibitors, and in Src-deficient and Src-overexpressing VSMC (Ishida et al., 1998).
4. Transactivation of Growth Factor Signaling by the AT1 Receptor,
In addition to Src, a calcium-dependent tyrosine kinase has been implicated in the Ang II-induced stimulation of the Ras/MAPK cascade in VSMC (Eguchi et al., 1996). This observation led to the recognition that transactivation of the EGF receptor contributes to the activation of MAPK by Ang II in these cells. This process involves the calcium-dependent phosphorylation of the EGF receptor to form docking sites for Src and the adaptor protein, Shc, which then recruits Grb2 and Sos to activate Ras (Eguchi et al., 1998).
These findings account for much of the similarity between the actions of Ang II and growth factors on the activation of PLC-γ, tyrosine kinases, and MAPKs, and increased expression of nuclear proto-oncogenes. They are also consistent with the observation that Ang and platelet-derived growth factor (PDGF) signaling cascades converge in VSMC, with activation of the PDGF-β receptor (Linseman et al., 1995). Ang II also stimulates the tyrosine phosphorylation and activation of the IGF-1 receptor, as well as insulin receptor substrate 1, in VSMC (Ali et al., 1997b). Recently, activation of the EGF receptor by Ang II has been implicated in the Ang II-induced synthesis of TGF-β and fibronectin in cardiac fibroblasts. This effect was mediated by downstream signaling from the EGF receptor, which was transactivated by Ang II via a Ca2+-dependent tyrosine kinase. The Ang II-induced expression of fibronectin mRNA was regulated at the transcriptional level by binding of the fos/jun complex to an AP-1 site and also by stabilization of the message through actions of TGF-β (Moriguchi et al., 1999).
These observations are relevant to the proposed role of AT1 receptor activation in the remodeling of cardiac interstitial tissue during ventricular hypertrophy. Ang II has been shown to stimulate the expression of collagen and fibronectin in cardiac fibroblasts, and to promote the release of newly formed collagen from the cells (Villareal et al., 1993; Crabos et al., 1994). At least one of the calcium-dependent tyrosine kinases that could mediate the transactivation of the EGF receptor by Ang II has been identified as Pyk2 (also termed CAKβ and RAFTK). However, it appears that Pyk2 does not fully account for this process and that other mechanisms are involved in AT1 receptor-mediated transactivation of the EGF receptor (Murasawa et al., 1993). Another example of cross-talk from the AT1receptor is its ability to influence the activity of the LOX-1 receptor for oxidized low-density lipoprotein in human coronary artery endothelial cells (Li et al., 1999). Since oxidized low-density lipoprotein has been implicated in the pathogenesis of atherosclerosis, and attenuates nitric oxide-induced dilatation, and promotes leukocyte deposition in the vascular wall, the ability of Ang II to up-regulate LOX-1 expression via AT1 receptors suggests another potential role for AT1antagonists in the management of cardiovascular disease.
5. Other AT1 Receptor-Mediated Signaling Pathways.
In addition to activating phosphoinositide/calcium signaling, tyrosine phosphorylation, and MAPK pathways, Ang II also acts through the AT1 receptor to stimulate the Jak/STAT signaling pathway. This was first observed in neonatal cardiac fibroblasts and transfected CHO cells, in which the AT1A receptor activates the STAT signaling pathway and stimulates sis-inducing factor-like DNA binding activity (Bhat et al., 1994). The stimulatory effect of Ang II on SIF/STAT activation was slower than that elicited by IL-6, and was preceded by an initial inhibitory phase that was associated with concomitant suppression of IL-6-induced STAT tyrosine phosphorylation and SIF responses (Bhat et al., 1995). The inhibitory action of Ang II on IL-6-induced Stat3 signaling was attributed to activation of the MAPK pathway and could be attenuated by the MAPK inhibitor, PD98059 (Bhat et al., 1996). Ang II was subsequently found to stimulate rapid serine phosphorylation of Stat3 via a MAPK1-dependent pathway (Bhat and Baker, 1997). In neonatal cardiac myocytes, the AT1 receptor is coupled through Jak2 kinase to the activation of Stat1 and Stat3, and to the formation of SIF complexes (McWhinney et al., 1997).
Ang II also causes rapid and transient activation of the Jak/STAT pathway in rat aortic smooth muscle cells (Marrero et al., 1995). This is initiated by tyrosine phosphorylation and increased catalytic activity of Jak2, and is dependent on its physical association with the agonist-activated AT1 receptor. This interaction occurs at a YIPP sequence that is located in the carboxy terminal tail of the receptor (Ali et al., 1997a,b), and is also present in the PDGF receptor. This motif has also been implicated in the binding of SH2 domains in PLC-γ to the AT1 receptor (Venema et al., 1998a). The binding of Jak2 to this motif has been attributed to the participation of SHP-2 phosphotyrosine phosphatase, which also contains an SH2 domain and functions as an adaptor protein in the association between JAK2 and the AT1receptor (Marrero et al., 1998).
In addition to ERK1 and ERK2, Ang II stimulates the activity of c-Jun N-terminal kinase (JNK) members of the MAPK family. In vascular smooth muscle cells, p21-activated kinase (PAK), which mediates JNK activation by IL-1 and tumor necrosis factor, was rapidly stimulated by Ang II before the activation of JNK. Ang II-induced activation of both PAK and JNK was dependent on calcium and PKC, and partially on a tyrosine kinase other than Src. These findings, and the ability of a dominant negative PAK to attenuate the JNK response to Ang II in transfected CHO and COS cells, indicated that PAK is a mediator of JNK activation by Ang II in VSMC (Schmitz and Berk, 1997). In this regard, Ang II again behaves like inflammatory cytokines such as IL-2 and tumor necrosis factor-α, by acting through PAK as an upstream mediator of JNK.
Ang II also promotes hypertrophy of VSMC through an oxidant stress-dependent mechanism. Reactive oxygen species such as hydrogen peroxide and O⨪2 appear to be involved in the pathogenesis of hypertension and atherosclerosis, and are released from endothelial cells and other vascular and circulating cells (Rajagopalan et al., 1996). These molecules have been implicated in vascular smooth muscle proliferation and in the development of hypertension (Laursen et al., 1997; Abe and Berk, 1998). The reactive oxygen species that are generated by xanthine oxidase are potent stimuli of VSMC proliferation (Rao and Berk, 1992). Conversely, antioxidants reduce the cell response to growth factors, and inhibit the proliferation of VSMC (Boscoboinik et al., 1995). In cultured rat VSMC, Ang II causes rapid increases in intracellular H2O2 and phosphorylation of p42/44 MAPK and p38 MAPK. Treatment with H2O2 activates only p38 MAPK, and an NADH/NADPH oxidase inhibitor reduced only p38 MAPK phosphorylation in Ang II-treated cells. Also, blockade of Ang II-induced p38 MAPK phosphorylation by transfected catalase inhibited solely p38 MAPK phosphorylation, and reduced Ang II-induced cell hypertrophy. These findings indicate that both the p38 MAPK and the p42/44 MAPK pathways are required for the hypertrophic response of VSMC to Ang II (Ushio-Fukai et al., 1998).
In addition to their role in Ang II-induced activation of p38 MAPK, reactive oxygen species mediate the activation by Ang II of Akt/protein kinase B (Akt/PKB) in VSMC (Ushio-Fukai et al., 1999b). This serine-threonine kinase has been implicated in protein synthesis and also appears to be an important component of a survival pathway that protects cells from apoptosis. Akt/PKB is activated by several growth factors via Ras and phosphatidylinositol-3 kinase, and also by heat shock and other stresses. In rat VSMC, the activation of Akt/PKB by Ang II is dependent on tyrosine phosphorylation and the activity of its upstream effector molecule, phosphatidylinositol-3 kinase (Takahashi et al., 1999). These findings suggest that Akt/PKB has an important role in the intracellular actions of Ang II and in particular in its effect on cell survival (Pollman et al., 1996).
NO inhibits several of the physiological actions of Ang II, and prevents its activation of ERK, JNK, and p38 MAPK (Wang and Murphy, 1998). In rat cardiac fibroblasts, NO inhibits the activation of PYK2 and causes a concomitant decrease in ERK phosphorylation in Ang II-stimulated cells. Since PYK2 is essential for Ang II-induced activation of ERK in VSMC (Sabri et al., 1998), these findings suggest that it could be a locus at which NO regulates calcium-dependent signaling by the AT1 and other Gq-coupled receptors (Lev et al., 1995).
J. Receptor Activation and Endocytosis
Agonist activation of many plasma-membrane receptors is followed by clustering of the complexes in clathrin-coated pits, and subsequent internalization in clathrin-coated vesicles to undergo degradation or recycling to the plasma membrane. Although nutrient receptors (e.g., for low-density lipoprotein and transferrin) frequently exhibit constitutive internalization, the hormone and growth factor receptors that evoke intracellular signals usually undergo ligand-induced endocytosis. Growth factor receptors with intrinsic tyrosine kinase activity and G protein-coupled receptors are internalized by a similar clathrin-coated vesicular endocytic process. There is increasing evidence that the intracellular signaling mechanisms that are activated by these receptors do not necessarily participate in the internalization process. Such a dissociation between Ang II-induced G protein activation and receptor internalization was observed in studies on mutant and wild-type AT1A receptors expressed in COS-7 cells (Hunyady et al., 1994b). Mutations of the third cytoplasmic loop revealed that the N-terminal part of this region is important for both receptor endocytosis and intracellular signaling. Also, three point mutations of the conserved Asp74 residue in TMD II, which has been implicated in signal transduction by the AT1Areceptor and other GPCRs (Bihoreau et al., 1993), significantly impaired G protein coupling and phosphoinositide hydrolysis. However, the Asp74 (D74N, D74H, and D74Y) showed markedly different internalization kinetics. The D74Y receptor showed the greatest impairment of internalization but retained the highest degree of inositol phosphate stimulation. In contrast, the D74N mutant, which showed the most impaired G protein coupling and inositol phosphate responses, had normal internalization kinetics. The combined mutant receptor containing the D74N substitution and deletion of residues 221–226 from the third cytoplasmic loop showed no G protein coupling or inositol phosphate response but was internalized about 60% as rapidly as the wild-type receptor. These data demonstrate that endocytosis of the AT1 receptor is independent of agonist-activated signal transduction and indicate that receptor internalization and activation phospholipase C have different structural requirements.
The demonstration that coupling to heterotrimeric G proteins and intracellular signaling responses is not required for internalization of the AT1 receptor indicated that the conformational changes that result from agonist-induced receptor activation exert largely independent effects on signal transduction and receptor endocytosis. However, since the structural requirements for signaling and internalization overlap, mutations that cause changes in G protein coupling are frequently accompanied by impairment of endocytosis. Thus, the N-terminal region of the third cytoplasmic loop is important for both receptor signaling and internalization. However, the ability of internalization-deficient mutant receptors with specific changes in the cytoplasmic tail or third loop to activate their respective G proteins and signaling responses indicates that G protein activation alone is not sufficient to induce receptor endocytosis. The sequences in these regions that are important for receptor internalization are frequently enriched in serine and threonine residues. For example, the rat AT1 receptor contains 12 serine/threonine residues in the 23-amino acid segment located between residues 326 and 348 of the cytoplasmic tail (Fig.4).
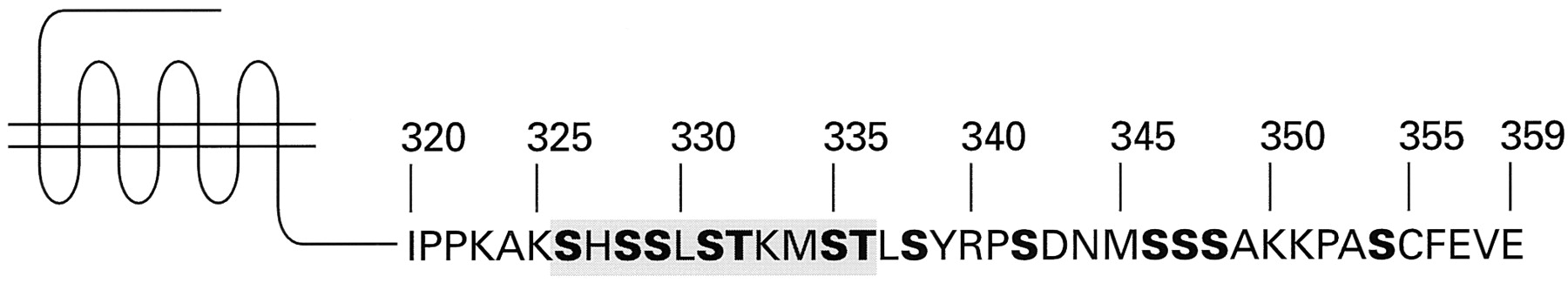
Location of agonist-induced phosphorylation sites in the AT1 receptor. The potential serine and threonine phosphorylation sites are shown in bold letters. The most highly phosphorylated region in the C-terminal cytoplasmic tail during Ang II action is indicated by the shaded box. Potential PKC sites are indicated by brackets, and the corresponding phosphorylated setrines are shown by asterisks. Recent evidence suggests that low agonist concentrations selectively induce PKC phosphorylation by other kinases is predominant at higher agonist concentrations (Quian et al., 1999).
A mutational analysis of this region of receptor revealed that deletion of the carboxyl-terminal 22 amino acids of the receptor did not affect agonist-induced endocytosis (Hunyady et al., 1994a). However, internalization was markedly impaired by removal of one additional residue (Leu337) and was reduced by 95% after removal of the adjacent Ser335 and Thr336. In addition, triple alanine replacement of the Ser-Thr-Leu residues reduced internalization to almost the same extent as the corresponding tail deletion mutant. The Ser-Thr-Leu motif is highly conserved in mammalian AT1 receptors but is not present in the noninternalizing AT2receptor. These findings demonstrate that a serine/threonine-rich region including Leu337 in the cytoplasmic tail of the AT1 receptor is a major requirement for endocytosis of the hormone-receptor complex, and support the concept that similar motifs in other G protein-coupled receptors are determinants of their agonist-induced internalization. An additional region in the cytoplasmic tail of the AT1receptor, involving hydrophobic and aromatic residues on a putative α-helix adjacent to the cell membrane, has also been found to be involved in agonist-stimulated endocytosis (Thomas et al., 1995). It is possible that these two regions in the cytoplasmic tail function cooperatively to trigger endocytosis, and that agonist-induced receptor activation leads to phosphorylation of the serine/threonine-rich region and causes a conformational change that exposes the more proximal hydrophobic residues for interaction with the endocytic mechanism.
K. AT1 Receptor Function in Selected Tissues
1. The AT1 Receptor and the Brain.
Ang II regulates numerous physiological responses through its central actions in the brain, where it functions as a neurotransmitter or neuromodulator to influence blood pressure, drinking behavior, salt appetite, and several neuroendocrine processes. Some of these responses are induced by the actions of circulating Ang II at the circumventricular organs and other specialized regions, and others are influenced by locally formed Ang II generated within the brain itself. Although circulating hormones are effectively excluded from most parts of the brain by the blood-brain barrier, neurons in the circumventricular organs are accessible to many circulating ligands via the fenestrated endothelial cells of their dense capillary circulation. Due to the absence of the blood-brain barrier at these sites, neurons within the subfornical organ (SFO), organum vasculosum lamina terminales (OVLT), and area postrema are exposed to circulating hormones, ions, and other potential regulatory factors. These three structures, sometimes called the sensory circumventricular organs (Johnson and Gross, 1993), are rich in AT1 receptors and have been implicated in numerous homeostatic processes. Ang II is but one of several peptides that bind to high-affinity receptors located on circumventricular neurons that project to hypothalamic nuclei and other brain regions. Other ligands with abundant circumventricular receptors include atrial natriuretic peptide, vasopressin, cholecystokinin, GLP-1, calcitonin, and relaxin (McKinley and Oldfield, 1998).
The pressor response to circulating Ang II is mediated by the area postrema and the SFO, and its stimulatory actions on water and salt ingestion, vasopressin secretion, and adrenocorticotropin (ACTH) release are mediated by the SFO. AT1-expressing neurons in the SFO send projections to the supraoptic and paraventricular nuclei of the hypothalamus to influence vasopressin and ACTH release (Oldfield et al., 1994), and to the median optic nucleus to stimulate water drinking (Cunningham et al., 1991).
Like several other peptide hormones, Ang II exerts effects in the central nervous system that are complementary to its primary physiological actions in peripheral tissues. This is well exemplified by the presence of brain AT1 receptors at sites that influence cardiovascular function, fluid and electrolyte homeostasis, and pituitary hormone secretion (Allen et al., 1998,1999). The existence of an endogenous brain renin-angiotensin system that is distinct from the systemic system comprised of the kidneys, liver, and lungs has been demonstrated by numerous studies over the last 30 years. Early evidence for a central mechanism that contributes to the development of Ang II-induced hypertension (Bickerton and Buckley, 1961) was followed by the demonstration of renin and angiotensin II in the brain (Fisher-Ferraro et al., 1971). These and other studies led to the recognition of an autonomous brain renin-angiotensin system that has significant actions in cardiovascular regulation and several other brain functions (Ganten and Speck, 1978; Ganong, 1984; Phillips, 1987).
The ability of centrally administered Ang II to increase blood pressure, fluid intake, and vasopressin release presaged the existence of specific receptors in the brain regions controlling these functions. This was confirmed by the selective effects of Ang II on brain structures that influence blood pressure, sympathetic nerve activity, drinking behavior, salt appetite, and pituitary hormone secretion (Phillips, 1987; Ganong, 1993; Wright and Harding, 1994). Effects of angiotensin on learning and memory, as well as sensory function, have also been observed. The number and scope of these central effects of angiotensin indicate that the renin-angiotensin system has a major role in the regulation of neural components that control multiple physiological functions. Most of these actions are mediated by AT1 receptors, which are expressed in brain regions known to be involved in the control of cardiovascular function and body fluid homeostasis.
At the cellular level, direct application of Ang II increases the firing rate of neurons in the supraoptic nucleus, subfornical organ, and paraventricular nucleus (Nicoll and Baker, 1971; Felix and Akert, 1974; Ambuhl et al., 1992a,b) as well as in the nucleus tractus solitarius, dorsal motor nucleus of the vagus, and thalamus (Wayner et al., 1973; Felix et al., 1988). In several of these studies, the angiotensin II (2–7) heptapeptide (Ang III) was found to be as or more effective than Ang II, raising the possibility that Ang III may be the centrally active form of Ang II (Harding et al., 1986). The application of topical autoradiography to receptor mapping in tissue sections revealed that angiotensin II receptors are present in many of the brain nuclei and other regions that are known to be involved in the central regulation of cardiovascular regulation and fluid homeostasis (Mendelsohn et al., 1984). The distributions of Ang II receptors in the brains of several mammalian species have been mapped by topical autoradiography with radioiodinated ligands, usually125I-[Sar1,IIe8]Ang II. Specific AT1 and AT2receptors were identified by inhibition of radioligand binding by selective AT1 (losartan) and AT2 (PD123177) receptor antagonists. The distribution of AT1 receptor messenger RNA in the rodent brain has also been analyzed by in situ hybridization using specific riboprobes to detect AT1A and AT1B receptor subtypes (Lenkei et al., 1998). Such studies have shown that all of the well defined physiological actions of Ang II in the brain are mediated by AT1 receptors.
In all species studied, AT1 receptors are most abundant in the hypothalamus and the circumventricular organs. In the hypothalamus, AT1 receptors are localized to the parvocellular region of the paraventricular nucleus, which has been implicated in the control of anterior pituitary hormone secretion, ingestive behavior, and autonomic regulation of the cardiovascular system. In contrast, AT1 receptor expression is low or absent in the magnocellular neurons of the paraventricular nucleus that control the release of vasopressin and oxytocin from the neurohypophysis. The circumventricular organs, which include the median eminence, arcuate nucleus, SFO, and OVLT, contain high densities of AT1 receptors. At each of these sites, the local deficiency in the blood-brain barrier renders them accessible to Ang II and other regulatory ligands in the circulation. Together with the interposed median preoptic nucleus, which also contains abundant AT1 receptors, the SFO and OVLT comprise the lamina terminalis of the forebrain. This structure has neural connections with the parvocellular neurons of the hypothalamus, as well as efferent projections to the supraoptic nucleus that subserve vasopressin secretion and other osmoregulatory responses including sodium excretion and thirst (McKinley et al., 1992).
In contrast to the relatively wide distribution of AT1A receptor expression in the CNS, the AT1B receptor is not detectable in most brain areas. However, AT1B receptors are the predominant subtype expressed in the anterior pituitary gland, where they mediate the direct actions of Ang II on pituitary function. Both Ang II and Ang III can elicit a variety of physiological changes, including pressor and dipsogenic responses, and increased salt appetite, when injected into the cerebral ventricles (Wright and Harding, 1992). This is accompanied by increased firing of magnocellular neurons in the supraoptic nucleus (Okuya et al., 1987;Yang et al., 1992). Injection of Ang II or Ang III into the supraoptic and paraventricular nuclei also stimulates magnocellular neurons, leading to the release of vasopressin from the posterior pituitary gland (Phillips, 1987).
2. Ang II-Induced Neuronal Signaling Pathways.
AT1 receptor-mediated signaling pathways in neurons are generally similar to those observed in other Ang II target cells. In addition to phosphoinositide hydrolysis, with formation of InsP3 and diacylglcerol, elevation of [Ca2+]i, and activation PKC, Ang II stimulates the ras/raf/MAPK cascade and translocation of MAPK into the nucleus (Lu et al., 1996). It also increases the expression of c-fos, c-jun, and other early response genes. The MAPK pathway mediates Ang II-stimulated norepinephrine synthesis and reuptake mechanisms in cultured neurons (Yang et al., 1996). In addition, Ang II-induced increases in [Ca2+]i activate calcium/calmodulin protein kinase II, which acts together with PKC to mediate the rapid actions of Ang II on neuronal membrane currents. These include a PKC-mediated increase in total neuronal Ca2+ current (ICa), as well as PKC- and calcium/calmodulin protein kinase II-mediated inhibition of delayed rectifier K+ current (Kv) and transient K+ current (IA) (Zhu et al., 1997a, 1998a). In rat supraoptic neurons, AT1 receptors mediate Ang II-induced activation of a nonselective Na+/Ca2+channel that promotes neuronal depolarization (Yang et al., 1992). Some of these effects of AT1 receptor activation in neurons, including MAPK activation, are under the inhibitory control of AT2 receptors via activation of serine/threonine type 2A (PP-2A) (Huang et al., 1996) and possibly tyrosine phosphatases such as MAPK phosphatase.
3. Role of Ang III in the Brain.
Ang II is rapidly cleared from the circulation with a half-life of 13 s and is extensively metabolized in its target tissues to form Ang III, Ang IV, and Ang (4–8) (Harding et al., 1986; De Silva et al., 1988). Although the Ang II octapeptide is the major active species acting at AT1 receptors in peripheral Ang II target tissues, the possibility that Ang III could be the predominant effector peptide in the brain has been recognized for several decades. This was first suggested by the observation that Ang III was twice as effective as Ang II in stimulating the firing rat of SFO neurons (Felix and Schlegel, 1978). This finding, and similar observations in paraventricular nucleus neurons, led to the suggestion that Ang III could be the centrally active form of Ang II (Harding et al., 1986). This proposal gained support from studies using nonselective aminopeptidase inhibitors such as bestatin, which increased the half-lives of Ang II and Ang III in the brain and potentiated the stimulatory action of Ang II and Ang III on paraventricular neurons (Harding and Felix, 1987).
The recent development of specific inhibitors of aminopeptidase A (APA) and aminopeptidase N (APN), which hydrolyze the N-terminal residues of Ang II and Ang III, respectively, has facilitated the investigation of the roles of Ang II and Ang III in the brain. The effects of i.c.v. administration of these compounds indicated that the central action of Ang II on vasopressin release in mice depends on its prior conversion to Ang III, confirming that Ang III acts as a major effector peptide of the renin-angiotensin system in the brain (Zini et al., 1996). Furthermore, the ability of centrally administered Ang II to stimulate the firing rate of magnocellular vasopressinergic neurons of the supraoptic nucleus (SON) of the rat was prevented by central administration of the selective APA inhibitor, EC33. Since the SON does not contain AT1 receptors, this effect of EC33 presumably results from its inhibition of APA in the subfornical organ, from which Ang II-sensitive neurons project to the SON (Zini et al., 1998).
Recent studies on the effects of selective APA and APN inhibitors on blood pressure in the rat have indicated that Ang III is also the main effector peptide of the brain RAS in the regulation of blood pressure (Reaux et al., 1999). Central inhibition of APA by EC33 not only blocked the pressor action of Ang II but also caused a fall in basal blood pressure comparable to that caused by i.c.v. administration of losartan, indicating a requirement for Ang III in the tonic regulation of blood pressure. Furthermore, the inhibition of APN activity by PC18 caused an increase in blood pressure that was blocked by losartan. This suggests that inhibition of Ang III degradation by PC18 causes an increase in Ang III levels that elevates blood pressure through an action on AT1 receptors. These observations indicate the potential applications of APA inhibitors as centrally acting compounds in the management of hypertension.
These observations raise the question of whether the central actions of Ang III are mediated by an as yet unidentified AT1-type receptor with higher affinity for Ang III over Ang II. This seems unlikely in view of the probability that such a receptor with similarity to the AT1 site would have been detected during isolation and cloning of the AT1 and AT2 subtypes. The alternative proposal, that blockade of the conversion of Ang II to Ang III by APA favors the activation of other Ang II-degrading pathways, appears to be a more likely explanation.
4. The AT1 Receptor and the Pituitary Gland.
The presence of Ang II receptors in the anterior pituitary gland was suggested by early reports of actions of the octapeptide on pituitary hormone secretion (Steele et al., 1981). This was later confirmed by binding studies with 125I-Ang II, which demonstrated specific, high-affinity binding sites in the anterior pituitary glands of rat, rabbit, and dogs. No receptors were found in the posterior pituitary gland or in GH3 pituitary tumor cells (Hauger et al., 1982; Mukherjee et al., 1982). The sites were nanomolar affinity, and bound the heptapeptide des-Asp1-Ang II with about one-tenth the affinity of the octapeptide. Ang II and the heptapeptide showed a similar potency ratio in their ability to stimulate ACTH release from pituitary cells (Capponi et al., 1982). Fractionation of rat pituitary cells revealed that Ang II receptors were associated with lactotrophs and stimulated prolactin as well as ACTH release. These findings suggested that Ang II contributes to the physiological control of prolactin secretion (Aguilera et al., 1982).
The stimulatory action of Ang II on prolactin production by pituitary lactotrophs is mediated by the AT1 receptor, which is coupled to phosphoinositide hydrolysis and PKC activation, and also affects cyclic AMP production (Audinot et al., 1991). In addition to these stimulatory actions, Ang II also exerts AT1-mediated inhibitory actions on adenylyl cyclase activity in pituitary cell membranes. In intact cells, blockade of AT1 receptors by losartan prevented AT1-induced prolactin release, as well as the phospholipase C and PKC-dependent increase in cyclic AMP production (Moreau et al., 1994). During development the responsiveness of rat pituitary lactotrophs to Ang II increases with age, and by 28 days was more prominent in female than in male rats (Diaz-Torga et al., 1994). The ability of the hypothalamic GnRH to stimulate prolactin release in perifused pituitary cells, but not gonadotropin release, was abolished by losartan, consistent with a paracrine role of Ang II in GnRH-stimulated prolactin release (Becu-Villalobos et al., 1994). Interestingly, the ability of Ang II to stimulate prolactin release from cultured pituitary cells from animals fed on an essential fatty acid-free diet was markedly decreased, although the stimulatory action of thyrotropin-releasing hormone, and the inhibitory action of dopamine were not affected (Alessio et al., 1994).
Ang II also increases the proliferation of cultured mammatrophs, and this effect is suppressed by tamoxifen and restored by estradiol-17β. Treatment with the peptide Ang II antagonist, saralasin, inhibited not only the Ang II-stimulated proliferative response but also that of GnRH, again indicating that GnRH-stimulated release of Ang II, together with estradiol, regulates lactotroph proliferation in the rat pituitary gland (Shinkai and Ooka, 1995). The stimulatory action of Ang II on prolactin release was associated with a rapid increase in intracellular calcium concentration that was abolished by losartan. However, in the same report no increase in [3H]thymidine incorporation was observed (Diaz-Torga et al., 1998). An analysis of the distribution of the AT1 receptor subtypes in the rat anterior pituitary gland, which expresses predominantly (80%) AT1B receptors, revealed that the majority of the AT1B receptor-expressing cells are lactotrophs, and the remainder are corticotrophs. In adult male rats, AT1B receptors are located in about 50% of the pituitary lactotrophs and about 25% of the corticotrophs. These findings suggest that Ang II synthesized in gonadotrophs can directly stimulate prolactin and ACTH release from lactotrophs or corticotrophs through activation of AT1Breceptors (Lenkei et al., 1999). This AT1B-mediated paracrine mechanism in the pituitary gland is subsidiary to its endocrine regulation by AT1A-mediated changes in the hypothalamic production of releasing hormones and neurotransmitters including dopamine and catecholamines (Denef, 1986; Ganong, 1993).
5. The AT1 Receptor and the Heart.
The role of the renin-angiotensin system in cardiovascular control and body water homeostasis is well documented (reviewed in Saavedra, 1992; Wright and Harding, 1994, 1997; Höhle et al., 1995; Mosimann et al., 1996; Unger et al., 1996). Ang II acts primarily at the AT1 receptor type in a variety of tissues including vascular smooth muscle, kidney, and adrenal gland to influence vasoconstriction and sodium reabsorption. Furthermore, it is known that Ang II acts locally within cardiac tissues to influence protein synthesis and cellular growth (Baker and Aceto, 1990;Lindpainter and Ganten, 1991; Baker et al., 1992; Dostal and Baker, 1992; Dostal et al., 1992, 1996). There is accumulating evidence that the AT1 receptor may be involved in the development of cardiac hypertrophy via the regulation of extracellular matrix accumulation (Weber and Brilla, 1991; Weber et al., 1993,1995a,b; Brilla et al., 1993, 1995b). Specifically, cardiac fibroblast stimulation appears to facilitate the accumulation of collagen in the extracellular matrix of the heart (Brilla et al., 1995a). Furthermore, Ang II appears to block MMP-1 activity, an enzyme directly involved in the degradation of fibrillar collagen. Thus, interference with MMP-1 results in a proliferation of extracellular matrix. Ang II-induced hypertrophy in cardiac myocytes and vascular smooth muscle cells follows the pattern of an initial increase in protein synthesis and cell size without an elevation in numbers of cells (Geisterfer et al., 1988; Berk et al., 1989). After prolonged exposure to Ang II, mitogenic changes were also observed (Weber et al., 1994). The ability of ACE inhibitors and AT1receptor antagonists to prevent the onset of cardiac hypertrophy in animal models (Zhu et al., 1997b), and to promote its regression in hypertensive patients (Thürmann et al., 1998), has also demonstrated the role of AT1 receptors in ventricular hypertrophy. These agents also prevent the onset of ventricular dilatation in rats after myocardial infarction by suppressing the expression of gene expression (ANF, β-myosin heavy chain, collagen) associated with cardiac remodeling (Yoshiyama et al., 1999). To determine whether this effect results from inhibition of the direct action of angiotensin on the heart, or reduction of pressure overload, Paradis et al. (2000) created transgenic mice over-expressing the AT1 receptor in cardiac myocytes. These animals spontaneously developed cardiac hypertrophy and remodeling, with increased expression of cardiac ANF and interstitial collagen deposition. They showed no change in heart rate or blood pressure, but died of heart failure at an early age. These findings indicate that the AT1 receptor mediates the direct actions of Ang II in the development of ventricular hypertrophy and heart failure. However, it should be noted that some of the beneficial effects of ACE inhibitors and AT1 antagonists on cardiac function could result from activation of B2 kinin and AT2 receptor-dependent pathways that modulate myocyte contractility and growth (Wollert and Drexler, 1999). The role of the bradykinin B2 receptor remains, however, disputed (Madeddu et al., 2000).
III. The Type 2 (AT2) Angiotensin Receptor
Whereas differential stability to dithiothreitol (Chang et al., 1982; Gunther, 1984; Chiu 1989b; Speth et al., 1991) suggested the presence of more than a single form of angiotensin II binding or receptor site (Chiu et al., 1989a; Whitebread et al., 1989; Chang and Lotti, 1990), cloning and expression of the receptor types AT1 (Murphy et al., 1991; Sasaki et al., 1991) and AT2 (Kambayashi et al., 1993b;Mukoyama et al., 1993) provided clear evidence for the presence of the second isoform, AT2. The identification and characterization of the biochemical and physiological functions of the AT2 receptor is still a matter of intense research.
A. Cloning, Purification, and Properties of the AT2Receptor
The AT2 receptor is clearly distinct from the AT1 receptor in tissue-specific expression, signaling mechanisms, and diversity in molecular weight. In the early phase of AT2 receptor research it was suspected that the AT2 receptor may not even be a G protein-coupled seven transmembrane domain receptor (Bottari et al., 1991). Clearly, its structure had to be determined by cloning and purification. Whether it was a single entity or had related isoforms was not apparent in the beginning. Undoubtedly, more fundamental studies were needed, which should be based on the cloning of the AT2 cDNA and purification of the AT2 receptor.
There are cell lines expressing the AT2 but not the AT1 receptor, such as PC12W, R3T3, and some lines of N1E-115. Fetal rats express a large amount of the AT2 receptor protein and its mRNA. However, the cloning of the AT2 receptor turned out to be difficult. Attempts to screen cDNA libraries with AT1 cDNA as a probe did not work, indicating a lack of homology between the AT1 and AT2 receptor gene. Thus, the expression cloning method had to be used. For some unknown reason, cDNA detected by autoradiographic identification of transfected COS-7 cells presumably expressing AT2 mRNA could not be recovered from these cells. After several technical refinements, a 4.5-kb rat AT2 cDNA was cloned from rat PC12W cells byKambayashi et al. (1993b), and from rat fetuses by Nakajima et al. (1993). It contained seven hydrophobic sequences compatible with the structural theme of the seven transmembrane domain receptors super family (or GPCR) (see Fig. 5) but showed only a 32% amino acid sequence identity with the rat AT1 receptor.

Amino acid sequences of rat AT1A and AT2 deduced from respective cDNA. Sequences in shaded boxes are identical. Of the overall 32% sequence identity, the transmembrane regions show a somewhat higher degree of homology.
Since targeted gene deletion can be performed only in the mouse, Ichiki et al. (1994) and Nakajima et al. (1993) cloned mouse AT2 cDNA from a fetal mouse cDNA library by a plaque hybridization method using rat AT2 cDNA as a probe. A mouse AT2 genomic 4.4-kb DNA fragment was cloned by Ichiki et al. (1994) and Koike et al. (1994). Since these studies show that the entire coding sequence is contained in one exon, the human AT2 coding sequence (Tsuzuki et al., 1994) or fragments containing more extensive sequences could be cloned (Martin et al., 1994; Koike et al., 1994). The genomic DNA of all three species consist of three exons with an uninterrupted coding region being confined to the third exon. The genes encoding the AT2 receptor are localized in human chromosome Xq22-q2, rat chromosome Xq3, and mouse chromosome X (Koike et al., 1994; Hein et al., 1995b; Tissir et al., 1995). The localization in the long arm of the X chromosome throughout species may have important implications in targeted gene deletion as well as in the genetics of the AT2 receptor gene.
The open reading frame of AT2 cDNA encodes 363 amino acid residues in all three species with 99% sequence identity between rat and mouse and 72% identity between rat and human. The divergence between rodent and human AT2 occurs mainly in the N-terminal region. AT1 and AT2 receptors have only 33 to 24% amino acid sequence identity (Kambayashi et al., 1993b; Nakajima et al., 1993). The homology was mainly localized in the transmembrane hydrophobic domains, which are believed to form seven transmembrane helical columns. Residues located in these helical column domains and considered to be essential for Ang II binding to the AT1 receptor are also preserved in the AT2 receptor. These include Lys118 (Lys102 of AT1) at the top of TM3, Arg183 (Arg167 of AT1) at the top of TM4 and Lys216 (Lys199 of AT1) and more.
Almost complete divergence between AT1 and AT2 receptors is seen in the third intracellular loop and more extensive differences in the carboxyl terminal tail (C-tail). The amino acid sequence of the rat AT2receptor indicates that it has five potentialN-glycosylation sites with the consensus amino acid sequences -Asn-X-Ser/Thr. It is possible that carbohydrate chains would give rise to increased and diverse molecular weights of the AT2 receptor. Southern hybridization analysis of restriction enzyme fragments of rat genomic DNA using rat AT2 cDNA as a probe did not produce evidence for a subtype of the AT2 receptor. Thus, the molecular weight divergence for the AT2 receptor ranging from 60 to 140 kDa may be due to a difference in the extent of glycosylation, as shown by Servant et al. (1994).
An earlier discriminator of the AT1 and AT2 receptors, before the isoform-specific antagonist became available, was the reducing agent dithiothreitol (DTT). The DTT sensitive Ang II receptor turned out to be the AT1 receptor, whereas DTT increased the ligand binding capability of the AT2 receptor and the AT2 receptor remained stable for hours (Chang, Lotti, Keegan, 1982; Whitebread et al., 1989; Chang and Lotti, 1990; Speth et al., 1991). This property was exploited for the direct purification of the AT2 receptor protein (see below). Another discriminator is Sar-p-benzoylphenylalanine8-angiotensin II, which can be used for selective labeling of AT2 by a photoaffinity technique. Although it binds to the bovine adrenal AT1 receptor with aK d value of 6.5 nM and human myometrium with a K d value of 0.39 nM, its covalent binding upon illumination takes place only with the AT2 receptor (Bosse et al., 1990). The selective labeling of the AT2 receptor is in contrast to another photoaffinity labeling peptide, Sar1-d-azido Phe8-Ang II, which is specific for the AT1 receptor (Guillemette et al., 1986). The 125I-radiolabeled peptide was used to visualize AT2 receptors in various cells to determine their molecular sizes by SDS gel electrophoresis. Cells known to contain the AT2 receptor produced radiolabeled AT2 receptor bands of 68 kDa (human uterine myometrium), 91 kDa (R3T3 cells), and 113 kDa (PC12 cells). However, endoglycosydase treatment reduced the myometrial 68-kDa receptor to 40- and 31-kDa proteins, the 91-kDa R3T3 receptor was reduced to 46- and 31-kDa and the 113-kDa PC12 AT2 receptor to 55 and 31 kDa. These results may involve artifacts of proteolytic digestion. More decisive evidence for glycosylation diversity came from the inhibition ofN-glycosylation by treatment of rat PC12 cells with tunicamycin (Servant et al., 1996). The 140-kDa AT2 receptor in untreated PC12 cells was reduced to 63- and 47-kDa glycosylated and 32-kDa unglycosylated receptors. An interesting byproduct of these studies is the observation of nonglycosylated receptor expression on the cell surface.
The structural diversity of the AT2 receptor is still a complex and unresolved issue. Attempts have been made to purify the AT2 receptor protein (Ciuffo et al., 1993a) by combining affinity chromatography on CGP42112-Sepharose and size separation by gel filtration and SDS gel electrophoresis from AT2 solubilized by CHAPS from neonatal rat kidney which had a 71-kDa molecular mass. The murine neuroblastoma cell line N1E-115 cell line contains both AT1 and AT2 receptors. The latter increases upon cellular differentiation. Ang II receptors were solubilized from membranes by the detergent CHAPS (1%). The labile AT1receptor could be removed by denaturation and the AT2 receptor was affinity purified on an Ang II-Sepharose affinity column by Siemens et al. (1991). They found that the AT2 receptor from N1E 115 can be separated to two different forms, peak I and peak III, by a Sepharose column chromatography. Peak III seemed to be an AT2receptor whose ligand binding activity is enhanced by the reducing agent DTT, but the stable GTP analog, GTPγS, did not suppress the binding. Ligand binding to a receptor under peak I was clearly reduced by GTPγS, but treatment with DTT reduced the ligand binding function (Siemens et al., 1994b). The peak I components consisted of two proteins 110 and 60 kDa. It has been proposed that the 110 kDa is a dimer of 60 kDa that is stabilized by the agonist (Yee et al., 1994). Interestingly, stabilization of the AT2 receptor on the cell membrane of R3T3 cells by an as yet undefined endogenous ligand, which is not identical with Ang II, has recently been described by Csikos et al. (1998).
B. Regulation of the AT2 Receptor
The AT2 receptor gene is expressed ubiquitously at a very high level in the fetus and it declines precipitously after birth in many but not all tissues. In the skin, for instance, it decreases to undetectable levels, but in certain tissues such as the adrenal and heart, the decline stops at certain levels, and AT2 receptor expression persists for the remainder of life. In uterine myometrium, the AT2receptor is expressed under nonpregnant conditions and declines during pregnancy, but returns to nonpregnant levels after parturition (de Gasparo et al., 1994). These few examples demonstrate clearly that the AT2 receptor expression is regulated. Theoretically, such regulatory changes may be controlled by transcription and/or by changing the stability of mRNA.
Post-transcriptional (translational) regulation also seems to take place. After confluency, the AT2 binding sites increase in R3T3 cells without an increase in mRNA (Dudley et al., 1991), whereas mRNA is low in the growth phase and increases in the preconfluent state. The AT2 receptor in these cells may signal to activate protein phosphotyrosine phosphatases (PTP), which inactivates mitotic or hypertrophic growth. Teleologically, its activity should then be expressed more or less constantly in normal nongrowing cells. That this receptor lacks an internalization and desensitization mechanism is compatible with its role to maintain differentiated cells in a quiescent state (Unger, 1999).
Attempts to investigate the transcriptional regulation have begun. AT2 genomic DNA have been cloned from mouse (Nakajima et al., 1993; Ichiki et al., 1994) and humans (Koike et al., 1994). The behavior of the AT2 receptor gene expression in rat vascular smooth muscle cells from the spontaneously hypertensive (SHR) and Wistar Kyoto or Sprague-Dawley rats are markedly different. This finding is in line with the observation that AT2 receptor binding in endothelial cells from SHR is markedly higher than in those from Wistar Kyoto rats (Unger, 1999). The 5′-flanking region of rat genomic DNA was also cloned to determine its functional elements (Ichiki and Inagami, 1995a).
The expression of the AT2 receptor was shown to be dependent on growth factors or growth states. In mouse R3T3 cells, AT2 mRNA is expressed when growth is arrested in the confluent state and is decreased in the vigorous growth phase (Dudley et al., 1991; Dudley and Summerfeldt, 1993). Prolonged serum depletion in the presence of insulin or IGF-1 stimulates the expression of AT2 receptor in vascular smooth muscle cells, whereas serum growth factors, like PDGF, lysophospatidic acid, and phorbol ester rapidly abolished AT2 receptor expression (Kambayashi et al., 1993a, 1996). Similar observations with respect to IGF-1 as well as interleukin-1β (IL-1β) were made in R3T3 fibroblast cells (Ichiki et al., 1995;Kambayashi et al., 1996). In contrast, growth factors (phorbol ester, lysophosphatidic acid, and basic fibroblast growth factor) markedly suppressed mouse AT2 mRNA expression in the fibroblast cells but did not completely eliminate it as they did in vascular smooth muscle cells (Ichiki et al., 1995a,b, 1996).
The transcriptional activity of the 5′-flanking region of the mouse AT2 receptor gene was investigated further by using constructs consisting of truncated 5′-flanking sequences (up to 1.2 kb) fused to the luciferase gene in R3T3 cells. An AP-1 site may transmit the suppressive effects of noninsulin growth factors. Thus, PKC-activating growth factors and phorbol esters rapidly down-regulate the AT2 receptor expression (Kambayashi et al., 1993a; Kizima et al., 1996). The C/EBP site may mediate the positive transcription effect of IL-1β as it stimulates the expression of C/EBPβ and γ (Ichiki and Inagami, 1995a,b, 1996).
AT2 receptor expression by insulin or IGF-1 is interesting in that the process has a very slow onset, taking several days. A putative insulin response sequence (IRS) is located at −126 to −117 of the 5′-flanking region of the rat AT2receptor gene. This sequence TAATGTTTTG is 80% homologous to the IRS in the phosphoenolpyruvate carboxykinase gene TGGTGTTTTG (O'Brien and Granner, 1991). An interesting aspect of the effect of insulin is that the aorta of obese (fa/fa) Zucker rats, which have about 10 times higher plasma insulin concentrations than Sprague-Dawley rats, expresses AT2 mRNA at a level clearly detectable by Northern blotting, whereas it was not detectable by the Northern blotting in the aorta of Sprague-Dawley rats (Kambayashi et al., 1996).
In these studies, an enhancer activity of the AP-1, C/EBP, NF/IL-6, IRS was not examined rigorously. Horiuchi et al. (1995)demonstrated by elegant methods that interferon regulatory factor IRF-2 attenuated AT2 receptor expression, both in the confluent and growing cells, whereas IRF-1 enhanced AT2 expression in confluent cells only. Several IRF binding motifs were found in the promoter in positions between −453 and −225. Whether these elements alone account for up-regulation is not yet known. Tissue-specific expression and widespread low level expression of the AT2 receptor rather point to complex mechanisms of AT2 receptor regulation.
C. AT2 Receptor Diversity
Although the AT2 receptor was cloned and purified, there remains a nagging question as to whether the AT2 receptor is a single entity, because there is evidence for divergence in the molecular and functional properties (Siemens et al., 1994a). As discussed above, the large disparity of molecular size was attributed to a difference in the size of carbohydrate structure (Servant et al., 1994) but it could be also due to dimerization or oligomerization (Yee et al., 1994). Although DTT enhances ligand binding of the AT2 receptor in most tissues investigated, in some this is not the case (Tsutsumi and Saavedra, 1992b). Most intriguing is the observation that agonist binding of typical AT2 receptors, including those that had been cloned and expressed, is not suppressed by GTPγS, or pertussis toxin. Some AT2 receptors in rat locus ceruleus and thalamic or geniculate nuclei are markedly affected by these reagents, which affect the heterotrimeric G proteins, Gi and Go. On the other hand, GTPγS has no effect on the AT2 receptor in R3T3 cells or the inferior olive (Table3). The lack of effect has also been seen in other cells expressing the AT2 receptor (Tsutsumi and Saavedra, 1991). Moreover, AT2 mRNA was not detected by in situ hybridization using a sequence of the cloned AT2 receptor as an antisense oligonucleotide probe (Jöhren et al., 1996). The question as to whether diversity in the functional properties of the AT2 receptor in different types of cells and tissues is due to another molecular species, or to additional factors associated with it, may be addressed by investigating various modes of the signal transduction mechanism of the AT2receptor associated with the molecular diversity.
Binding characteristics of the AT2receptor3-a
D. Targeted AT2 Receptor Gene Overexpression and Deletion
The use of AT2 receptor antagonists has provided evidence for a variety of functions of the AT2 receptor in several different types of cells and tissues including cardiovascular, renal, adrenal, central nervous, as well as dermal mesenchymal systems. Gene manipulation to increase, decrease, or abolish the expression of a gene in question are effective approaches to assess the pathophysiological roles of the gene or its product. Transgenic transfection of AT2 receptor expression vectors into COS-7 cells (Kambayashi et al., 1993b;Mukoyama et al., 1993), and vascular smooth muscle in vivo (Nakajima et al., 1995) in “gain of function” approach have proven to be efficient methods. Whereas cardiac targeted overexpression of the AT2 receptor in mice did not cause obvious morphological or functional changes, Ang II infusion paradoxically decreased blood pressure and produced a negative chronotropic effect (Masaki et al., 1998). Moreover, Tsutsumi et al. (1999) reported stimulation of bradykinin activity and nitric oxide production in AT2 transgenic mice following inhibition of the Na+/H+ exchanger.
The targeted gene deletion (AT2 knockout mouse or AT2 null mouse) method produced a very interesting strain of mice (Hein et al., 1995a; Ichiki et al., 1995a). In contrast to the hypertension and impaired vascular responses observed in AT1-deficient mice, knockout of the AT2 receptor leads to elevation of blood pressure and increased vascular sensitivity to Ang II (Hein et al., 1995a;Ichiki et al., 1995). This has suggested that the AT2 receptor may exert a protective action in blood pressure regulation by counteracting AT1receptor function. Such an action could be exerted in part by reduced expression of the AT1 receptor, which is increased in the vascular smooth muscle of AT2-deficient mice (Tanaka et al., 1999). However, the sustained hypersensitivity to Ang II in such animals is also attributable to loss of the counter-regulatory action of renal bradykinin and cyclic GMP formation (an index of NO production) (Siragy et al., 1999). The relative contributions of these two factors to AT2-dependent vascular regulation, and the extent to which AT2 receptor deficiency could contribute to sustained blood pressure elevation, as in human hypertension, have yet to be determined.
The AT2 receptor null mouse Agtr2(−/−) also showed behavioral changes and proclivity to have renal diseases. However, despite its massive expression in the mesenchymal tissues of skin, kidney, as well as in the brain and heart in fetal stages of development, the AT2 receptor null mouse does not show discernible gross anatomical defects or deformity to date. The early fetal form of the kidney mesonephros is programmed to disappear during the fetal development. Its tubular tissue is densely surrounded by mesenchymal cells expressing AT2 mRNA (Kakuchi et al., 1995). However, its disappearance also takes place in the fetus of the AT2 receptor null mouse. The medulla of the metanephros is also filled with AT2receptor-expressing mesenchymal cells, which undergo apoptosis as tubulogenesis proceeds (Kakuchi et al., 1995). Again, kidney formation in the AT2 receptor null mice takes place normally and newborn mice grew to adult mice with a normal pair of kidneys (Ichiki et al., 1995b). These studies with AT2 receptor gene deleted mice indicate that the AT2 receptor is not essential for apoptosis in nephrogenesis.
Functionally, the AT2 receptor seems to exert a hypotensive effect—although this function is still discussed controversially—and to blunt the sensitivity of AT1 receptors to Ang II. Its role in the regulation of cardiac, renal, and adrenal function is still under investigation. As for the renal function, the role of the AT2 receptor as a regulator (or modulator) of pressure natriuresis is still controversial (Keiser et al., 1992; Lo et al., 1995; Siragy and Carey, 1996). Whereas studies with AT2 receptor agonists or antagonists in intact rats suggested an antidiuretic and antinatriuretic effect of the AT2 receptor, results in AT2 receptor knockout mice showed the opposite effects (Siragy et al., 1999).
Although AT2 receptor null mice did not produce gross morphological abnormalities, vascular differentiation driving the expression of the constituents of the contractile apparatus was altered. The expression of h-caldesmon and calponin are delayed in the aorta, suggesting that the AT2 receptor enhances the differentiation of vascular smooth muscle cells and is involved in vasculogenesis (Yamada et al., 1999). In addition, more detailed studies with a larger population of AT2 receptor null mice are producing exciting evidence that a loss of the AT2 receptor in the AT2null mice results in the development of congenital urinary tract anomalies with a 23% penetrance.
1. Behavioral Changes in AT2 Receptor Null Mice.
In the brain, the AT2 receptor is expressed in distinct areas (Obermüller et al., 1991), prominently in the locus ceruleus (Rowe et al., 1990a,b;Saavedra, 1992). The locus ceruleus contains a high concentration of noradrenergic nerve endings that release norepinephrine upon treatment with Ang II (Sumners and Phillips, 1983) and sends out long noradrenergic projections to the cerebral cortex, hypothalamus, and hippocampus. The AT2 receptor is also expressed in the central amygdaloid nucleus (Song et al., 1992), and/or medial amygdaloid nucleus (Saavedra, 1992). Loss of the AT2 receptor in these areas could entail behavioral effects.
AT2 receptor null mice showed a markedly reduced exploratory behavior when placed in a new environment with ambulation reduced by 40% (Hein et al., 1995a; Ichiki et al., 1995). After water deprivation, AT2 null mice show a greater stimulation of dipsogenesis (Hein et al., 1995a).
E. Signaling Mechanisms of the AT2 Receptor
Studies on hormone receptor signaling mechanisms are aimed at explaining cellular and physiologic responses by intracellular biochemical processes. As Nahmias and Strosberg (1995) point out, the search for major physiologic functions of the AT2receptor is progressing and several interesting clues are at hand. The AT2 receptor seems to open a delayed rectifier potassium channel at least in hypothalamic neuronal tissues (Kang et al., 1994, 1995), to close a T-type Ca2+ channel (Buisson et al., 1992, 1995), to suppress tissue and cellular growth (Nakajima et al., 1995; Meffert et al., 1996; Munzenmaier and Green, 1996; Stoll et al., 1995; Tsuzuki et al., 1996a,b), to induce (neuronal) cell differentiation (Laflamme et al., 1996; Meffert et al., 1996; Gallinat et al., 1997; Stroth et al., 1998; Côté et al., 1999; Gendron et al., 1999) and to support apoptosis (Yamada et al., 1996; Chamoux et al., 1999; Gallinat et al., 1999). In addition, the AT2 receptor may exert hypotensive effects (Scheuer and Perrone, 1993; Ichiki et al., 1995) and appears to influence behavior (Hein et al., 1995a; Ichiki et al., 1995).
The consensus among investigators working with cells expressing the AT2 receptor exclusively without the AT1 receptor, such as R3T3 (Dudley et al., 1990,1991), PC12W (Bottari et al., 1991, 1992; Leung et al., 1992; Webb et al., 1992), ovarian granulosa cells (Pucell et al., 1991), and COS-7 cells expressing cloned AT2 cDNA (Kambayashi et al., 1993b; Mukoyama et al., 1993) is that the AT2 receptor does not modulate cytosolic Ca2+ or cyclic AMP, which are sensitive indicators of the heterotrimeric Gqprotein-coupled phospholipase Cβ activation and Gs or Gi-coupled activation or inhibition of adenylyl cyclase, respectively. Furthermore, agonist binding did not induce receptor internalization (Dudley et al., 1991;Csikos et al., 1998), nor did treatment of plasma membrane with stable GTP analogs (e.g., GTPγS or GDP-NH-P) result in a decrease of ligand binding. Heterotrimeric G proteins show a binding of a stable analog of GTP upon stimulation by agonist to their respective receptor. Stimulation of the AT2 receptor in membranes from human myometrium, bovine cerebellar cortex, and rat adrenal glomerulosa did not result in an increase in the binding of [35S]GTPγS (Bottari et al., 1991). These findings, obtained before the molecular structure of the AT2 receptor had been disclosed, led the authors to propose that the AT2 receptor was not coupled to a heterotrimeric G protein since the above are general criteria for the family of G protein-coupled receptors. Thus, even though the AT2 receptor, as we know today, has basic structural features commonly shared by GPCR (seven transmembrane domain receptors), it did not reveal any functional features ascribable to this class of receptors.
An elaborate study on the binding of the AT2labeled with 125I-Sar1-Ang II indicated that the AT2 receptor binds to Giα 2 or Giα 3 (Zhang and Pratt, 1996). However, the mode of interaction may be somewhat different from those seen with other Gi-coupling receptors.
There is evidence that the AT2 receptor inhibits the Gq-coupled phospholipase C activation by the AT1 receptor. Wounded rat skin expresses both AT1 and AT2 receptors (Kimura et al., 1992; Viswanathan and Saavedra, 1992). When rat skin slices are incubated with Ang II (10−7–10−4 M), the production of inositol-monophosphate is stimulated and is abolished by losartan. The AT1 receptor-mediated inositol monophosphate production is markedly potentiated by the AT2 antagonist PD123319 at 10−3 M (Gyurko et al., 1992). Although the results were obtained at high concentrations of Ang II and AT2 antagonist, these observations indicate that AT2 receptor could negatively modulate the AT1 receptor-mediated phospholipase C activation.
The effect of Ang II on cGMP in AT2receptor-expressing cells has been controversial (Dudley et al., 1991;Sumners et al., 1991; Sumners and Myers, 1991; Bottari et al., 1992;Leung et al., 1992; Webb et al., 1992; Brechler et al., 1993;Kambayashi et al., 1993b; Reagan et al., 1993b; Israel et al., 1995). The reason for this discrepancy is not yet clear. More recent in vivo studies on the effect of the AT2 receptor in the kidney indicate that the stimulation of the AT2receptor increases renal medullary and cortical interstitial cGMP by a bradykinin-dependent mechanism (Siragy and Carey, 1996, 1997a,b; Siragy et al., 2000). Similar observations were made by Golke et al. (1998) in the aorta of stroke prone SHR infused with Ang II, and some therapeutic effects of AT2 receptor blockade are clearly mediated by the AT2 receptor and kinin stimulation (Liu et al., 1997). Mice overexpressing the AT2 receptor have confirmed these findings (Tsutsumi et al., 1999). Thus, the signaling mechanisms of the AT2 receptor have been less clearly defined as yet compared with those of the AT1 receptor. In addition to the above-mentioned signaling pathways including Gi protein coupling and cGMP (NO) generation, activation of protein tyrosine phosphatase, serine/threonine phosphatase 2A (PP2A), protein kinase phosphatase (MKP-1) acting on both phosphotyrosine and phosphothreonine as well as SHP-1 tyrosine phosphatase have been reported (for details see Blume et al., 1999;Horiuchi et al., 1999; Millat et al., 1999).
1. Dephosphorylation and Inactivation of the Mitogen-Activated Protein Kinases ERK1 and ERK2.
MAPK plays a key role in cellular proliferation, and its activation can be detected by highly sensitive methods. VSMC transfected with an AT2-expressing vector responded to Ang II by a very slight increase in MAPK activity, whereas untransfected VSMC clearly increases MAPK. The AT2 antagonist markedly increased the MAPK activity (both ERK1 and ERK2 isoenzymes) close to the level attained in untransfected VSMC when stimulated with −10−7 M Ang II. These results indicate that stimulation of the AT2 receptor by Ang II suppresses the AT1 receptor-mediated activation of MAPK (Nakajima et al., 1995). Although adult aortic media do not seem to expresses AT2 at measurable levels, the carotid artery may express it to a limited degree (Viswanathan et al., 1991). AT2 receptor expressed, even in a limited quantity, at the edge of neointima, (or, for that matter, in a healing wound) may suppress MAPK in the growing edge. The serine/threonine phosphatase, PP2A, which is activated by Ang II stimulation of the AT2 receptor in the neonatal hypothalamic neurons, was recently shown to inactivate MAPK following the stimulation of the AT1 receptor. In the presence of the AT2 receptor antagonist, PD123319, MAPK activity was markedly enhanced. Thus, the AT2receptor is able to block MAPK activation either by the dephosphorylation of serine/threonine phosphate by PP2A or tyrosine phosphate by PTPs, most likely by MAPK phosphatase, MKP-1 (Huang et al., 1996).
The influence of the AT2 receptor on MAPK activity may also depend on the biological program engaged by the receptor under defined experimental conditions. Thus in PC12W cells expressing AT2, but not AT1, receptors, treatment with Ang II under conditions allowing for cell differentiation induced a short-lasting increase in ERK1 and ERK2 activity as was seen with NGF, a classical differentiation factor. Costimulation with NGF and Ang II led to initial MAPK stimulation but the subsequent NGF-induced plateau of ERK1 and ERK2 stimulation was suppressed by the AT2receptor (Stroht et al., 2000). Altered ERK activity was also observed in the heart of AT2 transgenic mice suggesting that the ERK inactivated by the AT2 receptor has a physiological role in vivo (Masaki et al., 1998).
Further evidence for a Gi-coupled activation of PTP was obtained in vascular smooth muscle cells in culture, even if the AT2 receptor was not expressed in these cells. Instead of using Ang II to stimulate Gi, it was activated by a 22 amino acid residue peptide with the sequence of the third intracellular loop of the AT2receptor protein, which was transferred into the cells expressing the AT receptor by LipofectAMINE liposome. Cells which received the AT2-related peptide showed small but significant attenuation of serum-stimulated MAPK and thymidine incorporation. The suppression of thymidine incorporation was reversed by pretreatment of cells with pertussis toxin, which indicates the role of a Gi or Go protein. The inhibition of MAPK activation by serum was also reversed by sodium orthovanadate, a tyrosine phosphatase inhibitor but not by okadaic acid, a serine/threonine phosphatase inhibitor, which indicates that the third intracellular loop of AT2 receptor activates a PTP via a Gi or Go protein (Hayashida et al.,1996). In the neuroblastoma cell line, N1E-115, the AT2receptor has been found to activate the catalytic activity of SH-PTP1, a soluble PTP implicated in termination of signaling by cytokine and growth factor receptors, resulting in ERK inactivation (Bedecs et al., 1997).
Recently, ceramide, which is linked to phosphatase activation, was proposed to be the second messenger in AT2receptor-mediated apoptosis, (Gallinat et al., 1999; Lehtonen et al., 1999), and pertussis toxin and orthovanadate blocked its production. A negative cross-talk appears to exist between AT2and AT1 receptors not only on a functional level as has been observed by various authors (see Unger, 1999) but also on the level of intracellular signaling. For instance, stimulation by Ang II, interferon, EGF, or PDGF of rat adult vascular smooth muscle cells transfected with AT2 receptor cDNA inhibits AT1 receptor-mediated ERK activation and tyrosine phosphorylation of STAT without influence on Janus kinase (Horiuchi et al., 1999). However, these results also indicate that the Gi-coupled activation of protein tyrosine phosphatase(s) or protein serine/threonine phosphatase is not targeted narrowly to specific channel proteins and that many other proteins can be their substrates.
2. Activation of Phospholipase A2 and Prostacyclin Generation.
In contrast to cells in which the AT2 receptor is the predominant or exclusive Ang II receptor, cardiac ventricular myocytes are interesting as they express both AT1 and AT2receptors (Busche et al., 2000). In the hypertrophic ventricles of SHR and two-kidney one-clip hypertensive rats, AT1receptor expression is increased along with AT2receptor density (Suzuki et al., 1993). Thus, the heart offers unique materials for studies on interactive regulation of AT1 and AT2 receptors. The AT2 receptor mediates sustained arachidonic acid release in isolated pure cardiac myocytes, and this effect is completely blocked by the AT2-specific antagonist PD123317. Losartan suppresses it by about 50%. On the other hand, the AT1 receptor supports largely the release of inositol phosphates. The fact that 1 mM DTT does not completely abolish arachidonic acid release, but completely eliminates inositol phosphate, supports the role of the AT2 receptor in the activation of phospholipase A2 but not phospholipase C (Lokuta et al., 1994).
Kohout and Rogers (1995) found evidence indicating that the AT2 receptor-mediated release of arachidonic acid may contribute to the activation of Na+/HCO− symporter system (NBC), which increases the pH value of myocytes by 0.08 pH unit. This stimulation of the symporter system by Ang II can be blocked by the AT2 blocker PD123319, but not by the AT1 blocker losartan. Furthermore, superfusion of myocytes with exogenous arachidonic acid (5 μM) mimicked the Ang II-mediated alkalinization. Thus, the arachidonate release and symporter activation is a unique function of the AT2 receptor in the heart. These observations were extended by Sandmann et al. (1998) in an ex vivo study in rats after myocardial infarction. Whereas pretreatment with an ACE inhibitor prevented the postinfarct up-regulation of both ion transporter systems, the Na+/H+-exchanger (NHE-1) and NBC, in cardiac tissue, the AT1 receptor antagonist, valsartan, selectively blocked the increase in NHE-1 and the AT2 antagonist, PD123319, selectively blocked the increase of the NBC.
AT2 receptor-dependent production of prostacyclin (PGI2) was reported in differentiated adipocytes Ob1771 in culture. The PGI2 formation is seen only in differentiated Ob1771 cells, and PGI2 thus formed induces differentiation of undifferentiated Ob1771 cells in coculture by a paracrine mechanism. This action is blocked by PD123177, but not by losartan, indicating that the prostacyclin formation is mediated by the AT2 receptor (Darimont et al., 1994). The mechanism involved in this signaling pathway remains to be clarified.
In summary, diverse AT2 receptor signaling pathways were unveiled, which include activation of serine/threonine phosphatase PP2A and subsequent opening of the delayed rectifier K+-channel, activation of cytosolic PTPs, which may lead to closing of the T-type Ca2+ channel, inactivation, but in some cases also transient activation, of MAPK, the inhibitory effect presumably through PP2A, MKP-1, or other PTP, and the activation of phospholipase A2 (Nouet and Nahmias, 2000). With respect to the specific pathways or substrates involved in AT2 receptor-mediated intracellular signaling, there are still numerous unresolved problems such as GTPγS sensitivity, PTP activation, or cGMP production.
F. Tissue Distribution of the AT2 Receptor
To identify the physiologic functions of the AT2 receptor, investigations were initiated on tissue-specific expression, changes in AT2expression in relation to ontogenic stages and tissue development, and identification of cell types in vivo and in cell lines carrying the AT2 receptor. In these studies, the use of isoform selective antagonists for the AT1 and AT2 receptors enabled investigators to detect biphasic binding of Ang II, which indicated the coexistence of AT1 and AT2 receptors. Later, cloning of AT2 cDNA allowed for specific identification of AT2 mRNA expressed in various tissues (Kakuchi et al., 1995; Shanmugam et al., 1995; Jöhren et al., 1996). Distribution of the AT2 receptor appears to be tissue- and species-specific. Both AT1 and AT2 receptors are expressed in the adrenal gland at varying ratios in different regions. In the rat adrenal medulla, the ratio of AT1 to AT2 receptors was approximately 20:80 (Chang and Lotti, 1990), whereas in rat, rabbit, monkey and human adrenal cortex, the AT2 receptor comprised ∼10 to 40% of the total angiotensin binding sites (Chiu et al., 1989a; Whitebread et al., 1989; Chang and Lotti, 1990). Other tissues that expressed the AT2 receptor at a high proportion, as compared to the AT1 receptor, were the nonpregnant human uterus (Whitebread et al., 1989; Criscione et al., 1990; Bottari et al., 1991; de Gasparo and Levens, 1994); sheep uterine myometrium (Cox et al., 1993); bovine cerebellar cortex (Bottari et al., 1991); and rat ovarian follicular granulosa cells (Pucell et al., 1991). Species dependence exists, as only 40% and 60% of Ang II binding sites are AT2 in rat and rabbit uterus, respectively (Dudley et al., 1990; Bottari et al., 1991). The proportion of the AT2 receptor in the kidney cortex is less than 10% of the AT1 receptor in the rat and rabbit, and ∼55% in monkey (Chang and Lotti, 1991). On the other hand, the heart in the rat, rabbit, and monkey contains the AT2 receptor in small but finite and measurable amounts, which comprises about 30% of the total Ang II binding sites (Chang and Lotti, 1991). The rat and primate pancreas were found to contain AT1 and AT2receptors (Chappell et al., 1992, 1994). Expression of the AT2 receptor was particularly high in pancreatic acinar cells (Chappell et al., 1995). Table4 summarizes adult and fetal tissues that express AT2 receptors.
Tissue distribution of the AT2 receptor and ontogenic change
Some fetal tissues express the AT2 receptor at high levels. As summarized in Table 4, in many of these tissues the AT2 receptor emerges on embryonic days 11 to 13 (E11–E13) and reaches a maximal level on E19. The AT2 receptor then rapidly declines in newborn animals to lower levels or to undetectable levels. As examined by autoradiographic techniques and/or in situ hybridization, expression of the AT2 receptor was particularly dense in differentiated mesenchymes, such as mesenchymal tissues in the tongue, subdermal, and s.c. regions of the skin and the diaphragm. In these tissues, the preponderant fetal Ang II binding sites are AT2 (>97%) (Grady et al., 1991; Feuillan et al., 1993; Grady and Kalinyak, 1993; Shanmugam et al., 1995). The skin and tongue are the tissues in which fetal expression of AT2 receptor is particularly intense, however, rapid postpartum disappearance is seen. In many other tissues where fetal AT2 receptor expression level is detectable, a precipitous lowering of the AT2receptor is observed. The very dense expression of Ang II receptors in rat fetal skin cells noted by Millan et al. (1989) seems to be AT2 receptor (Feuillan et al.,1993). These cells were isolated and cultured. Earlier passage cells express more AT2 than AT1 receptor. Switching of AT2 to AT1expression, or AT2-expressing cells to AT1-expressing cells seems to take place (Johnson and Aguilera, 1991). In some tissues, AT2receptors reach undetectable levels (submucosal cells of the stomach and intestine or trachea). On the other hand, in certain tissues the process of rapid postpartum decrease is arrested, and the AT2 receptor remains at detectable levels in the adrenal medulla, zona glomerulosa, (Shanmugam et al., 1995), pancreas (Chappell et al., 1994), uterus (Cox et al., 1993), and heart (see below).
In the fetal kidney, AT2 receptor expression is seen mostly in the mesenchymal cells of differentiating cortex and medulla, which are surrounding glomeruli in the cortex and tubular tissues in the medulla (Kakuchi et al., 1995). The mesenchymal cells will undergo apoptosis and will be replaced by tubular tissues. The role of the AT2 receptor in fetal development is not yet clearly established. Although the apoptosis of the AT2 receptor-expressing cells in fetal tissue development is an attractive concept, hemizygotic male AT2 receptor gene deleted mice (the AT2 receptor gene is localized to the X-chromosome) and homozygotic female AT2 receptor gene null mice undergo normal fetal development, and newborns and adults do not show any abnormality in the gross morphology of the skin, tongue, kidney, or adrenal. A delay in vasculogenesis was observed, however (Yamada et al., 1998).
Although in many tissues the AT2 receptor undergoes a unidirectional decrease in its expression, this receptor also undergoes reversible changes. The myometria of the human, sheep, and rat express measurable AT2 receptors at a high level (Criscione et al., 1990; Bottari et al., 1991; Cox et al., 1993; de Gasparo et al., 1994). In sheep uterus, for instance, the ratio of AT1:AT2 receptor is 15:85. In the pregnant ewe, the AT2 receptor is reduced by more than 90% and AT1 is reduced by 60% with the AT1 to AT2ratio being reversed to 80:20. However, upon parturition, the AT2 receptor rapidly returns to the high prepregnancy levels (Cox et al., 1993). Similar observation was made in human myometrium during pregnancy (de Gasparo et al., 1994). The AT2 receptor also emerges during wound healing of the skin (Kimura et al., 1992; Viswanathan and Saavedra, 1992). This increase occurs particularly in the superficial dermis. A low level of AT2 receptor expression was also seen in the neointimal tissue of rat carotid artery following balloon catheterization (Janiak et al., 1992; Viswanathan et al., 1994b). These changes in the uterus during pregnancy or in wound healing in the skin and brain indicate that the AT2 receptor has a definitive but not yet identified regulatory function in these tissues.
1. Brain.
Although circumventricular organs of the brain respond to circulating Ang II, an endogenous brain renin-angiotensin system can generate Ang II in tissues protected from circulating Ang II by the blood-brain barrier. Ang II generated by this system reacts with angiotensin receptors within the brain. The brain angiotensin receptors have been studied and reviewed extensively by Gehlert et al. (1991a,b),Rowe et al. (1992), Saavedra (1992), Song et al. (1992), andHöhle et al. (1995). An expression of the AT2 receptor in fetal (E18) brain tissues was reported in several areas: inferior olive, paratrigeminal nucleus, and hypoglossal nucleus. Saavedra (1992) determined AT1 and AT2 receptor expression in various regions of the brain, and its age dependence in 2- and 8-week-old rat brains. Although AT1receptor expression did not show marked age dependence, the AT2 receptor showed a significant decrease from 2 to 8 weeks of age (see Table 5).
Distribution of the AT2 receptor in the brain—effect of age
Obermüller et al. (1991) investigated the distribution of Ang II receptor types in adult rat brain nuclei using competitive radioligand binding. Whereas midbrain and brain stem contained AT1 and AT2 receptors in comparable concentrations, AT1 receptors were by far predominant in several hypothalamic nuclei, although a limited amount of AT2 receptors was detectable in most of them. Although minor differences may exist among these studies, they share the following observations: AT2 receptor expression is consistently high in the cerebellar nuclei, inferior olive, and locus ceruleus in the brain stem, which is rich in noradrenergic neurons. In contrast to the AT1receptor, which participates in various central cardiovascular functions and is expressed in the hypothalamus and in brain stem nuclei (nucleus of the solitary tract, dorsal motor nucleus of vagus at a low level), the AT2 receptor is much less present in distinct hypothalamic and brain stem nuclei associated with the regulation of cardiovascular functions. The presence of the AT2 receptor in the dorsal motor nucleus of vagus is not completely settled.
Although the distribution of AT2 receptors in the brain is well known, their effects are still not clear. As outlined above, brain AT2 receptors may play a role in cognitive functions and certain types of behavior such as exploration or drinking (Hein et al., 1995a). On the other hand, they may also antagonize the central effects of angiotensin peptides in osmoregulation mediated via AT1 receptors (Höhle et al., 1995, 1996). A possibly important role of AT2 receptors in neuroregeneration and neuroprotection will be dealt with below.
2. Heart.
Earlier studies suggested that myocytes of rabbit and rat hearts contain AT1 and AT2 receptors in comparable quantities as shown in Table 6 (Rogg et al., 1990; Scott et al., 1992; Sechi et al., 1992b). However, questions remained as to the cell-specific localization of these receptors in situ. A recent study using single cell RT-PCR has demonstrated that in the adult rat about 50% of cardiomyocytes contain the AT1receptor, whereas the AT2 receptor is much more scarce with only about 10% carrying this receptor type (Busche et al., 2000).
Coexistence of AT1 and AT2 in cardiac tissues
Autoradiographic studies of the rat heart show about a 2-fold increase in AT1 and AT2 receptors after birth, and their receptor numbers are about equal from E16 through 10 to 16 weeks of age (Sechi et al., 1992b). They are expressed in the myocardium of all four chambers, as well as in the vascular smooth muscles of the aorta and pulmonary arteries. However, very little ligand binding was seen in the coronary artery. The conduction system (the atrioventricular and sino-aortic nodes) was reported to contain both AT1 and AT2 receptors by Sechi et al. (1992b), whereas Saavedra et al. (1993 ), Brink et al. (1996), andWharton et al. (1998), showed only AT1 receptors in these tissues.
Primary culture of cardiomyocytes from neonatal rat left ventricles showed about a 50% decrease in the AT2 receptor from those of E19 fetuses, whereas the AT1receptor did not decrease (Matsubara et al., 1994). In contrast, the AT2 receptor in cultured fibroblasts collected from 1-day-old rat heart was less than 10% of the AT2 receptor in fibroblasts cultured from E19 fetal heart, whereas there was no change in AT1receptor number. In cardiac fibroblasts cultured from 7-day-old rats, AT2 receptor density was negligible and the majority of the Ang II receptor population was reported to be AT1, whereas in cultured cardiomyocytes AT2 receptor numbers are maintained at a finite and measurable level (about 50% of AT1). Bovine and human ventricular and atrial myocardium also contain both AT2 and AT1 receptors (Rogg et al., 1991; Nozawa et al., 1994; Regitz-Zagrosek et al., 1995). Although the AT2 receptor is usually expressed at low density in adult, it is up-regulated to different extents in pathological circumstances such as cardiac hypertrophy, myocardial infarction, cardiomyopathy, and congestive heart failure (Matsubara, 1998; Unger, 1999). Interestingly, both in nonfailing and explanted end-stage human heart the AT2 receptor population measured in a binding assay (65% of total Ang II receptor) is greater than AT1, and there may be a correlation between the density of the AT2 receptors and the severity of heart failure (Rogg et al., 1996). Wharton et al. (1998) also observed a significant increased density of high-affinity binding sites in endocardial, interstitial, perivascular, and infarcted regions of the ventricle of patients with end-stage ischemic heart disease or dilated cardiomyopathy, greater than in adjacent noninfarcted myocardium. The border zone between noninfarcted and infarcted myocardium was rich in microvessels with perivascular AT2 receptors. Ohkubo et al. (1997) reported that in the heart of cardiomyopathic hamster (Bio 146), both AT1 and AT2 receptors were increased during heart failure (153% and 72%, respectively). In human, the expression of the AT2 receptor was markedly (3-fold) increased in patients with dilated cardiomyopathy at both protein and mRNA levels compared with patients with acute or well organized old myocardial infarction (Tsutsumi et al., 1998). In contrast, the AT1 receptor expression was significantly down-regulated. The AT2 receptor sites were highly localized in the interstitial region in the fibrotic areas where fibroblasts are present. Collagen and fibronectin formation from fibroblasts were suppressed by the AT1antagonist TCV116, and increased by the inhibition of AT2 receptor with PD123319 in the cardiomyopathic hamster. These results are intriguing as they suggest the possibility that the AT1 and AT2receptors modulate extracellular matrix formation in an opposite way as has already been suggested from experiments using cardiac microvascular enothelial cells (Fischer et al., 1996).
Although much remains to be learned about the various functions of cardiac AT2 receptors, their distribution and time- and event-driven regulation suggest roles in cardiac development, repair and remodeling with actions opposing those mediated by cardiac AT1 receptors.
3. Kidney.
The kidney is a major target organ of Ang II. Ang II exerts its regulatory function on both hemodynamic and tubular functions, and most of its actions seem to be explained by AT1 receptor functions. The AT1 receptors are indeed abundantly expressed in small cortical arteries, glomeruli, proximal tubules, and interstitium. The AT2 receptor is, however, also expressed in the kidney. Ontogenic studies on rat and humans by autoradiography, in the presence of losartan (Sechi et al., 1992a; Ciuffo et al., 1993b), on rat (Shanmugan et al., 1995), and on mouse (Kakuchi et al., 1995) showed marked development-dependent changes in AT2 receptor expression in the kidney.
In the mouse (Kakuchi et al., 1995), on embryonic days 12 to 16 (E12–E16) AT2 mRNA was densely expressed in the mesenchymal cells of the mesonephros surrounding the mesonephric tubules. In this developmental period, the mesonephros begins regression, and a possible function of the AT2receptor in the apoptotic loss of mesonephros can be envisioned. On E14, the AT2 receptor emerges in the interstitial mesenchymes of the kidney, but not in the glomeruli or the S-body, whereas the AT1 receptor is richly expressed in the preglomeruli and S-bodies. On E16, AT2 mRNA is seen in the renal capsule and inner medulla where it is prominent along the papillary duct and between the collecting ducts. AT2 receptor on the medullary ray extend into the cortex, whereas the detection signals are reduced to undetectable levels after birth (Kakuchi et al., 1995; Shanmugam et al., 1995).
About 10 to 15% of the renal Ang II binding sites were blocked by CGP42112 (de Gasparo et al., 1990; Zhuo et al., 1992, 1993,1998). The outermost layer of the cortex outside of the glomeruli and inner layer of medulla seem to show signaling for the AT2 receptor, the proximal convoluted tubule is a candidate site for AT2 receptor localization, which is functional in the renal tubular system, as its existence has been reported by Dulin et al. (1994). The same group also reported the presence of the AT2 receptor in cultured rat mesangial cells by binding studies (Ernsberger et al., 1992). However, quantitative autoradiographic studies of rat renal glomeruli by Ciuffo et al. (1993b) did not detect the AT2receptor in rat renal glomeruli. Reviewing many of these studies, it appears that the competitive ligand binding autoradiography or in situ hybridization does not provide sufficient sensitivity for detecting a low-level expression of AT2, comprising only 10 to 20% of total binding. Along this line, earlier studies by a similar technique by Gibson et al. (1991) did not detect AT2 receptor in the rat renal cortex. By contrast, in rhesus monkey kidneys, AT2 receptors (PD12198 sensitive sites) were seen in the juxtaglomerular apparatus and in arterial smooth muscle cells.
Ozono et al. (1997) examined the expression of the AT2 receptor in the fetal, newborn, and adult kidney by immunohistochemistry using antibodies raised against the peptide epitope with the amino acid sequence of the N terminus of the rat AT2 receptor. Positive immunohistochemical staining of the AT2 receptor was observed in the mesenchymal cells and ureteral bands of the 14-day-old fetal kidney and in the glomeruli, tubules, and vessels in the 19-day-old fetal and newborn kidney. Glomeruli expressing the AT2receptor were localized mainly in the outer layers of the renal cortex. In the young and adult rat, AT2 receptors were present in glomeruli at a substantially diminished level. Lowering dietary sodium intake increased glomerular and interstitial AT2 receptors. In human kidney, the AT2 receptor is clearly present in large preglomerular vessels of the renal cortex (Grone et al., 1992) and in the tubular interstitium (Chansel et al., 1992; Goldfarb et al., 1994). Possible renal functions of the AT2 receptor have recently been reviewed (Carey et al., 2000).
4. Vasculature.
Although it is well known that the AT1 receptor, present in vascular smooth muscle, mediates the contractile and hypertrophic effects of Ang II, the presence of the AT2 receptor in the vasculature in vivo was seen only by autoradiography. Its exact site of localization and functional roles are yet to be clarified. In coronary endothelial cells derived from SHR, AT2 receptor mRNA as well as AT2 binding were present with a ratio AT1/AT2 of about 80%/20% (Stoll et al., 1995). In ontogenic studies on AT2 receptor expression in fetal (E18), 2-week-, and 8-week-old rat aorta autoradiographic determination of specific binding of 125I-Sar1-Ang II in the presence of losartan and PD123177 was performed (Viswanathan et al., 1991). Interestingly, the AT2 receptor was the major or almost exclusive Ang II receptor in the fetal aorta (E18), accounting for 85% of the total binding. Even at 2 weeks of age the AT2 was still the dominant form but it became a minor Ang II receptor at 8 weeks of age (25%). However, the total Ang II receptor sites decrease after birth and throughout the process of maturation from a very high level (300 fmol/mg of protein) in the fetus down to less than 10 fmol/mg of protein in 8 weeks. Autoradiography showed the AT2 receptor in the medial layer even at 8 weeks of age. Similar results were obtained by competitive binding of the vascular membrane fraction (Chang and Lotti, 1991). This small but finite population of AT2 receptors in the media of the vasculature may explain a rapid pressor and subsequent depressor effect of Ang III infused into rats pretreated with losartan. The AT2 blocker almost completely eliminated the depressor phase (Scheuer and Perrone, 1993). The presence of AT2 receptors even at a low level may be compatible with the increased sensitivity to the pressor action of Ang II at a low concentration (10-12-10-10M) under AT2 blockade. Thus, it has been speculated that the AT2 blockade unmasks the sensitivity of the vascular AT1 receptor to a low concentration of Ang II (Hong et al., 1994).
Formation of neointima following balloon catheterization of the rat carotid artery can be suppressed by angiotensin converting enzyme inhibitors or AT1 antagonists (see above). However, there was a report that an AT2 agonist was efficient in preventing neointima formation, but not an AT1 antagonist (Janiak et al., 1992). Autoradiographic studies of Ang II receptor expression indicated a marked increase in AT1 and a decrease in AT2 receptors in the balloon catheter-treated aorta, and equally elevated AT1 but no expression of AT2 receptors in the normal and injured carotid artery (Viswanathan et al., 1994b). However, the neointimal area of the vessels transfected with and expressing the AT2 receptor transgene was decreased by 70% compared to untransfected or control-vector transfected vessels. This inhibitory effect on the development of the neointima was blocked by an AT2 receptor antagonist, PD123319 (Nakajima et al., 1995). The in vivo localization of the AT2receptor in the aorta of rats may provide a basis for a possible interpretation of the interesting report that chronic blockade of AT2 rather than AT1receptor antagonizes Ang II-induced aortic hypertrophy and fibrosis (Levy et al., 1996). In a model of chronic Ang II infusion, Cao et al. (1999) reported on an increased mesenteric weight and wall-lumen ratio of the vessels as well as proliferation of smooth muscle cells. These vascular changes were attenuated by both AT1 and AT2 receptor antagonists. Moreover, AT1 receptor blockade was associated with down-regulation of both receptors, whereas AT1receptor blockade was associated with reduced Ang II binding to only the AT2 receptor. Although these observations suggest that vasotrophic effects of Ang II could, at least partially and under certain experimental conditions, be mediated by the AT2 receptor type, this topic is still controversially discussed. The marked AT2-mediated increase of aortic cGMP under Ang II infusion in SHR as observed by Gohlke et al. (1998) is just one example speaking against AT2 as a vasotrophic factor.
Despite the fact that the media of rat thoracic aorta express the AT2 receptor, smooth muscle cells derived from the aorta and cultured in vitro do not (Kambayashi et al., 1996;Ichiki et al., 1996). This is possibly due to down-regulation of the AT2 receptor gene by growth factors in serum-containing culture medium since AT2receptor expression can be regained upon prolonged serum depletion in the presence of insulin (Kambayashi et al., 1996).
Coronary endothelial cells cultured from adult SHR contain AT1 and AT2 receptors in a ratio of about 80/20% as determined by the demonstration of AT2 mRNA, AT2 binding and functional studies (Stoll et al., 1995). The same authors showed that in these cells, serum- or basic fibroblast growth factor-induced proliferation is dose-dependently inhibited by Ang II (Metsärinne et al., 1992; Stoll et al., 1995) and that this antiproliferative effect is sensitive to AT2 receptor blockade. Furthermore, that under nonstimulated conditions, the mitogenic activity of Ang II is suppressed by the AT1antagonist losartan but is markedly enhanced by the AT2 antagonist PD123177 (Stoll et al., 1995). This is presumably because the mitogenic action of AT1 receptor is partially suppressed by the AT2 receptor. The antiproliferative effect of the AT2 receptor has subsequently been confirmed in several laboratories and also extended to nonvascular cell systems (Meffert et al., 1996; Munzenmaier et al., 1999; Goto et al., 1997; Van Kesteren et al., 1997; Maric et al., 1998).
5. Pancreas, Lung, Thymus, and Other Tissues.
Canine and primate pancreas were found to contain the AT2receptor at a relatively high level (Chappell et al., 1992, 1994). This receptor was characterized by high binding affinity (K d ∼ 0.48 nM) for Ang II, a 2-fold enhancement of binding by DTT and inhibition of radioactive Ang II binding by AT2-specific antagonists. Of the total Ang II binding sites approximately 70% seems to be AT2. Autoradiographic studies showed a predominance of AT2 over AT1 receptors throughout the pancreas including islet cells, acinar and duct cells, as well as vasculature cells (Chappell et al., 1991, 1992). Subsequently, the rat pancreatic acinar cell line AR42J was shown to contain a high level of AT2 receptors (250 fmol/mg of protein) with concomitant AT1 receptors at ∼10 to 15% of the level of the AT2 receptor (Chappell et al., 1995). The pancreas also contains a large amount of angiotensinogen and its mRNA studied (Chappell et al., 1991).
The distribution of the AT1 and AT2 receptor in human lung was studied using immunohistochemistry with specific polyclonal antibodies and with in situ hybridization. The AT1 receptor mRNA and protein were localized on vascular smooth muscle cells, macrophages, and, in particular, in the stroma underlying the airway epithelium. In contrast, the AT2 receptor mRNA and protein was observed in the epithelium, with strong staining on the bronchial epithelial cell brush border and also on many of the underlying mucous glands. The AT2 receptor was also present on some endothelial cells (Bullock et al., 1999). The thymus was shown to contain sites able to bind Sar1-Ang II at a high affinity. This binding is inhibited by an AT2antagonist (Correa et al., 1994).
There are tissues or cells that are devoid of AT2receptor when using either 125I-Ang II in the presence of AT1 selective antagonists or the AT2-specific binding agent125I-CGP 42112. These tissues are the liver (Whitebread et al., 1989; Dudley et al., 1990) and the pituitary (Leung et al., 1991).
6. Cells in Primary Culture and Cell Lines Expressing the AT2 Receptor.
To clarify the signaling mechanism and pathophysiological significance of the AT2receptor, numerous types of cells derived from tissues known to contain the AT2 receptor were prepared or identified. They provided useful materials for studies on AT2receptor signaling, pathophysiologic roles, and regulation for cloning and purification.
Among primary cultured cells were neuronal cells dispersed from rat neonatal hypothalamus (Sumners and Myers, 1991), neonatal rat cardiomyocytes (Lokuta et al., 1994; Suzuki et al., 1993), adult rat aortic endothelial cells (Stoll et al., 1995), rat fetal skin cells (Tsutsumi et al., 1991), and rat fetal fibroblasts (Johnson and Aguilera, 1991).
Rat adrenomedullary pheochromocytoma PC12 cells express both AT1 and AT2 receptors reflecting their adrenal medullar origin where both AT1 and AT2 receptors are coexpressed. A subline, PC12W, which expresses a large amount of AT2 but not AT1 receptors (Speth and Kim, 1990), were extensively used for studies of AT2 signaling mechanisms and for AT2 cloning. A subline of the Swiss mouse 3T3 fibroblast cell line, R3T3, also expresses AT2without AT1 receptor (Dudley et al., 1991; Csikos et al., 1998). Although in both of these cell lines expression of the AT2 receptor is suppressed in the cellular growth phase, AT2 mRNA increases as they approach the subconfluent or confluent phase. When R3T3 cells are shifted to a quiescent condition by serum deprivation, the AT2receptor protein production increases without proportional increase in mRNA (Dudley and Summerfeldt, 1993).
An undifferentiated mouse neuroblastoma cell line, NG108-15, was shown to express the AT2 but not the AT1 receptor (Buisson et al., 1992) and was used in studies of the signaling mechanism: AT2receptor activation by Ang II resulted in phosphotyrosine phosphatase activation and closing of calcium T-channel (Buisson et al., 1995).
Another mouse neuroblastoma cell, N1E-115, expresses both AT1 and AT2 receptors (Reagan et al., 1990). Since AT1 receptors can be rapidly destroyed by dithiothreitol, AT2receptors could be preserved in fractions solubilized by the detergent CHAPS and were used as a source for purification of the AT2 receptor. The transformed rat pancreatic acinar cell line, AR42J, contains both AT1 and AT2 receptors and shows a marked rise in Ca2+ characteristic of the AT1 receptor response (Chappell et al., 1995). Thus, this cell line may not be very convenient for the study of the role of AT2 receptor. However, AR42J cells have an exocrine function that may or may not be regulated by AT2. Ovarian granulosa cells isolated to a high degree of purity and maintained in primary culture were also very useful to study AT2 receptor-dependent differentiation and apoptosis (Pucell et al., 1991; Tanaka et al., 1995). The cloned mouse preadipocyte cell line, Ob1771, expresses the AT2 receptor upon differentiation in a serum-free medium and responds to Ang II by producing prostacyclin, which promotes the differentiation of the preadipocytes by a paracrine mechanism (Darimont et al., 1994). Rat VSMC in culture do not express the AT2 receptor under regular culture conditions in the presence of fetal calf serum (Stoll et al., 1995; Ichiki et al., 1996; Kambayashi et al., 1996), but, as outlined above, the AT2 receptor can emerge in rat thoracic VSMC when cultured in serum-depleted medium supplemented with insulin or other insulin-like growth factors such as IGF (Ichiki et al., 1996;Kambayashi et al., 1996). Thus rat VSMC grown and maintained under ordinary culture conditions do not seem to provide convenient materials for the study of the role of the AT2 receptor. However, VSMC can be transfected with an artificial AT2 receptor gene consisting of an AT2 receptor coding region with a myosin heavy chain promoter (Nakajima et al., 1995).
G. Pathophysiological Aspects of AT2 Receptor Activation
The widespread distribution of AT2 receptors in various brain nuclei, heart, vascular tissues, adrenal, kidney, skin, and during wound healing suggests a physiological role for the AT2 receptor. The identification of several AT2 receptor signaling pathways as reviewed in preceding sections implies diverse pathophysiologic consequences. Studies to identify the role of the AT2 receptor in pathophysiology have been conducted using diverse approaches, which include the use of cultured cells, in vivo vascular tissues injured by balloon catheter, transgenic expression of AT2receptor, targeted AT2 receptor gene deletion (gene knockout), and chronic administration of AT2 antagonists and agonists (Horiuchi et al.,1998; Matsubara, 1998; Unger, 1999; Carey et al., 2000).
1. The AT2 Receptor Can Induce Apoptosis.
The effect of the AT2 receptor does not stop at the inhibition of cellular proliferation but is associated with cellular programs such as differentiation and regeneration (see below) and, under appropriate conditions, also with apoptosis.
In PC12W cells, expressing only AT2 but not AT1 receptors, nuclear condensation, fragmentation, and marginalization were observed in serum-free medium containing NGF (1 ng/ml) after treatment with 10−7 M Ang II for 2 days (Yamada et al., 1996;Gallinat et al., 1999). Characteristic internucleosomal DNA fragmentation was also observed. In R3T3 cells, serum depletion seemed to be the major contributor to apoptotic DNA fragmentation although activation of AT2 receptor has an additional effect.
Skin fibroblasts collected from mice embryos with genetic deletion of the AT2 receptor did not show any DNA fragmentation characteristic of apoptosis after stimulation with Ang II, whereas fibroblasts from wild mice did (Li et al., 1998b). Ovarian granulosa cells undergo apoptosis during follicular atresia. These cells contain the AT2 but not the AT1 receptor (Pucell et al., 1991). Tanaka et al. (1995) showed that these cells in culture underwent apoptotic DNA fragmentation when depleted of follicle-stimulating hormone. Although no direct effect of added Ang II on the apoptosis was observed, the AT2 receptor level in these cells was increased by the removal of follicle-stimulating hormone and the addition of Ang II, indirectly suggesting a possible role of the AT2 receptor in apoptosis and even perhaps in ovulation.
“Programmed cell death” is an important concept in developmental morphogenesis. The fetal kidney expresses the AT1receptor in the preglomerular S-bodies, whereas the AT2 receptor is expressed in fetal renal mesenchymal cells, which are replaced by tubular tissues in the later stages of renal development. Mesonephros also express the AT2 receptor in the mesenchyme surrounding its tubular system. Mesonephros are known to disappear by apoptosis (Kakuchi et al., 1995). However, the nephrogenesis occurs even in AT2 receptor null mice. Thus, triggering of the AT2 receptor-mediated activation of MKP-1 and apoptosis may be contingent on additional growth suppressive measures such as serum depletion.
The mechanism behind AT2-mediated apoptosis appear complex as both AT1 and AT2 receptors are involved in the process (de Gasparo and Siragy, 1999). The calcium flow between endoplasmic reticulum and mitochondria and the ratio Bcl2/Bax, which is modulated by both AT1 and AT2 receptors play a key role in homeostasis between cell growth and cell death (Kajstura et al., 1997; Berridge et al., 1998; Fortuno et al., 1998; Tea et al., 1998). Horiuchi et al. (1997b) reported that AT2 receptor stimulation in PC12W cells dephosphorylates Bcl2 by activation of MKP-1. In PC12W cells, AT2stimulation markedly lowered MAPK. This effect was suppressed by pretreatment with vanadate and pertussis toxin, indicating that a G protein-driven PTP decreased MAPK activity. The PTP was shown to be MKP-1, since the elimination of MKP-1 by an antisense oligonucleotide transfection abolished DNA fragmentation (Yamada et al., 1996). Activation of caspase may be involved in AT2receptor-induced apoptosis in umbilical venous endothelial cells (Dimmeler et al., 1997). A stimulation of ceramide, which replaces the JNK-stress-activated protein kinase pathway leads to caspase stimulation (Hayashida et al., 1996).
Gallinat et al. (1999) observed that in PC12W cells, kept under conditions allowing for apoptosis, the AT2receptor-induced apoptosis was associated with an Ang II concentration-dependent ceramide production. Because sphingomyelin concentrations were unaltered by Ang II these findings suggested a de novo synthesis of ceramide by AT2 receptor stimulation. This report was subsequently confirmed by Lehtonen et al. (1999).
Stretch also activates apoptosis following increased expression of the transcriptional factor p53 and up-regulation of the local renin-angiotensin system, whereas Bcl2 expression is decreased. p53 indeed binds to the promoter of the angiotensinogen and the AT1 receptor genes and stimulates production of Ang II and expression of the AT1 receptor (Pierzchalski et al., 1997; Leri et al., 1998).
Together, these reports clearly indicate that the AT2 receptor is able to induce apoptosis, although the signaling pathways await further investigation. It should be noted, however, that the proapoptotic features of the AT2 receptor can only be unveiled under specific experimental conditions, for instance serum deprivation/NGF dependence, which generally prepare the grounds for apoptosis.
2. Effects on Vascular Tone.
The presence of AT2 receptors in the vasculature has been discussed above. Whereas the vascular antiproliferative and neointima-reducing effects of AT2 receptor stimulation are now well established (Horiuchi et al., 1998; Unger, 1999), the participation of the AT2 receptor in the regulation of vascular tone is still controversial. When a bolus injection of Ang III was given i.v. into anesthetized rats, both pressor and depressor effects were observed. Under AT1 receptor blockade, Ang III exerted a dose-dependent depressor effect, which was abolished when both AT1 and AT2 receptors were blocked (Scheuer and Perrone, 1993). In rabbit abdominal aorta, the AT2 blocker PD123177 unmasked losartan-sensitive pressor effects of Ang II at a low-concentration range of 10−12 to 10−10 M where the peptide alone was not pressoric. These in vivo results suggested that a pressor action of the AT1 receptor was masked by the AT2 receptor (Hong et al., 1994). AT2 receptor gene null mice prepared by Ichiki et al. (1995) show an elevated systolic and diastolic pressure, indicating that the AT2 receptor has a depressor effect. In a renal wrap Grollman hypertension model, AT1receptor blockade normalized blood pressure, whereas AT2 receptor antagonism significantly increased blood pressure but decreased renal interstitial concentration of bradykinin and nitric oxide (Siragy and Carey, 1999). These results suggest that the AT2 receptor mediates counterregulatory vasodilation. Similarly, AT2receptor activation with CGP42112 facilitates in SHR the depressor response caused by an AT1 receptor antagonist (Barber et al., 1999). On the other hand, in adult conscious SHR a concomitant infusion of Ang II and the AT2antagonist, PD123319, did not result in blood pressure elevation, whereas the AT1 antagonist, losartan, consistently lowered blood pressure in the presence or absence of Ang II (Gohlke et al., 1998).
Thus, although the results of several studies suggest that the AT2 receptor exerts depressor effects upon stimulation by Ang II or III, there is also evidence against the idea of AT2 as a vasodilator. It appears that, under normal conditions, the pressor effect of the AT1receptor exerts a dominant effect, and overall effect of Ang II is directed to blood pressure elevation. A defect of the AT2 receptor, either genetic or otherwise, could lead to an elevation in blood pressure or increased sensitivity to Ang II or III. On the other hand, blockade of AT1receptor in juxtaglomerular cells by selective AT1 receptor antagonists increases circulating Ang II, which will then interact with the unopposed AT2 receptor. Stimulation of AT2 receptors could thus contribute to a further decrease of blood pressure. However, the exact mechanism of AT2 receptor-mediated depressor effect is still not clear.
3. Vascular Hypertrophy and Fibrosis and the AT2Receptor.
Arterial hypertrophy, remodeling, and fibrotic changes induced by chronic administration of Ang II have been believed to be due to diverse effects mediated by the AT1receptor. However, when normotensive rats received a chronic (3 weeks) s.c. infusion of Ang II (120 ng/kg/min), Ang II + losartan (10 mg/kg/day) or Ang + PD123319, it was the AT1receptor blocked (with losartan) rat that showed a marked increase in collagen and elastin in the thoracic aorta, despite normal blood pressure maintained by losartan. By contrast, collagen and elastin in the Ang II-PD123319-treated rats were at low control levels despite markedly elevated blood pressure. Media thickness was also increased in the AT2-blocked rats rather than AT1-blocked animals (Levy et al., 1996; Cao et al., 1999). A persuasive explanation for these intriguing observations, which were not confirmed by Li et al. (1998a) and are in disagreement with a host of reports on the antigrowth effects of the AT2 receptor in many tissues including the vasculature (Horiuchi et al., 1999; Unger, 1999) is not yet available.
4. Renal Tubular Function.
The presence of an AT2-like (termed AT1B) receptor in renal tubules particularly in the proximal tubules have been reported by Douglas and associates (Douglas, 1987; Ernsberger et al., 1992; Dulin et al., 1994). However, AT2 mRNA is not readily detected in adult rat kidney. On the other hand, the AT1 receptor is clearly expressed in the glomeruli and in the inner stripe of the outer medulla. Glomerular AT1 receptors may control the hemodynamic function of the kidney.
The AT2 receptor seems to have some detectable effect on renal tubular function. The AT2selective antagonist, PD123319, infused i.v. at 300 μg/kg/min into anesthetized dogs increased free water clearance 4-fold and sodium excretion 3-fold, whereas the AT1 selective losartan had only insignificant effects on the tubular functions (Keiser et al., 1992). In these experiments, renal blood flow was decreased only by 10%. On the other hand, salt replete dogs infused with PD123177 into the renal artery did not show appreciable changes in free water clearance and natriuresis (Clark et al., 1993). When the hemodynamic function and tubular function are dissociated by maintaining the renal blood flow constant using inflatable aortic cuffs above and below the renal artery of an unilaterally nephrectomized rat (Roman et al., 1984), infusion of the AT2blocker, PD123319, rapidly induces an increase in diuresis and natriuresis. As the extent of diuresis was perfusion pressure-dependent, it is considered to represent “pressure natriuresis”. CGP42112, an AT2 agonist, suppresses the diuretic response. The glomerular filtration rate remained remarkably constant (Lo et al., 1995). These results can be interpreted to mean that AT2 receptor stimulation by Ang II induces Na+ retention when kidney perfusion pressure is increased. In contrast, in conscious rats with a chronic microdialysis cannula in the renal medullary interstitium, AT1 receptor blockade showed a marked diuretic effect, whereas AT2 receptor blockade by PD123319 did not exert any actions on water clearance or natriuresis. When animals were placed on a low-salt diet, cGMP and PGE2 in the microdialysate from the kidney interstitial fluid were generally increased 2- to 3-fold over a 5-day period. Upon infusion of losartan, PGE2 was reduced to the basal level of rats on regular diet, whereas cGMP was not affected. PD123319 markedly increased (∼4-fold) PGE2 and reduced cGMP to basal levels. Losartan plus PD123319 reduced PGE2 to basal levels overriding the effect of PD123319, whereas cGMP was essentially regulated by PD123319. These results indicate that the AT2 blocker, PD123319, rapidly induces an increase in diuresis and production of cGMP, which is regulated primarily by the AT2 receptor, whereas the AT1 receptor has no effect. On the other hand, PGE2 production was abolished by losartan and was markedly increased by PD123319 (Siragy and Carey, 1996, 1997a,b). Similar microdialysis has indicated that tissue bradykinin, nitric oxide, and PGF2α formation were released upon stimulation of the AT2 receptor (Siragy et al., 2000).
In AT2 receptor knockout mice, a subpressor dose of Ang II inhibits natriuresis and diuresis suggesting that the AT2 receptor stimulation physiologically increase pressure natriuresis and that this effect is sustained over a prolonged period (Siragy et al., 1999). An explanation for the discrepancy with the data of Lo et al. (1995) is not obvious as yet.
5. Neuronal Cell Differentiation and Nerve Regeneration.
Cultured Schwann cells express both AT1 and AT2 receptors, and Ang II decreases the expression of the neurite-promoting protease nexin-1. Blockade of the AT1 receptor or stimulation of the AT2 receptor leads to a severalfold increase of nexin-1 favoring nerve regeneration (Bleuel et al., 1995). Similarly, treatment of nondifferentiated NG108-15 or PC12W cells with Ang II or nerve growth factor induces growth arrest and morphological differentiation of neuronal cells including neurite outgrowth and up-regulation of polymerized tubulin as well as microtubule-associated protein MAP2c expression and differential regulation of neurofilament M (Laflamme et al., 1996; Meffert et al., 1996; Gallinat et al., 1997; Stroth et al., 1998). These effects were mimicked by the AT2 receptor agonist, CGP42112A, and abolished by coincubation with the antagonist, PD123319. The underlying signaling mechanisms may include inhibition of p21ras as well as activation of MAPK (Gendron et al., 1999; Stroth et al., 2000). Repression of c-fos and c-jun expression does not seem to be involved in AT2receptor-mediated growth arrest and cell differentiation in PC12W cells (Steckelings et al., 1998). Moreover, there is a marked up-regulation of AT2 receptor mRNA in regenerating neurons in response to nerve crush coinciding, as an AT2expression wave from proximal to distal, with the regeneration of nerve fibers (Gallinat et al., 1998). Both in vitro and in vivo, Ang II promotes axonal elongation of postnatal retinal explants and dorsal root ganglia neurons and, in adult rats, axonal regeneration of retinal ganglion cells after optic nerve crush. This effect is completely abolished by an AT2 receptor antagonist but not affected by an AT1 receptor antagonist (Lucius et al., 1998). Thus, the AT2 receptor plays a role in neuronal cell differentiation and nerve regeneration via regulation of the cytoskeleton, whereas the AT1receptor may inhibit this process (Unger, 1999).
H. Summary
AT2 receptor expression is widespread. However, in several tissues it disappears after birth or when cells are transferred in culture in the presence of serum and growth factor. On the other hand, a sometimes dramatic up-regulation of the AT2 receptor can occur after tissue injury. Research on the AT2 receptor has revealed actions differing greatly from those of the AT1 receptor such as antiproliferative effects in several tissues, cellular differentiation, nerve regeneration, and apoptosis. It appears that the AT2 receptor often plays the role of a modulator of biological programs in tissue development or repair.
The signaling mechanisms of the AT2 receptor are diverse, and only a few of them have as yet been characterized reasonably well. In some cases they are coupled to Gi proteins. One pathway in neurons (and perhaps other tissues) involves activation of protein serine/threonine phosphatase PP2A, which leads to the activation of the delayed rectifier K+-channel (outward K+ current in neuronal cells). This will result in hyperpolarization of plasma membranes that suppress cellular activities stimulated by depolarization. A second signaling pathway involves the activation of protein phosphotyrosine phosphatases (PTPases), which are of pivotal importance to prevent or rapidly shut off undesired and uncontrolled growth of normal tissues.
The AT2 receptor lacks the mechanism of rapid desensitization, internalization, and low-affinity shift by GTP. Molecular mechanisms underlying such a unique signaling mechanism have not been clarified and warrant further intensive investigation.
Arachidonic acid release from cardiac myocytes leading to the activation of Na+/HCO3symporter may be a unique regulatory mechanism of cardiac myocytes. Release of nitric oxide with subsequent formation of cGMP appears to be another important intracellular effect of AT2stimulation occurring in vascular and renal tissues. In many adult tissues, the modulatory or suppressive actions of the AT2 receptor, often counteracting the AT1 receptor-mediated stimulatory actions, require measure of reduction in activities, which is technically more difficult than measurement of increased activity from a very low level. This may have been the reason for which the AT2receptor has sometimes escaped the attention of investigators. Chronic application of AT2 antagonists and AT2 receptor gene deletion are beginning to reveal the role of AT2 receptor in the maintenance of, or restoring normality not only in the cardiovascular system but also in other tissues e.g., the central nervous system. A possible role of AT2 receptor in apoptosis of mesenchymal cells in conjunction with other factors is beginning to be clarified as an important basis of organogenesis, failure of which may lead to some childhood diseases.
IV. The AT4 Receptor
In 1990, during the process of purifying and sequencing the AT1 receptor type, it was noticed that heat-denatured purified receptor from the bovine adrenal gland lost binding with125I-Sar1,Ile8Ang II, whereas 125I-Ang III binding persisted. At first it was suspected that an angiotensin receptor specific to Ang III was isolated. However, with sufficient peptidase inhibitors added to prevent the conversion of Ang III to shorter fragments, this binding activity was lost. This was puzzling given that the two known receptor types at that time, AT1 and AT2, each accepted Ang II and Ang III as ligands, albeit with different affinities. A fragment of Ang III was suspected to be acting at this new site because Sar1,Ang II, Sar1,Ile8Ang II (Sarile), Sar1,Ala8Ang II (Saralasin), DuP753 (losartan), PD123177, CGP42112A, Ang II(1–7) and Ang III did not serve as ligands (Harding et al., 1992; Swanson et al., 1992). In fact, 125I-Ang II(3–8)[Ang IV] did bind at this site reversibly, saturably, and with high affinity (K d = 1 nM) (Harding et al., 1992;Swanson et al., 1992; Sardinia et al., 1993). Having identified a ligand that bound at this site it was possible to determine its brain distribution using in vitro autoradiographic techniques. The brain distribution of this receptor site was unlike that of either the AT1 or AT2 receptors. The greatest concentrations of what has now been termed the AT4 site (IUPHAR Nomenclature Committee, de Gasparo et al., 1995) were in structures classically associated with cognitive processes and sensory and motor functions, not in structures associated with the functions of body water balance, cardiovascular regulation, and control of reproductive hormones and behaviors where the AT1 receptor is predominant. The following sections summarize what is presently known about this new Ang IV/AT4 system with emphasis on signaling mechanisms, tissue distributions, the development of analogs that act as agonists and antagonists, and the physiologies and behaviors associated with this AT4 site.
A. Signaling Mechanisms
Given that small peptides are able to activate the AT4 receptor and that the vast majority of small peptide receptors are G protein-linked, it would seem logical to predict that the AT4 receptor might be a serpentine, G protein-linked receptor as well. This, however, does not appear to be the case since the AT4 receptor exhibits a molecular weight between 160 and 190 kDa as determined by reduced SDS-polyacrylamide gel electrophoresis. Of particular interest is the observation that the adrenal AT4 receptor may be multimeric. This suggestion comes as a result of nonreducing gels that indicate a second specifically labeled band at 225 kDa. A similar molecular weight has been observed for other bovine tissues including heart, thymus, kidney, bladder, aorta, and hippocampus (Zhang et al., 1999). Bernier et al. (1995) have reported a similar molecular weight for the binding subunit of the AT4receptor in bovine aortic endothelial cells. Together these data all but preclude the linkage of AT4 receptors to G proteins. The lack of linkage to G proteins is further supported by the observation that GTPγS fails to alter 125I-Ang IV binding in rabbit heart (Hanesworth et al., 1993), guinea pig brain (Miller-Wing et al., 1993), and rat vascular smooth muscle (Hall et al., 1993). However, a single report by Dulin et al. (1995) indicates that GTPγS can inhibit binding in opossum kidney cells. As experienced with the AT2 receptor, a definite conclusion has to wait the cloning of the AT4receptor.
The initial events that characterize AT4 receptor intracellular signaling mechanisms are presently unknown. Nevertheless, downstream targets appear to include immediate early genes. Intracerebroventricular infusion of Ang IV in rats induces c-Fos expression in brain regions associated with cognition (Roberts et al., 1995). Angiotensin IV agonists can also stimulate c-Fos, c-Jun, and egr-1 in isolated, nonstimulated rabbit hearts (Slinker, personal communication). Studies carried out in the laboratory of Vaughan (Kerins et al., 1995) indicate that AT4activation increases the expression of plasminogen activator inhibitor, PAI-1, and is blocked by coapplication of an AT4receptor antagonist. However, a possible involvement of the AT1 receptor has been postulated both in vitro and in vivo (Brown et al., 1999; Chabielska et al., 1999; Goodfield et al., 1999; Sironi et al., 1999).
A final area of investigation indirectly associated with AT4 signaling is the identification of endogenous ligands for the AT4 receptor. The first putative ligand identified for the AT4 receptor was the hexapeptide, Ang IV. Although Ang IV is known to be present in the circulation (Semple et al., 1976) and is generated from Ang II or Ang III (Abhold and Harding, 1988), recent studies (Møeller et al., 1997) indicate that other peptides like LVV-hemorphin-7 are also capable of binding and activating AT4 receptors. This is not surprising given that the structural requirements for AT4 ligands are fairly minimal. Presumably, other putative ligands will be found in the future.
B. Tissue Distribution of the AT4 Receptor
1. Brain.
To date, brain distributions of the AT4 binding site using in vitro autoradiography have been completed in rat (Roberts et al., 1995), guinea pig (Miller-Wing et al., 1993), macaca fascicularis (Møeller et al., 1996a), rhesus monkey (Wright et al., 1995), and human (hippocampus only; Harding, unpublished observations), and there is cross-species consistency. The predominant brain distribution of AT4 receptor is presented in Table 6 by comparison with the AT1 and AT2 receptors. The highest densities of the AT4 site are located in regions involved in cognitive processing, and motor and sensory functions. Specifically, the AT4 receptor site is prominent in structures associated with the cholinergic system (Møeller et al., 1996a). This system is composed of two major pathways: one from the basal forebrain with cell bodies located in the nucleus basalis magnocellularis, which project primarily to the neocortex, whereas the other originates in the medial septum-diagonal band of Broca complex and projects primarily to the hippocampus (Wenk et al., 1980; Mesulam et al., 1983; Dutar et al., 1995). There are also additional projections to the amygdala and thalamus presumed to be involved in the integration of subcortical contributions to this system (Goldman and Cote, 1991), and the piriform cortex.
Equally impressive binding has been reported in structures associated with motor function including the ventral horn of the spinal cord (i.e., spinal motor nuclei) (Møeller et al., 1995, 1996a; Wright and Harding, 1995) inferior olivary nucleus, motor trigeminal nucleus, vestibular and reticular nuclei of the hindbrain, red nucleus, oculomotor nucleus, substantia nigra, and ventral tegmentum of the midbrain. In the forebrain, considerable binding is present in the globus pallidus, caudate-putamen, and nucleus accumbens (Miller-Wing et al., 1993; Møeller et al., 1995, 1996a). There are also high densities of AT4 receptors in the granular cell layer of the cerebellum and deep cerebellar nuclei (Miller-Wing et al., 1993), as well as Betz cells of the primary motor neocortex (Møeller et al., 1996a). AT4 receptors have also been identified in brain autonomic nuclei such as the dorsal motor nucleus of the vagus, nucleus ambiguus, rostral ventral lateral medulla, and paraventricular nucleus of the hypothalamus (Møeller et al., 1995,1996a).
Finally, less dense distributions of AT4 sites have been identified in sensory associated structures including spinal trigeminal nuclei, the colliculi, gracile and cuneate nuclei, and lateral geniculate nuclei, thalamic nuclei (anterior, lateral, and ventral), lateral olfactory tract, and primary sensory neocortex (Miller-Wing et al., 1993; Møeller et al., 1995, 1996a).
A comparison of the adult brain structures most densely distributed with AT1, AT2, and AT4 receptors reveal some overlap (Table7). Most notably in the dorsal motor nucleus of the vagus, inferior olivary nucleus, cerebellum, superior colliculus, lateral geniculate nucleus, and paraventricular nucleus (Höhle et al., 1995; Wright and Harding, 1995, 1997; Møeller et al., 1996a). However, there are structures rather uniquely distributed with AT4 receptors. These include: reticular formation (motor areas), motor trigeminal and vestibular nuclei, cuneate and gracile nuclei, ventral tegmental area, periaqueductal gray, caudate-putamen, medial habenula, nucleus basalis of Meynert, hippocampus, piriform cortex, Betz cells in neocortex, and granular layer of the cerebellum.
Predominant brain distributions of the three angiotensin receptor types identified in the mammalian brain7-a
2. Peripheral Tissue.
Table 8provides the binding constants for AT4 receptors located in several peripheral tissues. Bovine adrenal cortex revealed a mean (± S.D.) K d value of 0.7 (± 0.14) nM and a B max value of 3.82 (± 1.12) pmol/mg of protein for 125I-Ang IV (Harding et al., 1994). The binding of 125I-Ang IV at this AT4 site could not be inhibited with Ang II, Sar1,Ile8Ang II, Ang II(1–7), DuP753, CGP42112A, PD123177. The metabolically resistant form of Ang III, [d-Arg1],Ang III had a significantly lower apparent binding affinity for this site.
Binding constants for AT4 receptors in various species and tissues
Monkey kidney displayed a K d value of 1.5 (± 0.31) nM and a B max value of approximately 1.0 (± 0.21) pmol/mg of protein (Harding et al., 1994). Autoradiographic analyses of rat kidney indicated heavy concentrations of AT4 receptors in the outer stripe of the medulla and a few receptors in glomeruli and the core of the medulla (Harding et al., 1994; Coleman et al., 1998). The reverse pattern was observed for125I-Sar1,Ile8Ang II binding to the AT1 and AT2 receptor sites.
Rat, guinea pig, and rabbit hearts revealed heavy concentrations of AT4 receptors withK d values of 3.3(± 1.1), 1.33(± 0.02), and 1.75(± 0.5) nM, respectively. CorrespondingB max values were: 0.32(± 0.03), 0.14(± 0.02), and 0.73(± 0.16) pmol/mg of protein (Wright et al., 1995). Autoradiographic analyses indicated heavy [125I]Ang IV binding throughout the heart muscle. Guinea pig and bovine vascular smooth muscle evidenced considerable binding with K d values of 0.40(± 0.09) and 1.85(± 0.45) nM, respectively. TheB max values were: 1.04(± 0.24) and 0.96(± 0.1) pmol/mg of protein, respectively. Binding constants for other tissues of interest, such as guinea pig colon and spleen, human bladder and prostate, are also included in Table 8.
C. Development of Agonists and Antagonists
1. Binding Requirements of AT4 Receptor.
The characteristics of the Ang IV molecule that result in high-affinity binding at the AT4 type are much different than those described above for the AT1 receptor. Specifically, they include the following. 1) Removal of the N-terminal valine significantly reduces binding affinity as compared with Ang IV (K d ≈ 2.6 · 10−9 M) (reviewed in Wright et al., 1995). 2) Glycine substitution in positions 1, 2, or 3 of the Ang IV molecule or the use of d-isomers at these positions, greatly reduces affinity (Sardinia et al., 1993). 3) On the other hand, positions 4, 5, and 6 can accommodate several amino acid constituents without significantly impacting affinity. A detailed study of position 1 of Ang IV revealed the following specific characteristics. 1) High affinity binding necessitates the presence of a primary amine in position 1. Methylation, cyclization, or removal of the N-terminal amine yields a dramatic loss of affinity. 2) Hydrophobic residues in position 1 produce the greatest binding affinity. Thus, norleucine1-Ang IV with a 4-carbon straight chain aliphatic group in position 1 results in a very high affinity (K d ≈ 3,zmd.10−12 M). The addition of the amino group in position 1 significantly lowered affinity as compared with analogous straight chain analogs. 3) The amino acids substituted in position 1 must be in the l-configuration. For example, replacing l-norleucine withd-norleucine in position 1 significantly reduces affinity (K d ≈ 6 · 10−7 M). Reasonably high affinity can be achieved with a CH2-NH isostere substituted for the 1–2 bond. These isosteres permit increased rotation around the peptide bond (Sardinia et al., 1994). They also introduce a formal positive charge with a bond length similar to the normal peptide bond accompanied by increased metabolic stability. Position 2 of the Ang IV molecule requires an aromatic amino acid in thel-configuration to achieve high-affinity binding. Position 3 requires a hydrophobic amino acid also in thel-configuration. Thus, the requirements for significant occupancy and activation of the AT4receptor type include a cluster group of Val-Tyr-Ile-R1-R2-R3. Removal of phenylalanine from the C terminus of Ang IV yields a binding affinity, which is nearly equivalent with that of native angiotensin IV, but is physiologically inactive as measured by its influence on blood flow. With further successive removal of amino acids, binding affinity declines such that the K dvalue for Ang IV(1–4) ≈ 5.7 · 10−8 M, and for Ang IV(1–3) ≈ 4.8 · 10−7 M (reviewed in Wright et al., 1995). In sum, the minimal ligand requirements for an Ang IV-like peptide to bind with reasonably high affinity at the AT4 receptor type appears to be Val-Tyr-Ile-R1-R2, although an agonist appears to require the addition of phenylalanine at the C terminus yielding a required structure of Val-Tyr-Ile-R1-R2-Phe.
2. Antagonists of the AT4 Receptor.
Antagonists of the AT4 receptor type have been reasonably difficult to design and synthesize. Harding's laboratory has had success with the substitution of Val for Ile in the third position of Ang IV accompanied by reduced peptide bonds between Val-Tyr and Tyr-Val (i.e., Val-Tyr-Val-His-Pro-Phe). This compound called divalinal-Ang IV (Krebs et al., 1996) binds at the AT4 receptor type with high affinity to a similar number of binding sites as125I-Ang IV. It does not bind at the AT1 and AT2 receptor types and has been shown to prevent i.c.v. Ang IV-induced c-Fos expression in rats (Roberts et al., 1995), Ang IV-induced elevation in cerebral (Kramár et al., 1997) and kidney cortical blood flow (Coleman et al., 1998), and Ang IV-induced inhibition of proximal tubule Na+ transport (Handa et al., 1998).
D. Physiology Associated with the AT4 Receptor
1. Regulation of Blood Flow.
Haberl et al. (1991) have reported dilation of rabbit pial arterioles with topical application of Ang IV using the closed cranial window technique (Haberl, 1994). Ang IV-induced dilation was dependent upon pretreatment with exogenousl-arginine. Application of l-arginine alone resulted in only minimal vasodilation (+4% at 10−5 M), whereas coapplication ofl-arginine and Ang IV produced a 20+% vasodilation. Pretreatment with methylene blue blocks vasodilation suggesting that it is dependent upon an endothelial-dependent factor, probably nitric oxide (Riedel et al., 1995). A degradation product of Ang II could therefore induce this endothelium-dependent dilation (Haberl et al., 1991).
Laser Doppler flowmetry was used to measure Ang IV-induced increase in blood flow within the cerebral cortex (Kramár et al., 1997) and kidney cortex (Swanson et al., 1992; Harding et al., 1994; Coleman et al., 1998). Angiotensin IV was intra-arterially administered via the internal carotid or renal arteries, respectively. Angiotensin II induced decreases in blood flow in these preparations. Pretreatment with DuP753 blocked subsequent Ang II-induced reductions in cerebral or renal cortical blood flow, whereas both PD123177 and divalinal-Ang IV failed to inhibit these responses. In contrast, pretreatment with the antagonist divalinal-Ang IV blocked the Ang IV-induced elevation in cerebral or renal cortical blood flow, whereas pretreatment with DuP753 or PD123177 failed to influence this response. In a related study,Kramár et al. (1998) have noted that pretreatment with the NO synthase inhibitor,N ω-nitro-l-arginine (l-NAME) blocked the vasodilatory effect of Ang IV suggesting that this Ang IV-induced elevation in cerebral blood flow is dependent upon the synthesis and release of NO from vascular endothelial cells. Similar results have been obtained byColeman et al. (1998) concerning kidney cortical blood flow.
Finally, Näveri et al. (1994a,b) have noted that i.v. administration of Ang IV reversed the decreases in cerebral blood flow that accompanies experimentally induced subarachnoid hemorrhage. Pretreatment with the nonspecific AT1and AT2 receptor antagonist, Sar1-Ile8 Ang II, failed to influence this Ang IV-induced effect suggesting that the elevation in blood flow was mediated by the AT4 receptor type. Taken together these results argue in favor of important roles for angiotensins II, III, and IV in the control of blood flow. In contrast with the above findings, Gardiner et al. (1993) have noted Ang IV-induced reductions in renal and mesenteric blood flows and vascular conductances in conscious rats using implanted pulsed Doppler flow probes. Pretreatment with DuP753 blocked these responses suggesting mediation by the AT1 receptor type. The authors concluded that Ang IV acts as a weak agonist at the AT1 receptor type. In agreement with these findings, Fitzgerald et al. (1999) infused Ang IV via the left renal artery and noted dose-dependent reductions in renal artery blood flow via transit-time flow probes placed around the artery. Once again these responses could be blocked by pretreatment with DuP753. Infusion of LVV-hemorphin-7 failed to alter blood flow. Presently it is difficult to resolve these conflicting results. Those reports noting Ang IV-mediated vasoconstriction determined that it was mediated by the AT1 receptor type. Those reports noting Ang IV-induced vasodilation attributed it to the AT4receptor type. These Ang IV responses have been further separated into direct and indirect effects. Specifically, pretreatment withl-NAME prevented subsequent Ang IV-induced elevations in cerebral (Kramár et al., 1998) and renal cortical blood flow (Coleman et al., 1998). Nossaman et al. (1995) have shown thatl-NAME and the cyclooxygenase inhibitor, meclofenamate, shift the Ang IV-induced vasoconstrictor dose-response curve in the pulmonary circulation. These researchers concluded that Ang IV could be promoting the release of vasodilators such as prostaglandins and nitric oxide. Consistent with these results, Yoshida et al. (1996)found the vasoconstriction induced by Ang IV to be initially facilitated by l-NAME, and by l-NAME and indomethacin (another cyclooxygenase inhibitor). In contrast, Li et al. (1997) have reported that indomethacin failed to impact the Ang IV dose-response curve in isolated human saphenous veins. Recently, Patel et al. (1998) and Hill-Kapturczak et al. (1999)determined that blockade of AT1 and AT2 receptor types failed to influence Ang II-induced NO release, or the release of NO synthase (NOS). Ang IV stimulated significantly greater NO release and greater endothelial-type constitutive NOS activity than the equivalent dose of Ang II. Divalinal-Ang IV blocked these Ang II and Ang IV-mediated NO effects and NOS activation. The authors concluded that the Ang IV/AT4 system is primarily responsible for the Ang II facilitation of NO release and endothelium-dependent vasorelaxation. This endothelial cell NOS activation appears to be regulated by intracellular Ca2+ release and increased expression of calreticulin (Patel et al., 1999). Resolution of these issues awaits further investigation.
2. Cardiac Hypertrophy.
Autoradiographic analyses utilizing125I-Ang IV reveals considerable binding in guinea pig and rabbit heart membranes that is distinct from AT1 or AT2 receptor types (Hanesworth et al., 1993). Angiotensin IV binding has also been shown using cultured vascular smooth muscle cells (Hall et al., 1993), coronary microvascular endothelial cells (Jarvis et al., 1992; Hall et al., 1995), and cultured rabbit cardiac fibroblasts (Wang et al., 1995b). Baker et al. (1990, 1992) have reported that Ang IV blocks increases in RNA and protein synthesis initiated by Ang II in chick cardiocytes. Rats, predisposed to left ventricular hypertrophy via abdominal aortic coarctation, revealed significant accumulations of collagen that is facilitated with the AT4receptor antagonist, divalinal-Ang IV, and significantly reduced when treated with the AT4 receptor agonist norleucine1-Ang IV. These results point to a potentially important role for the Ang IV/AT4system in collagen accumulation in hypertrophied hearts.
3. Renal Tubular Reabsorption.
Autoradiograms utilizing125I-Ang IV demonstrate heavy distributions of AT4 sites in the outer stripe of the outer medulla with diffuse labeling of the cortex (Coleman et al., 1998). This binding of Ang IV was not influenced by the specific AT1 receptor antagonist losartan, or the AT2 receptor antagonist PD123177. However, the AT4 receptor antagonist, divalinal-Ang IV, or Ang IV, completely displaced this binding. Emulsion autoradiography determined that these AT4 receptors are localized on cell bodies and apical membranes of convoluted and straight proximal tubules (Handa et al., 1998). Activation of these receptors by Ang IV produced a concentration-dependent decline in sodium transport as measured by rate of tissue oxygen consumption (Handa et al., 1998). Thus, these results provide strong evidence that Ang IV acts on tubular epithelium to inhibit sodium reabsorption. This natriuretic effect of Ang IV has been confirmed using in vivo infusion via the renal artery in anesthetized rats. Urine sodium concentration was found to significantly increase, whereas urine volume and glomerular filtration rate were not affected. This Ang IV-induced natriuresis was unaffected by pretreatment with DuP753 but was blocked with divalinal-Ang IV. These results support an important role for Ang IV in the control of sodium transport by the kidney (Ardaillou and Chansel, 1996).
4. Electrophysiological Analysis.
Albrecht and colleagues (1997a) have investigated the ability of microiontophoretic administration of Ang IV to influence neurons in the hippocampus and in the dorsal lateral geniculate nucleus (Albrecht et al., 1997b). Of 43 hippocampal neurons tested, Ang IV produced a greater than 40% increase in firing frequency in 16 of these cells (37%) with a decrease in three neurons. Of the 72 hippocampal neurons tested with Ang II, 21 evidenced a greater than 40% elevation in firing rate (29%), with a decrease in eight cells. With both Ang II and Ang IV the excitation effects were seen primarily in neurons that evidenced slower spontaneous discharge rates, whereas decreases in firing rate were usually seen in cells with higher spontaneous discharge rates. These changes in firing rates induced by Ang II could be blocked with the specific AT1 receptor antagonist DuP753 (eight of ten neurons examined), however the specific AT4receptor antagonist divalinal-Ang IV was ineffective. On the other hand divalinal-Ang IV was effective at blocking the Ang IV-induced increases in discharge rate in six of nine neurons tested, while DuP753 was ineffective. These results suggest that Ang II and Ang IV are acting at different receptors, i.e., AT1 and AT4, respectively. Along these lines, the authors saw no evidence of cross-desensitization between Ang II and Ang IV. The authors suggest that the presumed colocalization of AT1 and AT4 receptor types on the same hippocampal neurons supports the notion that both Ang II and Ang IV are capable of influencing hippocampal neurons.
Similar findings were obtained with angiotensin-sensitive geniculate neurons (Albrecht et al.,1997b). These investigators further reported that Ang II application produced a potent inhibition ofN-methyl-d-aspartate- and kainate-induced facilitation of firing rates in some neurons, whereas increased discharge frequencies were observed in others. Ang IV was also found to block such glutamate receptor excitation in three neurons, whereas this excitation was facilitated in two neurons. Finally, these investigators found the iontophoretic application of Ang II onto these geniculate neurons suppressed light-evoked excitation (nine neurons) in some neurons, although other units also revealed facilitation (eight neurons). A similar evaluation of Ang IV indicated a facilitation of this light-evoked excitation in ten neurons and a suppression in seven neurons. These angiotensin effects could be blocked by the appropriate AT1 or AT4 receptor antagonists. Furthermore, the suppression effect of Ang II and Ang IV on light-evoked activity could itself be blocked by γ-aminobutyric acid receptor antagonists. Albrecht et al. (1976) suggested that Ang II and Ang IV appear to act as neuromodulators in the dorsal lateral geniculate nucleus. Determination of the precise role of each ligand will require further investigation.
5. Role of Ang IV in Learning and Memory.
Intracerebroventricular injection of Ang IV leads to c-Fos expression in the hippocampus and piriform cortex, whereas similar injection of Ang II failed to induce c-Fos-like immunoreactivity in these structures but did activate c-Fos expression in circumventricular organs (Roberts et al., 1995) as well as paraventricular nucleus, SON, and the medial preoptic nucleus (Zhu and Herbert, 1996, 1997). Pretreatment with losartan prevented this Ang II-induced c-Fos-like immunoreactivity, whereas pretreatment with divalinal-Ang IV blocked the Ang IV-induced c-Fos expression (Roberts et al., 1995). There were no crossover effects demonstrated by these antagonists. Along these lines, Braszko and colleagues (1988, 1991) established that i.c.v. injected Ang II and Ang IV (1 nmol) were equivalent at facilitating exploratory behavior in rats tested in an open field, and furthermore, improved recall of passive avoidance conditioning and acquisition of active avoidance conditioning. Intracerebroventricular treatment with divalinal-Ang IV (10 nmol), disrupted recall of this response (Wright et al., 1995). Metabolically resistant analogs of Ang IV injected i.c.v. can be used to facilitate acquisition of successful search patterns in a circular water maze task in scopolamine-treated rats (Pederson et al., 1998). Taken together, these results suggest an important role for the Ang IV/AT4 system in learning and memory processes. Recently Møeller et al. (1997) have isolated the globin fragment LVV-hemorphin-7, a decapeptide (Leu-Val-Val-Tyr-Pro-Trp-Thr-Gln-Arg-Phe) that binds at the brain AT4 receptor type, which could be the endogenous ligand that acts at this receptor.
Finally, Delorenzi and colleagues (1997) have reported Ang IV facilitation of long-term memory of an escape response in the crabChasmagnathus. Angiotensin II was also shown to facilitate long-term memory in this species, although not as robustly; however, this Ang II effect could not be blocked by losartan or PD123177. These results suggest that the Ang IV/AT4 system may have been involved in invertebrate memory processes long before the emergence of mammals.
E. Summary
Recently a new angiotensin receptor type, AT4, has been discovered and characterized that preferentially binds Ang II(3–8), a fragment of angiotensin II, now referred to as Ang IV. This receptor site is prominent among brain structures concerned with cognitive processing, motor and sensory functions. Specifically, high densities of AT4sites have been localized in neocortex, piriform cortex, hippocampus, amygdala, and nucleus basalis of Meynert. Major motor structures with high levels of AT4 receptors include basal ganglia, red nucleus, substantia nigra, ventral tegmentum, vestibular and reticular nuclei of the hindbrain, motor trigeminal nucleus, Betz cells of primary motor neocortex, cerebellum, and ventral horn of the spinal cord. Significant sensory structures include thalamus, the colliculi, gracile and cuneate nuclei, lateral geniculate, lateral olfactory tract, and primary sensory neocortex. Peripheral tissues that reveal heavy distributions of AT4 sites are kidney, bladder, heart, spleen, prostate, adrenals, and colon.
The primary functions thus far associated with this Ang IV/AT4 system include memory acquisition and recall, the regulation of blood flow, inhibition of renal tubular sodium reabsorption, and cardiac hypertrophy. There are preliminary indications that this system may also be involved in neurite outgrowth (Møeller et al., 1996b), angiogenesis, and stimulation of endothelial cell expression of PAI-1 (Kerins et al., 1995). The identification of additional functions awaits further elucidation of this new receptor system. Cloning of the AT4 receptor will certainly substantiate its potential role in pathophysiology of the renin-angiotensin system.
V. General Conclusions
Most of the known effects of Ang II are mediated through the AT1 receptor, e.g., vasoconstriction, aldosterone and vasopressin release, salt and water retention, and sympathetic activation without neglecting the important autocrine and paracrine effects of Ang II on cell proliferation and migration and on extracellular matrix formation. The function of the AT2 receptor has become unraveled over the last few years owing to various sophisticated approaches including gene transfection and deletion. Accumulated published data suggests that the AT2 receptor counterbalances the effect of the AT1 receptor in vitro as well as in vivo. There is an inactivation of MAPK, antiproliferation, promotion of apoptosis, differentiation and regeneration, opening of delayed-rectifier K+ channels and closing of T-type Ca2+ channels. The re-expression of the AT2 receptor in various diseases suggests a role of this receptor in pathophysiology.
The AT4 receptor appears to be involved in memory acquisition and recall. Like the AT2 receptor, it may also oppose the effect of the AT1 receptor as it regulates renal blood flow, inhibits tubular sodium reabsorption and affects cardiac hypertrophy.
Cloning of the described angiotensin receptors and the ability to express these clones in mammalian cells will allow exhaustive structure/function studies. Further pharmacological and molecular studies will allow for a better and more complete understanding of the role of the renin-angiotensin system in pathology.
Footnotes
-
↵1 Address for correspondence: Marc de Gasparo, Novartis Pharma AG, Metabolic & Cardiovascular Diseases, WKL 121-210, P.O. Box 4200, Basel, Switzerland. E-mail:marc.de_gasparo{at}pharma.Novartis.com and m.de_gasparo{at}bluewin.ch(after October 1, 2000)
-
↵3 Members of the NC-IUPHAR Subcommittee on Angiotensin Receptors: R. Wayne Alexander, Kenneth E. Bernstein, Andrew T. Chiu, Theodore Goodfriend, Joseph W. Harding, Ahsan Hussain, Pieter B. M. W. M. Timmermans.
Abbreviations
- ACE
- angiotensin converting enzyme
- NC-IUPHAR
- International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification
- GPCR
- G protein-coupled receptor
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- PKC
- protein kinase C
- kb
- kilobase(s)
- bp
- base pair(s)
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- IGF-1
- insulin-like growth factor 1
- NO
- nitric oxide
- NOS
- NO synthase
- VSMC
- vascular smooth muscle cell(s)
- MAPK
- mitogen-activated protein kinase
- EPO
- erythropoietin
- CHO
- Chinese hamster ovary
- TMD
- transmembrane domain
- PKB
- protein kinase B
- EGF
- epidermal growth factor
- GAP
- GTPase-activating protein
- PDGF
- platelet-derived growth factor
- JNK
- c-Jun N-terminal kinase
- PAK
- p21-activated kinase
- PLC
- phospholipase
- SFO
- subfornical organ
- OVLT
- organum vasculosum lamina terminales
- ACTH
- adrenocorticotropin
- APA
- aminopeptidase A
- APN
- aminopeptidase N
- GnRH
- gonadotropin-releasing hormone
- DTT
- dithiothreitol
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- MKP-1
- MAPK-phosphatase-1
- PTP
- phosphotyrosine phosphatase
- PGE2
- prostaglandin E2
- IL
- interleukin
- IRS
- insulin response sequence
- IRF
- interferon regulatory factor
- NGF
- nerve growth factor
- NBC
- Na+/HCO− symporter system
- NHE
- Na+/H+-exchanger
- PAI
- plasminogen activator inhibitor
- Ang
- angiotensin
- l-NAME
- Nω-nitro-l-arginine
- AP-1
- activator protein-1
- ERK
- extracellular signal-regulated kinase
- TMD
- transmembrane domain
- JAK
- Janus cytosolic protein kinase
- STAT
- signal transducers and activators of transcription
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵




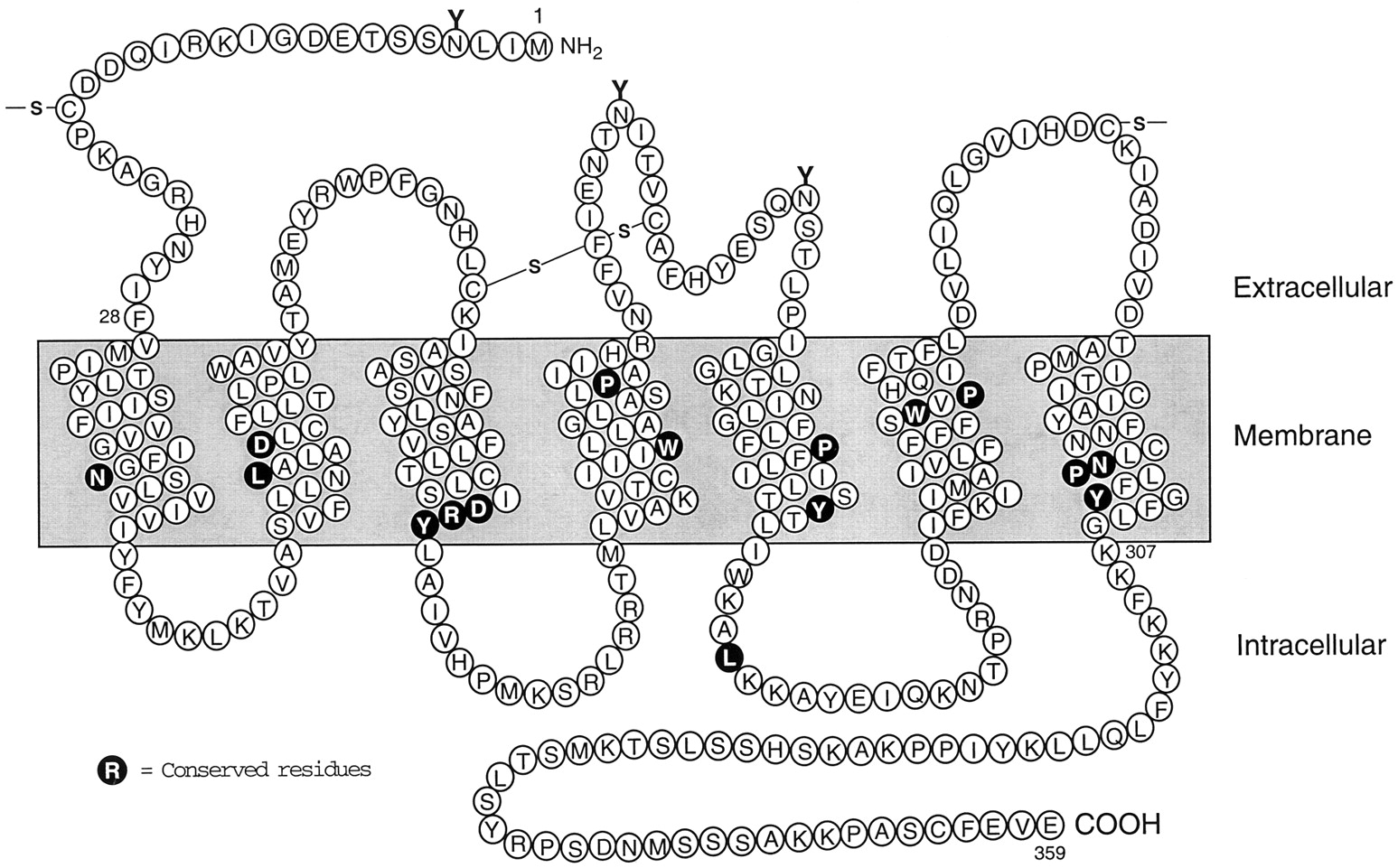{kind=link}
{kind=link}
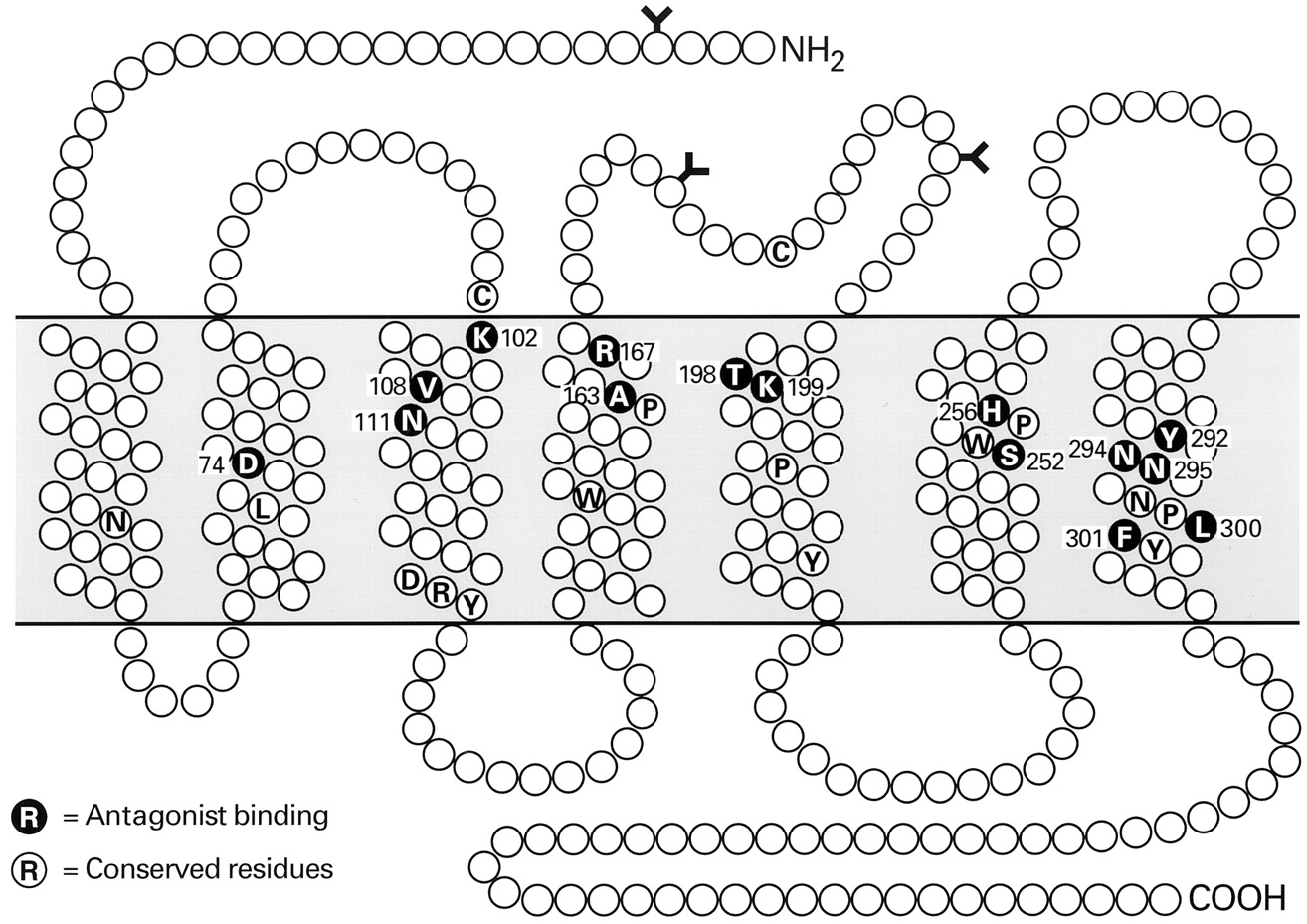{kind=link}
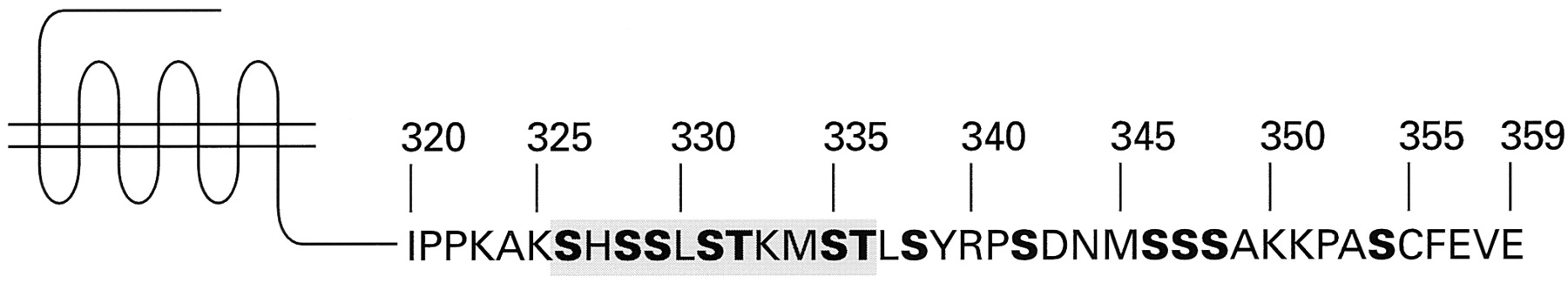{kind=link}
{kind=link}