


Visual Overview

Abstract
Adolescence is a developmental period when physical and cognitive abilities are optimized, when social skills are consolidated, and when sexuality, adolescent behaviors, and frontal cortical functions mature to adult levels. Adolescents also have unique responses to alcohol compared with adults, being less sensitive to ethanol sedative–motor responses that most likely contribute to binge drinking and blackouts. Population studies find that an early age of drinking onset correlates with increased lifetime risks for the development of alcohol dependence, violence, and injuries. Brain synapses, myelination, and neural circuits mature in adolescence to adult levels in parallel with increased reflection on the consequence of actions and reduced impulsivity and thrill seeking. Alcohol binge drinking could alter human development, but variations in genetics, peer groups, family structure, early life experiences, and the emergence of psychopathology in humans confound studies. As adolescence is common to mammalian species, preclinical models of binge drinking provide insight into the direct impact of alcohol on adolescent development. This review relates human findings to basic science studies, particularly the preclinical studies of the Neurobiology of Adolescent Drinking in Adulthood (NADIA) Consortium. These studies focus on persistent adult changes in neurobiology and behavior following adolescent intermittent ethanol (AIE), a model of underage drinking. NADIA studies and others find that AIE results in the following: increases in adult alcohol drinking, disinhibition, and social anxiety; altered adult synapses, cognition, and sleep; reduced adult neurogenesis, cholinergic, and serotonergic neurons; and increased neuroimmune gene expression and epigenetic modifiers of gene expression. Many of these effects are specific to adolescents and not found in parallel adult studies. AIE can cause a persistence of adolescent-like synaptic physiology, behavior, and sensitivity to alcohol into adulthood. Together, these findings support the hypothesis that adolescent binge drinking leads to long-lasting changes in the adult brain that increase risks of adult psychopathology, particularly for alcohol dependence.
I. Introduction
Adolescence is a period of developmental transition, encompassing physical, mental, emotional, and social aspects. The development of both physical and interpersonal skills required to successfully integrate into society is essential for living in groups, and these skills improve through adolescence to adult levels. In addition, adolescence is a time when talents, reasoning, and other abilities are formed. Adolescence in humans and other social animals is characterized by high expression of risk taking, exploration, novelty and sensation seeking, social interaction, and play behavior that contributes to this transition. Recent discoveries using human brain imaging provide strong evidence that these characteristics are linked to maturation of brain structure (Lenroot and Giedd, 2006; Bava and Tapert, 2010). Although much of development involves programmed sequences of change in gene expression related to cellular differentiation and protein expression, experience and environment during adolescence also contribute to lifelong adult abilities and characteristics. Nutrition, alcohol exposure, and multiple other environmental factors are known to impact both prenatal and postnatal physical development.
Adolescent development of abilities, social skills, and other complex processes is difficult to define and quantitate. However, in general, training and acquisition of skills in adolescence are important for developing both highly-skilled human and animal individuals. Training during adolescence improves abilities involving cognition, like playing chess or training to be a guide dog, as well as physical abilities. Training at all ages improves performance, but the improvement is often much faster and greater during adolescence. During adolescence, physical abilities improve in parallel to the development of self-control, consideration of future consequences, planning, and socialization skills, and eventually reductions in risk taking and sensation seeking. Frontal cortical synaptic refinement and increased myelination in adolescence most likely contribute to maturational changes in reasoning, goal setting, impulse control, and evaluation of consequences. Other adolescent brain changes include increased hippocampal neurogenesis, maturation of brain regulatory neurotransmitters (e.g., their receptors and transporters), as well as hormonal maturation during puberty. Each of these maturation processes is driven by innate programming that responds to environmental stimuli. Adolescent development is common to humans and rodents, allowing controlled preclinical studies to focus on those environmental factors that create resilience or risk for long-lasting changes in adult characteristics.
The complex interactions of nature and nurture, intermixed with adolescent resilience and sensitivities, confound discernment of what characteristics are highly sensitive versus insensitive to environment. Many mental disorders emerge during adolescence, perhaps due to genetically programmed dysfunctional development, environmental disruption of developmental programs, or more likely a combination of both (Paus et al., 2008; Davidson et al., 2015). In humans, family structure and socioeconomic status, adolescent choice of peer group, and other individual selections create unique environments that confound a clear understanding of their impact on maturation of adult characteristics and skills. Animal studies have the advantage of control over environmental and genetic factors and can better elucidate the impact of specific environmental events on adolescent development. This review presents findings that support adolescence as a unique period of brain maturation that is characterized by increased vulnerability to binge alcohol-induced alterations in brain maturation and adult neurobiology due to the distinct adolescent responses to alcohol relative to adults. Preclinical studies from the Neurobiology of Adolescent Drinking in Adulthood (NADIA) Consortium, funded by the National Institute of Alcohol Abuse and Alcoholism, are presented and related to human findings when possible. Together, they support the hypothesis that adolescent binge drinking produces long-lasting effects in the brain that increase the risk for the development of psychopathology in adulthood, including alcohol-use disorders.
The adolescent period is marked by behavioral and hormonal changes that are common across species. Adolescents are highly tuned to the environment and peers, and adolescence is a critical period of social development and integration into society. In the rat, the adolescent period has been conservatively demarcated as postnatal day (P) 28–P42 (Spear, 2000), although some have suggested a more liberal range from P21 to P60 (Laviola et al., 2003). More recently, the adolescent period has been divided into early (P25–P42) and late (P43–P55) adolescence in rats, with the early and late periods corresponding to approximately 10–18 and 18–25 years of age in humans, respectively (Spear, 2015). Puberty, the hormonal and physiologic change associated with sexual maturation, takes place within the broader adolescent period. Although there are species-specific behavioral and hormonal responses, adolescence and puberty are general developmental periods that are shared across mammalian species. As in humans, complete pubertal maturation of the rat occurs earlier in females than males (approximately P36 and P44, respectively) (Vetter-O’Hagen and Spear, 2012). Importantly, adolescent-typical behavioral characteristics are also conserved across species, such as increased reward and sensation seeking, social interactions with peers, and risk taking, and reduced responses to aversive stimuli, which are all observed during adolescence, even beyond the peripubertal period (for review, see Spear, 2000, 2011). For instance, increased time spent engaging in social behaviors is common in human adolescents (e.g., increased communication with peers) (Csikszentmihalyi et al., 1977; Steinberg, 1989) as well as in adolescent rodents and nonhuman primates (e.g., increased levels of play and affiliative behaviors, such as huddling and grooming) (Ehardt and Bernstein, 1987). In rodents, increased social interactions influence food choices (Galef, 1977) and sexual and aggressive behaviors (Fagen, 1976; Smith, 1982). Rodent adolescents also find peers (Douglas et al., 2004) and novelty (Douglas et al., 2003) more rewarding than adults do. These adolescent-typical characteristics are important during the transition from dependence to independence. These characteristics also result in increased possibility of environmental exposures and influences. As discussed below, the recent discovery of epigenetic mechanisms under environmental regulation may represent a significant portion of the genetic aspects of adolescent maturation. Adolescent high novelty-seeking and risk-taking behaviors contribute to the increased propensity for experimentation and initiation of drug and alcohol use during this developmental period. Furthermore, the ability to learn and acquire new skills or habits can combine with initiation of drug use to increase the risk of long-lasting adult pathology. Given that adolescence is a unique period of brain and behavioral development that is highly sensitive to environmental influences, clinical and preclinical studies focused on adolescent development to understand what factors best promote individual success for all in the community are of great importance.
II. Brain Maturation and Adolescence
Brain development coincides with improvement in abilities. One example is the maturation of visual and auditory sensory processing. The sensory cortex has unique developmental periods that are highly responsive to enriched or deprived environments that drive synaptic rearrangements and cortical response pattern plasticity far more than are found at other times across the life span. These highly plastic periods of sensory cortical maturation are referred to as critical periods of experience-dependent plasticity, and some of these critical periods occur during the adolescent age (Gordon and Stryker, 1996). Visual cortex maturation involves optimizing visual acuity and discrimination through activity-dependent synaptic pruning of inactive synapses as well as maintenance and strengthening of active synapses. Maturation of the visual cortex precedes the critical period of the auditory cortex, which is characterized by acquisition of tonal specificity and maturation of auditory cortical responses. During plasticity of the cortical critical periods, γ-aminobutyric acid (GABA) interneuron synapse formation and regulation of pyramidal neuronal responses stabilize, and then plasticity subsides. Synaptic rearrangements in the developing cortex are dependent upon neuronal activity that triggers microglial–neuronal signaling. For example, in developing mouse visual cortex at P28 near the peak of the critical period of visual cortical experience-dependent plasticity and synapse formation, light deprivation and re-exposure regulate microglial–synaptic interactions (Tremblay et al., 2010). Microglial activity-dependent synaptic pruning involves complement receptor signaling between immature synapses and microglia (Schafer et al., 2012). In addition, microglia regulate the formation and degradation of extracellular matrix—secreted noncellular molecules that support cells and in brain stabilize synapses and form neuronal nets primarily on GABAergic neurons (Celio and Blumcke, 1994; Celio et al., 1998; Frischknecht et al., 2009; see Coleman et al., 2014). Thus, adolescent brain maturation involves neuronal and glial signaling that regulates synapses, particularly interneuron–projection neuron synaptic fields that are tuned during development to more stable and less plastic adult brain synapses.
Synapses are functional elements of the brain that are very small—most are less than 0.1 μM3—whereas brains are 1012–1014 times that size (e.g., human brain is about 1200 cm3 and adult rat brain about 2100 mm3) (Oguz et al., 2013). Interestingly, overall brain structure changes during adolescence, with decreases in gray matter and increases in white matter shown in both human (Giedd et al.,1999; Gogtay et al., 2004; Bava et al., 2010) and rodent studies (e.g., Oguz et al., 2013; Mengler et al., 2014). These changes are far larger than can be explained by changes in synapses, and they are thought to be associated with the processes of synaptic pruning, extracellular matrix formation, and increased myelination. The developmental trajectory of brain regional volumes in humans has been studied (Giedd et al., 1996; Sowell et al., 1999; Gogtay et al., 2006; Demaster and Ghetti, 2013) and is generally similar to that found in rats (Calabrese et al., 2013; Oguz, et al., 2013). For instance, subcortical limbic structures, such as the hippocampus and amygdala, mature during adolescence in humans (Giedd, et al., 1996; Sowell et al., 1999; Suzuki et al., 2005; Gogtay et al., 2006; Uematsu et al., 2012; Demaster and Ghetti, 2013) at a relatively faster pace than the prefrontal cortex (PFC) (see Casey et al., 2005 for review). The PFC is the last structure to mature, and development of PFC structural and functional connectivity continues into late adolescence and early adulthood in humans (Lebel et al., 2008; Petanjek et al., 2011) and rodents (Cunningham et al., 2002; Markham et al., 2007). An immature PFC, along with more developed limbic regions, may lead to an imbalance or disruption of top-down control, which is thought to underlie particular adolescent-typical behavior such as impulsivity and risk taking (Andersen and Teicher, 2008; Casey et al., 2008; Ernst and Fudge, 2009; Casey and Jones, 2010). PFC development and connectivity parallel the appearance of adult executive functions.
Late youth and adolescence are also when mental diseases commonly emerge (Paus et al., 2008; Davidson, et al., 2015), with some clearly related to alterations in the patterns of gray and white matter that exemplify the adult brain (Giedd, 2004). Indeed, white matter structures mature hierarchically and become more organized in parallel with the development of cognitive faculties (Asato et al., 2006; Lenroot and Giedd, 2006; Bava and Tapert, 2010). Myelin increases efficient neural transmission throughout the brain, and it is thought to contribute to the enhanced brain-regional connectivity, processing speed, and cognitive function that occur during childhood and adolescence (Casey et al., 2008). In a study of 885 individuals between 3 and 20 years of age, magnetic resonance imaging brain scans accurately distinguished biologic age, primarily by using diffusivity indices of white matter maturation (Brown et al., 2012). Recent studies have related the development of white matter along an accumbofrontal tract connecting the orbitofrontal cortex (OFC) and nucleus accumbens to the maturation of developmental models of decision making (Karlsgodt et al., 2015). Exercise, as assessed by fitness among adolescents, is associated with increased white matter microstructure and frontal and motor fiber connectivity, consistent with the postulate that environment and experience impact white matter development and connectivity (Herting et al., 2014). In rats, whole brain volume increases by approximately 20% from P28 to P80 (that is, from early adolescence to young adulthood), whereas white matter, including the corpus callosum and external capsule, increases by about 30% (Oguz et al., 2013). In rats, there are maturational changes in corpus callosum anisotropy found with diffusion tensor imaging (Vetreno et al., 2016a), and diffusion tensor imaging has been used to detect anisotropic changes in the human adolescent brain that are consistent with increased myelination (Zhu et al., 2012).
The PFC is particularly dynamic during adolescence. Human histologic studies find that the dendritic spine density of PFC synapses is two- to threefold higher in youth and declines through adolescence and into the third decade of life before stabilizing at adult levels (Petanjek et al., 2011). These findings are consistent with delayed maturation of PFC and its regulation of mesolimbic, amygdala, and behavioral control, resulting in the thrill-, novelty-, and sensation-seeking behavior that is characteristic of adolescence (Ernst and Fudge, 2009; Pattwell et al., 2012). Human adolescents also show an exaggerated amygdala response to fear that matures with the development of connections between the amygdala and ventromedial PFC in humans and infralimbic PFC in mice (Malter Cohen et al., 2013). This is consistent with studies that find attenuated extinction of fear conditioning in adolescent humans (Pattwell et al., 2012) that matures in parallel with frontal cortical circuits important for fear extinction (although see Broadwater and Spear, 2013a). As discussed above, activity-dependent plasticity in the PFC involves responsiveness of both GABAergic interneurons and glutamatergic pyramidal projection neurons, as well as consolidation of circuitry within other regions, to produce the development of executive functions during adolescence. Maturation of cortical GABAergic and glutamatergic synapses occurs in parallel with ongoing adolescent-specific changes in several major neuromodulatory neurotransmitter systems, such as acetylcholine, serotonin (5-HT), norepinephrine, and dopamine (see Guerri and Pascual, 2010; Spear, 2000, 2010 for review). Neuromodulatory neurotransmitters integrate GABAergic interneuronal and glutamatergic pyramidal neuronal firing, synchronizing firing and connectivity. Thus, both human and animal studies are consistent with adolescence being a critical period of frontal cortical activity-dependent plasticity. Furthermore, it is thought that adolescent frontal cortical integration underlies the maturation of adult emotion and reasoning. As PFC circuits mature, reflections on long-term consequences start to guide behavior, an important adult characteristic that may blunt the impulsive thrill seeking that is often seen during adolescence.
III. Adolescent Alcohol Sensitivity
A. Developmental Insensitivity to Ethanol
Numerous studies have found that adolescents are less sensitive to certain adverse effects of ethanol relative to adults (see Spear, 2011, 2014; Novier et al., 2015 for review), perhaps contributing to a propensity for adolescents to binge drink (Johnston et al., 2015). [TheNational Institute of Alcohol Abuse and Alcoholism definition of binge drinking is 4+ or 5+ drinks in a row for females or males, respectively, or achieving blood ethanol concentrations (BECs) of greater than 0.08 g/dL.] For example, adolescent rats are generally less sensitive to ethanol-induced sedative/hypnotic effects (Moy et al., 1998; Silveri and Spear, 1998; Draski et al., 2001), social inhibition at high ethanol doses (Varlinskaya and Spear, 2002), motor impairment (Hollstedt et al., 1980; Silveri and Spear, 2001; White et al., 2002a), conditioned taste aversion (Anderson et al., 2010; Schramm-Sapyta et al., 2010), and acute ethanol withdrawal (i.e., hangover) (Doremus et al., 2003; Varlinskaya and Spear, 2004; Doremus-Fitzwater and Spear, 2007). Thus, adolescents are less sensitive to several factors that may serve as feedback cues to limit alcohol consumption. A low sedative response to alcohol is a risk factor for development of alcohol-use disorder in humans (Schuckit et al., 2004) and is an adolescent characteristic that crosses species (Spear, 2011). Furthermore, low sensitivity to the perception of alcohol, as measured by the Subjective High Assessment Scale, has been established as one of the most significant risk factors for the development of heavy drinking and alcoholism (Schuckit et al., 2014). Studies relating blood alcohol to behavior have suggested that adolescent humans are less sensitive than adults (Day et al., 2013), although this is more clearly established in animal studies (Spear, 2014). Another index of alcohol sensitivity may be the amount of alcohol consumed, and studies find that both adolescent humans and rodents consume about twice as much as adults (Spear, 2014). Although the mechanisms of adolescent low alcohol sedative response or tolerance-like ethanol responses are not known, adolescent binge drinking in humans is predictive of adult alcohol-use disorders (for review, see Patrick and Schulenberg, 2013), and studies in rodents that control for genetic and environmental differences find adolescents are less sensitive to alcohol sedative/hypnotic effects (Silveri and Spear, 1998; Spear, 2014) and adolescent alcohol exposure of rats leads to long-lasting changes in adult rats that support hypotheses on long-lasting changes in adult human brain due to adolescent drinking.
The mechanisms underlying age-specific ethanol sensitivity are not fully understood, but one possibility is that adolescents are less susceptible to many ethanol effects because they metabolize ethanol faster. Although some studies have found that rodent adolescents metabolize ethanol slightly faster than adults (Hollstedt et al., 1980; Brasser and Spear, 2002), this is not a consistent finding (Kelly et al., 1987; Silveri and Spear, 2000). Furthermore, enhanced sensitivity to certain ethanol effects observed in adolescents (detailed below) argues against metabolic rate being the primary mechanism for age-related differences in ethanol sensitivity. Lastly, several studies have directly compared developmental responses to various ethanol concentrations in vitro when metabolism is not a factor (e.g., Swartzwelder et al., 1995a,b; Li et al., 2003). Another potential mechanism is that the functional properties of the neural systems underlying ethanol responses are fundamentally different between adolescents and adults. As suggested by Spear (2014), altered sensitivity to ethanol during adolescence may be due to age-related differences in excitatory glutamate [particularly at N-methyl-D-aspartate (NMDA) receptors], inhibitory GABAergic, and modulatory opioid systems. Relative immaturity of these neurotransmitter systems, which are directly targeted by alcohol, may alter brain excitatory–inhibitory balance during adolescence, perhaps contributing to age-related differences in ethanol effects (for review, see Spear and Varlinskaya, 2005; Spear, 2014). For example, adolescent rats differ from adults in electrophysiological properties, with reduced sensitivity to GABA type A (GABAA) receptor-mediated inhibition in hippocampus (Li et al., 2003, 2006; Yan et al., 2010; but see Yan et al., 2009), yet enhanced sensitivity to ethanol-induced inhibition of NMDA-mediated excitatory postsynaptic currents (Swartzwelder et al., 1995a). Thus, altered responsivity of these neurotransmitter systems during adolescence may underlie differential alcohol sensitivity, perhaps increasing risks of excessive drinking. However, additional research is needed to clearly define the unique aspects of the adolescent response to alcohol.
B. Developmental Sensitivity to Ethanol
Adolescents also show enhanced sensitivity to certain effects of ethanol (for review, see Spear, 2011, 2014; Novier et al., 2015). For instance, adolescent rats show ethanol-induced social facilitation at low ethanol doses, an effect not observed in adult rats (Varlinskaya and Spear, 2002, 2006), and greater ethanol-mediated reinforcement than adults (Pautassi et al., 2008). Increased sensitivity to the positive and/or reinforcing effects of ethanol may promote alcohol intake, although some would argue that elevated alcohol consumption is due to decreased sensitivity to the rewarding effects in adults (e.g., Koob and Le Moal, 1997). In animal and human studies, multiple factors impact behavior, making unequivocal conclusions on reinforcement difficult (for review, see Stephens et al., 2010). In the case of adolescent alcohol consumption, humans (SAMHSA, 2006) and rodents (Brunell and Spear, 2005; Doremus et al., 2005; Vetter et al., 2007) have been reported to consume up to 3 times more ethanol than adults, which may be related to altered ethanol sensitivity.
Adolescents are also more sensitive to some memory-impairing effects of ethanol. For example, adolescent rats show greater memory impairment than adults when assessed on the Morris water maze and in discrimination tasks (Markwiese et al., 1998; Land and Spear, 2004), but the opposite is observed in fear conditioning, another learning and memory paradigm; specifically, adolescent rats are less sensitive to memory-disrupting effects of ethanol (Land and Spear, 2004; Broadwater and Spear, 2013b). Also, people in their early 20s have been found to be more sensitive to the effects of ethanol on multiple memory tasks than those in their late 20s; however, tolerance due to prolonged alcohol use in the older age group cannot be definitively ruled out in this study (Acheson et al., 1998). When measuring the hippocampal electrophysiological response in adolescent rats relative to adults, ethanol more potently inhibits adolescent NMDA receptor-mediated synaptic activity (Swartzwelder et al., 1995a) and the induction of long-term potentiation (Swartzwelder et al., 1995b), perhaps contributing to enhanced sensitivity to memory-impairing effects of ethanol during adolescence. Adolescent rats are also more sensitive to frontal cortical brain damage in binge-ethanol models (Crews et al., 2000), consistent with the hypothesis that developing brain regions are more sensitive to ethanol toxicity than mature brain regions.
Although not assessed in the aforementioned studies, others have reported that adolescents do not show higher brain or blood ethanol concentrations compared with adults. Ethanol is typically administered at doses relative to body weight to account for the large differences in body mass between adolescent and adult rodents, but it distributes preferentially into watery, nonfatty tissues (Kalant, 1971). Body composition changes across the life span, and factors that might contribute to adolescent–adult distribution of ethanol include decreases in water content in lean tissue as well as increases in percentage of body fat from adolescence into adulthood (for review in humans, see Veldhuis et al., 2005). Consistent with an increase in percentage of body fat, adult rodents tend to have higher blood ethanol concentrations and a more prolonged ethanol clearance relative to adolescents (Doremus et al., 2003), making the possibility of higher ethanol exposure contributing to enhanced sensitivity to cognitive effects of ethanol during adolescence unlikely. Taken together, these findings suggest that adolescents are more sensitive to some effects of ethanol than adults, perhaps due to enhanced sensitivity of NMDA-mediated ethanol responses.
IV. Adolescents Binge Drink
Differing from the adult and alcoholic patterns of daily, heavy drinking, adolescents generally drink in social groups on weekends. Moreover, human and rodent adolescents drink about 2–3 times more alcohol than adults per drinking occasion (SAMHSA, 2006; Doremus et al., 2005). Adolescent binge drinking is a problem in many countries. The percentage of students in 2003 who reported being drunk 10 times or more in the last year were 40% in Denmark, 25% in the United Kingdom, and 8% in the United States (Andersson et al., 2002). In the United States 2014 Monitoring the Future Survey, 11%, 30%, and 50% of 8th, 10th, and 12th graders reported having been drunk in their lifetime, and 19% of 12th graders reported binge drinking (5+ drinks in a row) within the past 2 weeks (Johnston et al., 2015). Binge drinking peaks between the ages of 18 and 25 years of age, with males reporting binge drinking four to five times per month (2003 National Survey on Drug Use and Health). In fact, many adolescents drink more, as 1 in 10 high school seniors reported drinking 10 or more drinks in a row, and 5.6% of high school seniors reported consuming 15 or more drinks in a row (Patrick et al., 2013). Longitudinal studies of adolescent and young adult men and women (ages 18 and 24) find that 15–20% report 15–20 maximum drinks per occasion in the 6 months prior to each follow-up (Schuckit et al., 2014). The low sensitivity to alcohol sedation, combined with high risk taking and social reward seeking, most likely contributes to the extreme heavy drinking found in some adolescents.
Heavy binge drinking can result in a blackout, or loss of memory of events that took place during a drinking episode. Blackouts are based on the amount of alcohol consumed and are more common in adolescents than adults. BECs of over 0.30 g/dL, or about 4 times the legal BEC limit for driving in the United States (0.08 g/dl), are associated with 60% of alcohol-related blackouts (Hartzler and Fromme, 2003; Wetherill and Fromme, 2009; Rose and Grant, 2010). Blackouts are common in alcoholics and adolescents, consistent with these groups drinking to the very high BECs that can result in blackouts. For example, one study found that college student males who experienced blackouts reported consuming nine drinks on average (Zeigler et al., 2005). Among a sample of US college students, 51% report having experienced an alcohol-related blackout—40% within the last year and 9% within the past 2 weeks (White et al., 2002b). In another study that determined maximum drinks per occasion in subjects from ages 18 to 24, most subjects endorsed 5 as maximum, but about 15–20% endorsed 15–22 drinks as maximum per occasion (Schuckit et al., 2014), which would produce very high BECs. Magnetic resonance imaging studies find lower GABA in frontal cortex in 18- to 24-year-old binge drinkers compared with light drinkers, and binge drinkers with blackouts additionally had lower levels of frontal cortical glutamate (Silveri et al., 2014). In rats, equivalent binge models induce significantly more frontal cortical damage in adolescents than in adults (Crews et al., 2000). Thus, alcohol-related blackouts are common among human adolescents, and rat studies find the adolescent-maturing frontal cortex is uniquely sensitive to damage from binge-drinking levels of alcohol.
A lasting impact of adolescent binge drinking is suggested by associations of age of drinking onset with a number of lifelong risks. Adolescents who start drinking before 15 years of age are 4 times more likely to develop alcohol dependence in their lifetime than those who start drinking after 20 years of age (Grant and Dawson, 1997). A young age of drinking onset is also associated with increased risk for lifetime violence and fights and injuries associated with alcohol use (Grant and Dawson, 1997; Sher and Gotham, 1999; DeWit et al., 2000; Dawson et al., 2008; Hingson et al., 2009). Individual genotype and/or personality factors (such as sensation seeking) most likely contribute to early drinking, although peer use and alcohol-abusing parents are environmental factors that also contribute to an earlier onset of alcohol and substance use (Siqueira and Smith, 2015). Population studies of 9- to 20-year-old individuals find that a 10% delay in age of drinking initiation leads to a 35% decrease in subsequent alcohol consumption (Pedersen and Skrondal, 1998). For example, individuals who started drinking before age 13 consumed an average of 7 L alcohol/yr, whereas those who started after age 17 consumed 3.8 L/yr, suggesting that delaying onset of alcohol use can markedly reduce later alcohol consumption (Pedersen and Skrondal, 1998). Twin studies of 10- to 28-year-old subjects also find that early drinking increases risks for alcohol dependence, and that the risk for development of alcohol dependence declines by 21% for each additional year that drinking onset is delayed (Prescott and Kendler, 1999). Moreover, these authors find females to have higher risks than males from early drinking, and they attributed risks to familial factors related to genetics (Prescott and Kendler, 1999). Other studies have linked drinking onset and increased risks of alcohol dependence to familial density of alcoholism, extroversion, event-related brain potentials, and high posture sway (Hill and Shen 2002), supporting genetic components. More recent studies on familial factors have proposed that alcohol may promote unique induction of genes in adolescents that underlies the strong familial associations with an early age of drinking onset (Agrawal et al., 2009). Another recent study found that youth sipping alcohol in the 6th grade, often at home with parents, greatly increased the chances of getting drunk and drinking heavily by 9th grade when compared with nonsippers, even controlling for temperament and other behavioral problems (Jackson et al., 2015), suggesting an environmental familial influence. Thus, the strong familial contribution to early onset drinking and risks of alcohol dependence include both genetic and environmental components that are hard to untangle.
As mentioned earlier, extreme binge drinking of 10–15 or more drinks in a row was reported among 5–10% of 12th graders in the past 2 weeks (Patrick et al., 2013). This may represent a group that is at particularly high risk of later alcohol problems (Patrick and Schulenberg, 2013). Regardless, the high prevalence of alcohol binge drinking among school children indicates that many are drinkers (Table 1). Large longitudinal population studies find that the younger the age of drinking onset, the greater the prevalence of lifetime alcohol dependence. When these are combined with assessments of adolescent drinking, they support the idea that a large percentage of those who develop alcohol-use disorder do so, in part, due to adolescent binge drinking. However, other confounding factors are the adolescent emergence of conduct disorder or antisocial personalities that may identify themselves with early onset of alcohol drinking and that later develop into alcohol dependence. Alternatively, heavy binge drinking might disrupt adolescent brain development, altering maturation in complex ways. One study (White et al., 2011) following boys from 8 to 18 years of age found that impulsivity generally declined with increasing age, as mentioned above. Among a subgroup with intermediate impulsivity, heavy drinking at age 14 increased impulsivity at 15, but not older ages. However, continued heavy drinking at 14, 15, and 16 increased impulsivity within the binge group at each age, although both binging and nonbinging individuals showed decreased impulsivity with increasing age (White et al., 2011). These longitudinal findings indicate that the emergence of specific personality traits, such as impulsiveness, thrill seeking, and anxiety, are all adolescent traits, as well as traits associated with risk for alcohol dependence, and that there may be a bidirectional influence between alcohol use and the expression of these traits. Along these lines, impulsivity among university students has been found to predict the quantity of alcohol consumed per month (Caswell et al., 2016).
The prevalence of lifetime adult alcohol use disorders is related to age of alcohol drinking onset
The value in the last column is the percentage of the population with lifetime alcohol dependence (AD) related to adolescent drinking. It is calculated from the percentage having been drunk (Johnston et al., 2015), the prevalence of lifetime alcoholism related to age of initiation of drinking (Grant and Dawson, 1997), assuming having been drunk would be considered initiation of drinking. The last column calculates the prevalence of lifetime alcohol dependence related to adolescent drinking as a percentage of whole population studies of the prevalence of alcohol dependence in the United States (Hasin et al., 2007). These estimates suggest about one third to three quarters of alcohol dependence in the United States could be related to adolescent drinking.
Studies in animals are an important strategy to disentangle genetic and environmental contributions to alcohol use and its consequences. Whereas animals cannot model all aspects of alcoholism (Leeman et al., 2010; Stephens et al., 2010), there are many similarities between animal and human alcohol use. For example, impulsivity is greater in adolescent human binge drinkers and mice with high alcohol consumption (Sanchez-Roige et al., 2014a). Recent studies have also indicated that alcohol can change gene expression through epigenetic mechanisms in a manner that is inherited, representing an environmental alcohol-induced genetic change that was previously unexpected (see Pandey et al., 2015). Indeed, mouse studies find that exposure to alcohol epigenetically alters neuroendocrine and immune gene expression for at least three generations (Sarkar, 2016). Studies in rhesus monkeys have found that drinking in young adulthood strongly disposes individuals toward heavy drinking in adulthood, and this effect is independent of the sociocultural factors present in humans (Helms et al., 2014). Furthermore, studies in mice (Alfonso-Loeches and Guerri, 2011) and rats (Alaux-Cantin et al., 2013) have found that adolescent exposure to alcohol increases later voluntary alcohol drinking. These findings and those described below support the hypothesis that the age of drinking onset contributes to risks of alcohol dependence later in life at least in part via biologic consequences of alcohol exposure.
V. Modeling Adolescent Alcohol Drinking
Human alcohol abuse and dependence (Leeman et al., 2010), as well as sensitivity to alcohol response (Crabbe et al., 2010), can be difficult to model in rats and mice. Humans will drink far more alcohol by choice than rodents, although alcohol drinking preference, positive reinforcement, and negative reinforcement can be modeled in animals. Furthermore, components of alcohol dependence, alcoholic liver disease, and fetal alcohol syndrome are modeled by exposing animals to alcohol via various routes of administration, including ethanol vapor chambers, intragastric gavage, and i.p. injections, all of which can be used to reach high BECs like those associated with human binge drinking and blackouts. Models of adult alcohol abuse and alcohol dependence often involve long-lasting alcohol exposures, but human adolescent drinking is not typically characterized by continuous daily drinking. Generally, adolescent drinking is heavy binge drinking separated by periods of abstinence, as it often involves social events clustered around weekends or holidays when alcohol is available.
Due to commonalities of adolescent development across mammalian species (as described above), we can use animal models to explore the impact of heavy binge drinking during adolescence on the maturation of adult characteristics. Adolescent intermittent ethanol (AIE) exposure is a model that incorporates adolescent age with intermittent ethanol administration, most commonly 2 days of ethanol exposure followed by 2 days off (no exposure). Although all ethanol exposure regimens (vapor, gavage, i.p.) are compared with an appropriate vehicle control exposure, there is the potential for high levels of ethanol to be aversive. Guerri and colleagues first used this model (Pascual et al., 2007), and others have adopted it to investigate adolescent underage drinking in preclinical studies (e.g., Pascual et al., 2009; Vetreno and Crews, 2012; Alaux-Cantin et al., 2013; Ehlers et al., 2013b; Coleman et al., 2014). Some studies directly compare adolescent and adult responses, exposing adolescents to AIE and adults to an identical adult chronic intermittent ethanol (CIE) exposure, and this AIE-to-CIE comparison provides insight into adolescent-specific maturational or age-dependent responses. A major focus of the NADIA Consortium is on AIE-induced changes in behavior and physiology that persist into adulthood. The AIE models used by the NADIA Consortium encompass the adolescent period, include intermittent exposure, and achieve binge-like BECs (>0.10 g/dL). Below we describe studies largely from the NADIA Consortium finding that AIE leads to a persistent increase in neuroimmune gene expression, loss of cholinergic and other neuronal markers, reduced neurogenesis and brain-derived neurotrophic factor (BDNF), as well as persistence of adolescent-like responses to alcohol in adulthood, increased adult anxiety, increased adult alcohol drinking, and epigenetic signaling—all of which suggest that heavy binge drinking in adolescence has long-lasting effects on adult brain and behavior.
VI. Lock-In—Persistence of an Adolescent Phenotype in Adulthood, Including an Adolescent-Typical Response to Ethanol
Several preclinical studies have supported the hypothesis of a lock-in effect: that is, the idea that adolescent-typical ethanol sensitivities are retained into adulthood following a history of AIE (see Spear and Swartzwelder, 2014 for review). As mentioned earlier, adolescents are less sensitive to certain adverse effects of ethanol. Interestingly, several studies have found a similar adolescent-typical attenuated ethanol sensitivity in adults exposed to AIE, such as decreased sensitivity to ethanol-induced motor impairment (White et al., 2002a), conditioned taste aversion (Diaz-Granados and Graham, 2007; Sherrill et al., 2011; Saalfield and Spear, 2015), social inhibition (Varlinskaya et al., 2014), acute withdrawal (Boutros et al., 2014), and sedative/hypnotic effects (Matthews et al., 2008; Quoilin et al., 2012). The rewarding effects of ethanol may also be enhanced in adulthood after adolescent ethanol exposure, with evidence for greater motivation to consume ethanol on an operant task (Alaux-Cantin et al., 2013) and increased ethanol-induced social facilitation (Varlinskaya et al., 2014). Just as in adolescence, the maintenance of these adolescent-like phenotypes may allow and/or promote greater ethanol consumption in adulthood by attenuating sensitivity to adverse effects of ethanol and enhancing sensitivity to rewarding effects. Indeed, evidence is mounting to suggest that adolescent alcohol exposure in rats increases alcohol intake in adulthood (Pascual et al., 2009; Maldonado-Devincci et al., 2010; Gilpin et al., 2012; Alaux-Cantin et al., 2013; Milivojevic and Covault, 2013); this is described in more detail below.
Other long-lasting effects of adolescent ethanol exposure that appear to lock in an adolescent-like phenotype are, for example, a lack of an event-related potential response to ethanol (Ehlers et al., 2014a), increases in impulsivity (although this effect was unmasked after re-exposure to a chronic ethanol procedure in adulthood) (Mejia-Toiber et al., 2014), and greater risk preference (Boutros et al., 2014; Sanchez-Roige et al., 2014a,b; Schindler et al., 2014). Adults with a history of AIE also show adolescent-like increases in sensitivity to the deleterious effects of acute ethanol, such as impairment in hippocampal-dependent memory (White et al., 2000; Broadwater and Spear, 2013b; Risher et al., 2013), and there is evidence of an immature pattern of learning in a fear-conditioning paradigm (Broadwater and Spear, 2014a). Thus, adolescent ethanol exposure produces a variety of long-lasting consequences, many of which are reminiscent of adolescent-like ethanol responses.
Although the mechanisms of AIE-induced changes in ethanol responses are poorly understood, Spear and Swartzwelder (2014) propose that synaptic maturation of excitatory and inhibitory balance may be altered after adolescent ethanol, thereby contributing to the retention of an adolescent-like phenotype in adulthood. For example, persistent alterations in GABAA subunit expression have been observed after adolescent ethanol (Centanni et al., 2014; Risher et al., 2015), a receptor system that undergoes considerable reorganization during adolescence (Yu et al., 2006). Furthermore, there is evidence for enhanced sensitivity of GABAergic tonic current (Fleming et al., 2012) and increased propensity for induction of long-term potentiation (LTP) at lower levels of stimulation in the adult CA1 region of the hippocampus (Risher et al., 2015) after AIE. This lowered threshold for hippocampal LTP induction is indicative of an AIE-induced hyperplastic state across the hippocampal circuit, leading to interference in memory processes, and is reminiscent of an adolescent-like hyperexcitability, at least in the hippocampus. AIE exposure also alters adult dendritic spine density in amygdala and hippocampus in a manner consistent with blunted synaptic maturation, although the precise findings differ across brain regions, perhaps due to differences in stage of development. In hippocampus, AIE-exposed adult rats showed an increased number of dendritic spines, typical of immaturity as well as LTP sensitization (Risher et al., 2015). In amygdala, AIE caused a decrease in dendritic spine density in adulthood that was associated with decreased expression of BDNF and increased anxiety-like behavior and alcohol drinking (Pandey et al., 2015). The differences in projection neurons and interneurons as well as the development of synapses in these various brain regions require additional studies. However, as mentioned above, cortical maturation involves changes in interneuron GABAergic synapses regulating pyramidal neuronal inputs, with immature synapses being associated with a low alcohol response. Glial extracellular matrix deposition appears to stabilize synaptic structure and reduce plasticity, and AIE was found to increase frontal cortical extracellular matrix proteins (Coleman et al., 2011). Thus, it is possible that AIE-induced extracellular matrix deposition and/or microglial priming would stabilize immature synapses, resulting in the persistence of adolescent-like responses in adulthood, although more studies are needed to test this hypothesis.
Neuronal activation to an ethanol challenge appears to be altered after AIE in a brain-region–specific manner. Immediate early genes, such as cFos and egr1, rapidly increase in expression following neuronal firing and thus provide an indirect measure of neuronal response. Acute ethanol challenges increase cFos and egr1 expression in PFC, amygdala, nucleus accumbens, and ventral tegmental area of adult rats (Liu and Crews, 2015). However, adults with a history of AIE have a markedly reduced expression of immediate early genes in response to ethanol challenge in the PFC (both prelimbic and OFC portions; Fig. 1), and the adult neuronal response in the amygdala is slightly blunted by AIE. In contrast, the nucleus accumbens, a brain region associated with reward and reinforcement, shows an exaggerated cFos neuronal activation to ethanol challenge after AIE. These data support the interpretation that adolescent binge drinking (i.e., AIE) results in increased activation of reward circuitry and inactivation of frontal cortical executive functions during adult binge ethanol, even after long periods of abstinence. Together, these findings indicate that AIE alters adult brain responses to ethanol as well as other adolescent-typical characteristics in a manner consistent with increased risks of alcoholism.

AIE alters adult brain regional responses to an alcohol challenge in adulthood. Adult rats previously exposed to AIE exhibit altered neuronal responses to an ethanol challenge in adulthood as indexed by expression of the immediate early gene cFos, an indirect marker of neuronal activity. Comparison of cFos immunoreactivity (+IR) in adult Wistar rats that received an ethanol challenge (4.0 g/kg, i.g.) in adulthood (P80) revealed that prior AIE exposure (5.0 g/kg, i.g., 2 days on/2 days off from P25 to P55) significantly reduced cFos + IR in the orbitofrontal cortex (OFC; ↓57%), prelimbic cortex (PrL; ↓48%), ventral tegmental area (VTA; ↓50%), and basolateral amygdala (AMG; ↓33%), relative to ethanol-challenged control (CON) subjects. In contrast, previous AIE exposure increased neuronal activity in response to ethanol challenge in the nucleus accumbens core (NAcc; ↑43%) relative to CON subjects. These studies reveal that adolescent binge ethanol exposure causes long-lasting reductions in frontal cortical reactivity in areas involved in executive function and increased activation in reward circuitry in response to ethanol challenge in adulthood, indicative of an enduring alteration in the adult brain response to ethanol. Data are presented as mean ± S.E.M. *p < 0.05, ***p < 0.001, relative to CON. This figure is adapted from (Liu and Crews, 2015).
VII. AIE Increases Ethanol Self-Administration in Adulthood
Human studies report that earlier initiation of alcohol drinking is associated with an increased likelihood of developing an alcohol-use disorder across the life span (Grant and Dawson, 1997; DeWit et al., 2000). Preclinical models of binge AIE have also revealed increased voluntary ethanol drinking in adulthood in rodents (Pascual et al., 2009; Alaux-Cantin et al., 2013; Broadwater et al., 2013c; Gass et al., 2014; Pandey et al., 2015). When assessed by two-bottle, free-choice drinking with increasing ethanol concentrations (3%, 7%, and 9% every 3 days) beginning in adulthood, an i.p. AIE exposure led to a twofold increase in voluntary ethanol self-administration in male Sprague–Dawley rats (Pandey et al., 2015). Similarly, Alaux-Cantin et al. (2013) found that early (P30–P43), but not late (P45–P58), i.p. AIE exposure to male Sprague–Dawley rats increased voluntary ethanol consumption and preference in adulthood by approximately 75%, also assessed by two-bottle, free-choice drinking. In the same study, increasing the ethanol concentration (i.e., from 10% to 20% ethanol) and limiting the two-bottle choice to every-other-day access led to a larger, twofold increase in drinking and greater escalation of ethanol intake in adulthood. Finally, assessment of operant self-administration of 10% ethanol in adulthood revealed an approximate 70% increase in ethanol intake. These AIE-exposed adults also displayed a higher breakpoint across progressive ratio sessions, indicating that AIE-exposed rats will expend more effort to obtain ethanol. In another study, exposure of male Long–Evans rats to AIE vapor inhalation (P28–P42) increased ethanol intake by approximately 30% in adulthood when assessed via operant self-administration (Gass et al., 2014). Interestingly, these AIE-exposed rats later required approximately 33% more sessions to extinguish the learned ethanol-seeking behavior (Gass et al., 2014). In an adolescent self-administration model involving sole-source 10% ethanol in a sweet solution (0.125% saccharin/3% sucrose) for 30 minutes from P28 to P42, adult Sprague–Dawley rats increased voluntary consumption of sweetened ethanol by approximately 30%, but not consumption of 20% ethanol, relative to control subjects drinking the sweet-only solution (Broadwater et al., 2013c). A caveat of this study, however, was that control rats exposed to the sweet-only solution during adolescence drank relatively more sweet-only solution in adulthood, indicating greater adolescent responding for all rewards as well as the exposure effect increasing familiarity—the adult rats preferred whatever solution they experienced in adolescence. In another study (Pascual et al., 2009), male Wistar rats with a history of i.p. AIE exposure (P25–P38) that were assessed in adulthood on a two-bottle, free-choice model with 10% ethanol every other day for 10 days exhibited a twofold increase in both ethanol preference and resulting BECs in adulthood, and AIE-exposed adults continued to drink more ethanol than controls during a subsequent limited access to ethanol (1-hour access to 10% ethanol at the end of the light phase). Taken together, these rodent studies are consistent with human data and support the hypothesis that early initiation of binge drinking during adolescence increases ethanol seeking and drinking in adulthood, contributing to the development of alcohol-use disorders later in life.
VIII. AIE Results in Decreased Behavioral Flexibility
Behavioral flexibility refers to the ability to change a previously learned reinforced behavioral response to a new response in light of changing task demands or reinforcement. In a practical sense, behavioral flexibility may represent the ability to adjust to the responsibilities of emerging independence and parenthood. A consistent finding of the NADIA Consortium is that AIE exposure leads to impairments in behavioral flexibility in adulthood. In the section that follows, the long-term effects of AIE exposure on behavioral flexibility will be reviewed.
A. Flexibility in Spatial Tasks
Spatial learning is often assessed using maze tasks such as the Morris water maze or the Barnes maze. The Morris water maze consists of a circular tub filled with an opaque liquid containing a submerged platform, which is solved when the animal learns to locate the hidden platform by using spatial cues to escape the water. The Barnes maze is a large, brightly illuminated circular platform with multiple holes situated around the edge. An escape box is located under one of the holes, and the rodent uses spatial cues to locate the escape box. These tasks are ideal for assessing not only spatial learning, but also behavioral response to a subsequent challenge, such as moving the platform or escape hole, that would require a flexible strategy. Several studies have shown that AIE exposure does not affect spatial learning in adult mice (Coleman et al., 2011, 2014) or rats (Vetreno and Crews, 2012; Acheson et al., 2013) when assessed on the Morris water maze or the Barnes maze. Similarly, AIE exposure does not alter acquisition of a radial arm maze or operant task (Risher et al., 2013). However, when the learned location of the escape platform or hole is moved, adult AIE-treated mice and rats require significantly more trials to learn the new location or rule (Coleman et al., 2011, 2014; Vetreno and Crews, 2012). Perseveration of previously learned behaviors or difficulties breaking previously learned habits appear to underlie some of this poor performance. Indeed, AIE-exposed rats also exhibited perseverative behaviors, such as spending more time in the area of the original escape platform (Coleman et al., 2011; Vetreno and Crews, 2012), and behavioral inefficiency, such as taking longer and traveling farther to reach the same goal as control rats (Acheson et al., 2013). Interestingly, neuroimmune-signaling molecules have been shown to correlate with these behavioral deficits: increased expression of Toll-like receptors (TLRs) and high-mobility group protein B1 (HMGB1; discussed in more detail below) was associated with reduced behavioral flexibility and increased perseverative behavior on the Barnes maze (Vetreno and Crews, 2012) and may contribute to deficits in behavioral flexibility. These findings suggest that AIE-induced changes in neuroimmune signaling contribute to AIE alterations in PFC synaptic maturation, increased perseveration, and blunted ability to adapt to changes in the environment.
B. Flexibility on Operant Tasks
Instrumental conditioning involves training an animal to perform a specific action (such as a lever press or nose poke) to obtain a reward, which reinforces the operant action. Several studies have determined that AIE exposure does not alter acquisition of operant self-administration of a reward (Semenova, 2012; Risher et al., 2013; Gass et al., 2014; Mejia-Toiber et al., 2014; Boutros et al., 2016). It also does not change the preference for a large reward (Mejia-Toiber et al., 2014) or performance on a progressive ratio schedule (Gass et al., 2014). However, similar to AIE effects on spatial learning tasks, AIE deficits can emerge when the operant behavior is challenged, such as by changing the contingency between the operant and the reward. In a set-shifting study, Gass et al. (2014) trained rats to use a visual cue to determine which lever to press to receive a reward. Then they changed the rule so that the rat would use location cues and ignore the previously informative visual cue (i.e., set shifting). AIE exposure impacted learning this new rule—rats took longer to perform to criterion and made more errors than control rats (Gass et al., 2014). In a separate group of rats, Gass et al. (2014) trained rats to self-administer a 20% alcohol solution and found that AIE-exposed rats self-administered more alcohol than controls, similar to other reports (Alaux-Cantin et al., 2013). However, when the alcohol reward was withheld (i.e., extinction training), control rats learned to stop pressing the lever much faster than AIE-exposed rats (Gass et al., 2014). In humans, a similar resistance to extinction or abstinence of alcohol drinking after adolescent binge drinking could increase alcohol consumption in adulthood, as well as make it more difficult for individuals to discontinue drinking once initiated. Interestingly, the deficits in both set-shifting and extinction learning were reversed by treatment with the positive allosteric mGluR5 modulator 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl) benzamide, a putative cognitive-enhancing agent. The procognitive effect of 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl) benzamide may be due, in part, to its effects on the medial PFC (Fowler et al., 2013), a brain region particularly vulnerable to the neurotoxic effects of adolescent binge ethanol exposure (Crews et al., 2007). Thus, AIE disrupts frontal cortical control, increases repetitive habit-like responding, and reduces the ability to adapt to changes in reinforcement.
IX. Adolescent Alcohol Effects on Anxiety and Negative Affective Behavior
Adolescents can be highly emotional, with some suggesting that adolescents drink alcohol to enhance positive emotional states (e.g., enhancement motives), which has been related to heavy drinking and is linked to certain adolescent personality characteristics, such as sensation seeking, low inhibitory control, and impulsivity (Siqueira et al., 2015). Adolescents often exhibit high emotional and impulsive decision making, associated with negative affective states and low distress tolerance (Ernst and Fudge, 2009), especially among teens who misuse alcohol or drugs (Clark et al., 2008). For example, among Caucasian adolescents, negative affect and low distress tolerance are associated with increased probability of alcohol use (Daughters et al., 2009). Furthermore, protracted heavy drinking may provoke negative affect (Brown et al., 1995; Liappas et al., 2002) and diminish problem-solving abilities (Brown et al., 2000; Goudriaan et al., 2007). Youth who engage in heavy episodic drinking have greater recent and lifetime alcohol consumption, more frequent alcohol-induced blackouts, and more withdrawal symptoms, with all being associated with increases in negative affect (Winward et al., 2014). These studies are consistent with the hypothesis that binge levels of alcohol drinking during adolescence result in more negative affect in adulthood. Although emotional responses are difficult to quantitate in animal models, multiple assessment methods of affect have been developed to determine negative affect and/or anxiety-like behavior in rodents. In general, studies suggest that adolescent ethanol exposure induces long-lasting increases in adult negative affect, although there are some caveats to this conclusion.
A. Rodent Models of Anxiety
Many methods of assessing anxiety in rodents involve measuring locomotion in an experimental chamber, and relative locomotion in risky versus safe aspects of the environment provides an index of anxiety. Such tests include the light–dark box (consisting of a brightly illuminated compartment and a dark compartment) and the elevated plus maze (EPM; consisting of a plus-shaped maze with two open arms and two enclosed arms). Similarly, the open-field test can be used to index anxiety as highly anxious rodents display thigmotaxic behavior, in which they remain close to the walls of the chamber and do not venture into the center. All of these tests involve a conflict between the rodent’s tendency to explore a new environment with the discomfort of being in a bright, elevated, or otherwise unsafe environment (Bourin and Hascoet, 2003). Anxiolytic drugs increase time in the illuminated compartment of the light–dark box and the open arms of the EPM, whereas drugs that reduce time in the illuminated compartment are thought to reflect anxiogenic activity (Pellow et al., 1985; Lister, 1987; Onaivi and Martin, 1989; Bourin and Hascoet, 2003; Prut and Belzung, 2003). Young adolescent rats (P34) move more quickly out of the light compartment into the dark compartment in the light–dark box, consistent with adolescent high anxiety-like behavior, but by late adolescence (P55) behavior is comparable to adult performance (Desikan et al., 2014). Acute ethanol is anxiolytic, and, similar to other ethanol responses, adolescent rats required a higher dose of alcohol to increase open arm times in the EPM than adult rats (Varlinskaya and Spear, 2002; Sakharkar et al., 2012, 2014; Pandey et al., 2015). When examining the long-term effects of adolescent alcohol, Sakharkar et al. (2016) found that AIE exposure led to increased anxiety-like behavior in adulthood, as indicated by a significant reduction from about 65% to 35% time spent exploring the illuminated compartment of the light–dark box. Likewise, AIE exposure of Sprague–Dawley rats resulted in heightened anxiety-like behavior in the EPM, specifically, a decrease in open arm entries from about 45% to 30% at 24 hours after AIE that persisted for at least 50 days (Pandey et al., 2015). In the open-field test, AIE-exposed mice exhibited reduced center exploration when assessed in adulthood (Coleman et al., 2014), and AIE-exposed rats displayed longer latencies to enter the center (i.e., thigmotaxis) when assessed over 100 days later (Vetreno et al., 2014). Consistent with the findings that AIE enhanced anxiety in adulthood, other studies reported persistent increases in immobility in the Porsolt swim test. This test assesses the latency of the rodent to become immobile following placement into a cylinder of water and is a screen for antidepressant drugs, which increase the latency to immobility. Adult animals exposed to AIE exhibited both faster latency to immobility as well as more sinking episodes than controls (Slawecki et al., 2004; Ehlers et al., 2011).
B. Anxiety or Disinhibition?
As mentioned above, these common tests of anxiety measure the locomotion arising from the conflict of innate fear of brightly illuminated areas contrasted with the drive to explore novel environments. Consequently, these tests are known to vary across sites and can be confounded by impulsivity, poor behavioral control, and hyperactivity. In light of this, it may not be surprising that some studies have reported results that do not support enhanced anxiety when using the same tests. For example, Ehlers et al. (2013b) found that adult AIE-exposed animals exhibited shorter latencies to enter the light box as well as more vertical movements (rears) in the light compartment, which they interpreted as evidence that the AIE-exposed adult animals were more aroused and disinhibited. Other studies found that AIE exposure increases open arm time in the EPM in adulthood, suggesting arousal, disinhibition, and/or impulsivity, as well as anxiolytic responses (Ehlers et al., 2011; Gilpin et al., 2012; Gass et al., 2014). The interpretation of these data as disinhibition is supported by findings from the modified open-field conflict test. This test provides a measure of disinhibition by assessing a rodent’s contact with a food pellet in the center of a brightly illuminated test chamber. Relative to control subjects, adult animals exposed to AIE spent significantly more time approaching and consuming the food pellet, suggestive of disinhibitory behaviors (Ehlers et al., 2011). A potential mechanism for disinhibition could involve AIE-induced alterations in the maturation of the PFC. Indeed, Shah et al. (2004) found that inactivation of the PFC results in increased exploration of the open arms on the elevated plus maze.
Thus, anxiety and disinhibition appear to be confounds in these tests of anxiety, and the assessments of AIE exposure most likely reflect relative effects between these outcomes. One factor that may contribute to the disparate findings is the strain of rat, as rat strains are known to differ in baseline anxiety measures. Specifically, some reports of AIE-induced anxiety in adulthood used Sprague–Dawley rats (Pandey et al., 2015; Sakharkar et al., 2016), whereas those reporting disinhibition or impulsivity used Long–Evans or Wistar rats (Ehlers et al., 2011; Gass et al., 2014), although AIE enhanced thigmotaxis (consistent with enhanced anxiety) in adulthood in Wistar rats (Vetreno et al., 2014). Another potential factor is the AIE regimen, as the studies reporting enhanced anxiety used bolus administration routes (intragastric, i.p.) and those reporting disinhibition or anxiety applied the ethanol via vapor. A critical difference in these regimens is that the bolus administration will produce more dynamic BEC that rapidly rise and then fall, whereas vapor results in more stable, high BEC. Although all these routes achieve binge levels of alcohol, the different dynamics may shift the balance from enhanced anxiety to enhanced disinhibition. Thus, evidence from multiple laboratories indicates that AIE can promote both anxiety and disinhibition, but the nature of rodent assessments prevents a clear determination of how AIE impacts these two traits.
C. Rodent Models of Social Anxiety
Another measure of anxiety and negative affect in the rodent is the social interaction test. Human studies of adolescent development show that adolescents spend more time interacting with their peers than any other age group (Hartup and Stevens, 1997), and these peer interactions become highly significant and motivating (Steinberg and Morris, 2001; Spear, 2010). In a developmentally similar manner, adolescent rats engage in substantially more social activity with age-matched rats, typically in the form of play fighting (Vanderschuren et al., 1997; Varlinskaya and Spear, 2002, 2008). The rodent social interaction test can be used to measure these adolescent-typical behaviors by assessing social motivation as well as play fighting and social investigation (Varlinskaya et al., 1999) and to provide an index of anxiety-like behavior in social settings (File and Seth, 2003). In adolescent rats, low-dose acute ethanol challenge (e.g., 0.50 g/kg) in familiar, nonanxiogenic environments leads to increases in social behavior characterized by increased play fighting that is not observed in adults (Varlinskaya and Spear, 2002, 2006, 2007; Willey et al., 2009), which may be related to enhanced sensitivity to the rewarding effects of ethanol during adolescence (as discussed above). However, higher doses of ethanol (e.g., 1 g/kg) cause social inhibition, albeit to a lesser degree in adolescent relative to adult rats (Varlinskaya and Spear, 2002). These behavioral changes are not simple locomotor effects; the same doses of ethanol do not alter measures of nonspecific locomotion in novel test environments (Varlinskaya and Spear, 2002). Early AIE exposure (P25–P45) significantly decreases social preference and social investigation in adult male but not female rats, indicating that AIE-induced social anxiety is sex-specific. Interestingly, this effect appears to be specific to early adolescence, as intermittent ethanol exposure during late adolescence (P45–P65) did not affect social measures in adulthood. Furthermore, a history of AIE, regardless of the timing of exposure, altered the adult male responses to an acute ethanol challenge—specifically, an alcohol challenge increased social investigation and play fighting displayed by AIE-exposed males that were reminiscent of behaviors typically observed during adolescence, an effect that was not observed in control-exposed rats (Varlinskaya et al., 2014). These data suggest that early adolescence, more than late adolescence, is a critical period for establishment of age-appropriate social consequences in male rats.
X. Adolescent Alcohol-Induced Neuroimmune Gene Induction
As mentioned above, immune-signaling molecules and microglia, the brain monocyte-like cell, are involved in synaptic plasticity and brain development. During brain development, microglia undergo dramatic changes in morphology, being rounded and amoeboid in the early postnatal period and attaining an adult-like morphology by approximately P20–P30 in rat cortex (Orłowski et al., 2003; Harry and Kraft, 2012). Immune-signaling molecules, such as TLRs, HMGB1, receptor for advanced glycation end products (RAGE), proinflammatory cytokines, and other immune-signaling molecules, contribute to brain development (Boulanger and Shatz, 2004; Barak et al., 2014). Although their precise developmental role is poorly understood, TLRs undergo dynamic changes in expression during brain development (Kaul et al., 2012) and regulate neuroprogenitor cells (Barak et al., 2014). TLR and HMGB1 expression are increased in human developmental cerebral cortical dysplasia (Zurolo et al., 2011), consistent with involvement in cortical development. During maturation of rat PFC from late adolescence (P56) to adulthood (P80), there is an age-associated reduction in expression of immune-signaling receptors (TLR3, TLR4, and RAGE) that parallels the maturational loss of cholinergic and other neurotransmitter receptors (Vetreno and Crews, 2012; Vetreno et al., 2013). In contrast, HMGB1 shows a developmental increase in expression in PFC during maturation (Vetreno and Crews, 2012). There are also developmental increases and subunit changes in GABA and glutamate receptors that most likely reflect maturation of synapses, as discussed above. Interestingly, studies in mice find that microglia play an important role in maturation of brain synapses and function (Paolicelli et al., 2011; Paolicelli and Gross, 2011). Brain neuronal development involves overproduction of neurons and synapses that are later pruned, and elimination of nonintegrated neurons and silent synapses is associated with improved brain function (Paolicelli et al., 2011) and brain regional connectivity (Paolicelli and Gross, 2011).
Neuroimmune signals and HMGB1 activate microglia as well as release glutamate from astrocytes (Pedrazzi et al., 2006). Signaling between neurons, microglia, and astrocytes contributes to synaptic excitation (Fig. 2). Neuronal excitation can release HMGB1 from neurons, activating microglia, and astrocytes that in turn increase synaptic glutamate and other molecules to impact synaptic signaling. Moreover, alcohol activates microglia and astrocytes (Guerri and Pascual, 2010) through neuroimmune signaling, possibly via HMGB1 release from neurons (Zou and Crews, 2012). Postmortem brains of humans with alcohol-use disorder exhibited elevated microglial markers (He and Crews, 2007) and increased expression of HMGB1, TLR2, TLR3, and TLR4 (Crews et al., 2013), as well as proinflammatory cytokines and other neuroimmune-signaling molecules (Crews and Vetreno, 2016). A recent study reported that heavy binge-drinking adolescents have increased blood cytokines (Ward, et al., 2014). These results and others have led to the hypothesis that ethanol induces neuroimmune-signaling molecules and microglial activation, and that this induction in adolescence disrupts synaptic maturation.

Spreading proinflammatory signals across neurons and glia contributes to innate immune gene induction and hyperexcitability following AIE exposure. Left: Alcohol, glutamate, and other inflammagens cause the nuclear release of HMGB1 from neurons that cause microglia to become hyper-ramified, resulting in further release of HMGB1 and other proinflammatory signals. As a consequence, astrocytes reduce glutamate reuptake (Zou and Crews, 2005), thereby increasing extracellular glutamate levels that induce neuronal excitability, and leading to further release of HMGB1 in a positive feedback cycle. Right: Simplified schematic depicting innate immune induction of hyperexcitability. (1) Ethanol administration leads to neuronal release of HMGB1 into the extracellular space. (2) Extracellular HMGB1 binds to TLRs on microglia and RAGE, leading to the release of TNF-α and other innate immune signals. (3) TNF-α binds to TNF receptors on astrocytes, leading to glutamate sensitivity and reduced reuptake through glutamate transporters. (4) Increased glutamate in the synapse activates N-methyl D-aspartate receptor subtype 2B (NR2B), culminating in hyperexcitability. Neuronal hyperexcitability can contribute to alterations in neuronal connectivity as well as causing excitotoxicity. Figure adapted from (Crews et al., 2011).
In rats, AIE exposure increases HMGB1, TLR4, and RAGE expression compared with controls, and each of these signaling molecules remains elevated in abstinence and into adulthood (Vetreno and Crews, 2012; Vetreno et al., 2013, 2014). These studies are consistent with others indicating a vulnerability of the adolescent brain to AIE, producing long-lasting changes that persist into adulthood. Indeed, we found that expression of TLRs, RAGE, and HMGB1 was negatively correlated with behavioral flexibility; specifically, greater upregulation of innate immune receptor genes was associated with greater impairments in Barnes maze performance in adulthood (Vetreno et al., 2013). The persistence of innate immune gene induction most likely contributes to continuous neurodegeneration (discussed below), as well as to more specific insults to key neurotransmitter systems during adolescent maturation (Crews and Boettiger, 2009; Vetreno et al., 2014).
Although this review highlights HMGB1–TLR4 signaling, there are multiple other proinflammatory genes and proteins increased after AIE exposure in the rat, many of which we have also observed in postmortem brains of individuals with alcohol-use disorder. Our first human brain studies looked at microglia and the proinflammatory cytokine monocyte chemoattractant protein-1 (MCP-1; CC chemokine ligand 2), which is the cytokine induced most robustly by ethanol among those measured in brain slice cultures (Crews et al., 2006a; Zou and Crews, 2010). We found that postmortem brains from subjects with a history of alcohol-use disorder contain increased levels of MCP-1 protein and the microglial marker Iba-1 in hippocampus, ventral tegmental area, nucleus accumbens, and amygdala (He and Crews, 2007). In later studies, we focused on the OFC, a component of the PFC, and determined that postmortem alcoholic OFC has more expression of HMGB1 as well as TLRs and RAGE (Crews et al., 2013; Vetreno et al., 2013). We also observed increased interleukin (IL)-1B inflammasome markers in postmortem alcoholic hippocampus that could contribute to loss of neurogenesis (Zou and Crews, 2012). In addition, NADPH-oxidase is increased in human alcoholic OFC (Qin et al., 2013), consistent with increased oxidative stress, as found in the mouse brain after ethanol exposure (Qin et al., 2013). These findings show that neuroimmune-signaling pathways are upregulated in alcohol-use disorder, which may be an important aspect of the neurobiology of the disease (Fig. 3). Indeed, work from the Harris laboratory found that activation of the innate immune system increases alcohol consumption in mice (Blednov et al., 2011). Studies by multiple laboratories find that TLR, HMGB1, and other neuroimmune-signaling molecules are increased by alcohol and/or alter responses and preference for drinking alcohol, suggesting a bidirectional relationship between neuroimmune signaling and alcohol drinking.

Innate immune-signaling cascades and evidence for upregulation in brain following AIE exposure. A simplified schematic of the TLR and RAGE signaling cascades. Stimulation of TLRs and RAGE with their endogenous agonist HMGB1 and other inflammagens [e.g., lipopolysaccharide (LPS)] leads to the generation of proinflammatory oxidases and reactive oxygen species (ROS) and downstream activation of NF-κB. Nuclear translocation of NF-κB leads to the secretion of proinflammatory gene expression, innate immune gene induction, cell death, and addiction-like behaviors. AP-1, activator protein-1; CD14, cluster of differentiation 14; ERK, extracellular signal-regulated kinase; IKK, inhibitor of nuclear factor κ-B; JNK, c-Jun N-terminal kinases; MyD88, myeloid differentiation primary response gene 88; Src, proto-oncogene tyrosine-protein kinase; TIRAP, Toll/interleukin-1 receptor domain-containing adaptor protein.
As adolescent drinking is known to increase the risk of developing alcohol dependence during one’s lifetime, we investigated the relationship between alcohol drinking and neuroimmune gene expression across control and alcoholic postmortem brains (Vetreno et al., 2013). Interestingly, two forms of correlations were found linking neuroimmune gene expression to alcohol consumption and alcoholism. First, we found that HMGB1–TLR4 expression in OFC was negatively correlated with age of drinking onset—that is, expression was higher in individuals who initiated alcohol use early. Second, total lifetime alcohol consumption across groups was positively correlated with OFC expression of HMGB1, TLR4, TLR3, TLR2, and RAGE. This persistent relationship between cumulative alcohol use and HMGB1 and TLR gene induction in brain provides support to the hypothesis that alcohol-induced neuroimmune signaling results in long-term changes in brain function and neurodegeneration.
The critical role of neuroimmune gene induction in the persistent effects of adolescent alcohol exposure on neurobiology is stongly supported by Guerri’s studies in both rats (Pascual et al., 2014) and mice (Alfonso-Loeches and Guerri, 2011). AIE exposure in rodents insults PFC, hippocampus, cerebellum, white matter, as well as cognition and reward. Guerri’s laboratory finds that alcohol exposure increases neuroimmune protein expression, as assessed by both in vitro and in vivo methods. Guerri’s studies describe adolescent alcohol-induced changes in the dopaminergic system, white matter, and myelination, as well as synaptic and epigenetic factors, all of which may contribute to changes in adult alcohol reinforcement, anxiety, and cognition dysfunction, and other behaviors consistent with alcohol addiction (e.g., Pascual et al., 2007, 2009, 2012, 2014; Montesinos et al., 2015, 2016). Multiple studies have found that transgenic mice lacking TLR4 do not show adolescent brain neuroimmune gene induction following adolescent alcohol exposure (Montesinos et al., 2015, 2016; Alfonso-Loeches et al., 2016). Furthermore, these mice lacking TLR4 do not show the changes in anxiety, alcohol drinking, cognitive dysfunction, reduced myelination, glial activation, glutamate, and GABA receptor protein expression or epigenetic marker expression typically found following AIE treatment of control mice. Taken together, these studies support the hypotheses that the long-lasting pathology associated with adolescent alcohol abuse is linked to alcohol-induced neuroimmune activation and its resulting pathologic changes in brain.
XI. Brain Electroencephalography and Sleep
Brain function can be assessed using electroencephalography (EEG), an electrophysiological method that records the electrical activity across the brain to evaluate function. EEG rhythmic activity or event-related potentials (ERP) that measure brain responses to a specific sensory, motor, or cognitive event can be studied in both rats and humans to investigate how the brain processes sensory information (Handy, 2005). The P300 or P3 component of the ERP is an electrophysiological measure commonly studied in both humans and rats (Bauer and Hesselbrock, 1999; Porjesz et al., 2005; Ehlers and Criado, 2010). The P3 is a positive potential that occurs approximately 300 ms after unexpected and task-relevant sounds or lights (Gratton et al., 1988). In humans, the amplitude and latency of the visual P3 reduce across adolescence until stabilizing in early adulthood (Hill and Shen, 2002). Adolescent humans and rats have higher amplitude and longer auditory P3 latency compared with adults of their species (Polich et al., 1990; Ehlers and Criado, 2010). A low P3 amplitude in youth with a family history of alcoholism has been suggested to represent impaired inhibitory regulation or disinhibition, possibly due to a developmental delay (Hill and Shen, 2002; Bauer and Hesselbrock, 2003; Berman et al., 2006; Tremere and Pinaud, 2006). Studies of young adult southwestern California Native Americans with a history of adolescent binge drinking reported that low P3 amplitude was related to ethanol dependence (Criado and Ehlers, 2007; Ehlers et al., 2007). Similarly, rats exposed to AIE for 10 days (P30–P40) and assessed as adults 6–7 weeks after ethanol exposure display a reduced P3 ERP amplitude in the dorsal hippocampus (Criado and Ehlers, 2007; Ehlers et al., 2007). Adults tend to have increased ERP amplitude as compared with adolescents. The reduced hippocampal ERP amplitude following AIE exposure is consistent with disruption of hippocampal maturation of function (Ehlers and Criado, 2010). Additional studies are needed to determine how the lasting changes in ERP may be related to alterations in hippocampal neurogenesis, cholinergic signals, glutamate excitatory synapses, and/or other AIE-induced changes in adult hippocampus.
The effect of ethanol challenge on ERP responses in adult rats is also altered by AIE treatment. Similar to humans, adolescent rats (P32) have longer latency P3 components compared with adults. In rats, a dose-dependent increase in the latency of the P3 auditory ERP was observed after ethanol (1.5 and 3.0 g/kg) in both adolescents and adults. In adult rats (P99), the change in P3 latency due to ethanol challenge was smaller in rats with a history of AIE compared with age-matched controls not exposed to ethanol during adolescence (Ehlers et al., 2014a). These findings are consistent with other AIE findings supporting long-lasting decreases in adult response sensitivity to ethanol and retention of the adolescent phenotype (Ehlers et al., 2014a). These P3 ERP studies support the hypothesis that AIE alters brain information processing in adulthood, particularly after ethanol challenge, in a manner that reflects behavioral disinhibition and persistence of adolescent-like responses to ethanol.
The EEG also assesses rhythmic neural activity, with rhythmic activity divided into frequency bands known as alpha (8–15 Hz), beta (16–31 Hz), theta (4–7 Hz), and delta (<4 Hz) (Ehlers and Criado, 2010). Event-related oscillations (EROs) within and between different brain regions are thought to reflect neural networks and can provide insight into brain maturation in both humans and rodents (Ehlers et al., 2014b). Higher ERO energy and lower synchrony are found in adolescent humans and rats as compared with their adult counterparts. During early adolescence, humans have higher ERO energy in all frequency ranges (alpha, beta, theta, delta) across cortical regions compared with adults. Similarly, early adolescent rats have higher ERO energy in all frequency ranges in parietal cortex and in all frequencies except beta in frontal cortex as compared with adult rats. Early adolescent humans and rats also have lower synchrony within and across cortical regions (Ehlers et al., 2014b). EEG under wake and sleep conditions undergoes large changes in characteristic amplitude and frequency during adolescence. For example, EEG amplitude and frequency of the posterior alpha rhythm are increased in the adolescent brain. Slower waves in the waking EEG also decline across adolescence (Niedermeyer and Lopes da Silva, 1999; Ehlers and Criado, 2010). These findings are consistent with adolescent remodeling of the brain to increase brain regional connectivity, decrease ERO energy, and increase synchrony during maturation of local and regional neurocircuits in both rats and humans. Interestingly, the adult EEG theta response to acute ethanol following AIE was blunted in parietal cortex (Ehlers et al., 2013a). Thus, similar to the P3 ERP studies described above, adolescent waking EEG is less sensitive to ethanol than adult responses, and AIE blunts the sensitivity of waking EEG to ethanol challenge in adult rats.
The EEG has been used to study sleep in both rats and humans. EEG is used in sleep studies with other monitors of eye movements and muscle activity that divide sleep stages. A well-studied EEG pattern is the oscillatory theta rhythm of 6–10 Hz, which is prominent in the rat hippocampus, but is also observed in other cortical and subcortical brain structures. Hippocampal theta is observed during a variety of activities, including locomotion and active sniffing, as well as during rapid eye movement (REM) sleep. Theta rhythm in the hippocampus requires cholinergic-GABAergic circuits between the medial septal area and the hippocampus. Most sleep in humans is nonrapid eye movement (NREM or non-REM sleep), and theta disappears in NREM sleep. NREM sleep is divided into three stages, N1, N2, and N3, with the latter called delta sleep or slow-wave sleep (SWS). More SWS occurs earlier in the night, whereas REM sleep increases proportionally in the last cycles before natural awakening. The effects of alcohol on sleep have been studied extensively in adults (Roehrs and Roth, 2001). For example, chronic alcohol abuse in adults produces abnormal sleep patterns that are evident up to 2 years following the last use of alcohol (Drummond et al., 1998). Furthermore, during abstinence, EEG peak frequencies increase in individuals recovering from alcohol dependence (Irwin et al., 2000) with increases in REM sleep associated with relapse. Thus, stages of sleep EEG change during alcohol dependence and recovery.
During adolescence, sleep EEG follows a maturational trajectory. For example, waking delta and theta power decline by about 65% between early adolescence (e.g., ages 9–12) and 17 years of age. The maturational decreases in delta and theta sleep EEG are unrelated to pubertal maturation, but are strongly linked to age (Feinberg and Campbell, 2010). The age-related adolescent decline in EEG power is associated with an increase in brain regional interconnectivity and functional specialization of neural networks that underlie the cognitive improvements during maturation to adulthood (Quartz and Sejnowski, 1997; Tarokh et al., 2010). Acute ethanol challenge in naive adolescent rats alters subsequent sleep; for example, 20 hours after ethanol treatment during the rats’ next sleep cycle, ethanol withdrawal decreases SWS frequencies (1–4 Hz) more in adolescents than adults, suggesting that adolescents are more susceptible to hangover disruption of SWS (Ehlers et al., 2013a). AIE exposure followed by 5 weeks of abstinent maturation to adulthood also caused a significant reduction in episode duration and total amount of SWS in rats as compared with controls. According to spectral analysis, AIE significantly increases cortical peak frequencies in the 2–4 Hz, 4–6 Hz, and 6–8 Hz bands during SWS. These findings indicate that AIE exposure reduces adult SWS, consistent with the interpretation that AIE has altered brain maturation of the processes regulating sleep. Poor quality sleep is associated with family history of alcohol dependence, diagnoses of alcohol-use disorders or major depressive disorders across a lifetime, and acculturation stress. As mentioned above, EEG peak frequencies increase in alcohol-dependent individuals in recovery (Irwin et al., 2000), and increases in REM sleep may be an indicator of alcohol relapse (Irwin et al., 2009). Thus, changes in EEG during adolescent maturation as well as during alcohol dependence and recovery are consistent with EEG, providing insight into the mechanisms of brain maturation and the development of alcohol dependence.
Although the function of sleep is poorly understood, changes in sleep during maturation and in individuals with psychopathology have helped unravel some sleep-related mechanisms (Feinberg and Campbell, 2010). REM sleep is initiated by cholinergic neurons and inhibited by monoamines such as 5-HT (Brown et al., 2012). REM sleep has been referred to as paradoxical sleep because high-frequency EEG waves that are similar to a waking state occur, yet awakening an individual during REM is more difficult than any other sleep stage. The functions of sleep include links to increased clearance of metabolic waste products via the glymphatic system (Xie et al., 2013) as well as alterations in immune signaling. Sleep-deprived rats show a 20% decrease in white blood cell count and significant alterations in the immune system (Zager et al., 2007). Cytokines, such as IL-1 and tumor necrosis factor (TNF), play a role in the regulation of normal mammalian NREM. Electrophysiological, biochemical, and molecular genetic studies find that blocking IL-1 or TNF systems reduces spontaneous NREM sleep of healthy animals. Furthermore, antigenic challenge to the immune system increases brain IL-1 and TNF as well as NREM. Because sleep deprivation impairs immune function and immune challenge affects sleep, it has been hypothesized that sleep may be considered a component of the acute-phase response to infection and functions in host defense (Krueger and Majde, 1990; Opp, 2009). More recent studies have found that sleep alters monocyte–macrophage immune cell phenotypes, such as M1-proinflammatory macrophages or M2-trophic wound-healing macrophages (Hakim et al., 2014). For example, sleep deprivation reduces the healing of burns in rats (Gümüştekín et al., 2004) and enhances tumor growth in mice (Hakim et al., 2014). Depriving mice of sleep suppresses proinflammatory signals that promote tumor growth. Sleep deprivation shifted macrophages to M2 phenotypes with more TLR4. As discussed above, TLR4 molecules are signaling molecules for immune system activation that are also altered during brain development and by ethanol. Transgenic mice lacking TLR4 were resistant to the effects of sleep deprivation, consistent with sleep contributing to normal immune-signaling processes and overall health. In alcohol-dependent individuals, increased markers of inflammation coincide with more REM sleep, which is thought to predict alcohol relapse. Pharmacologic neutralization of TNF-α, a proinflammatory cytokine, significantly reduces REM sleep in abstinent alcohol-dependent subjects, linking circulating levels of TNF-α and REM sleep disruptions to the neuropathology of alcoholism (Irwin et al., 2009). Thus, innate immune signaling influences sleep cycles and maturation of sleep, and the enhanced innate immune signaling observed in adult rodents after AIE exposure may be one mechanism by which AIE disrupts adult sleep.
XII. Cholinergic System Development and Effects of AIE
Cholinergic neurons of the basal forebrain play a major regulatory role in learning and memory and are the primary source of cholinergic innervation to the hippocampus (Mesulam et al., 1983; Smith and Pang, 2005). They are generated early in embryonic development (Gould et al., 1989, 1991; Dinopoulos et al., 1992; Linke and Frotscher, 1993) and continue to undergo maturational consolidation of projections during adolescence (Matthews et al., 1974; Nadler et al., 1974; Zahalka et al., 1993). Cholinergic neurons begin to extend their axons toward the hippocampus during embryonic development (Linke and Frotscher, 1993), and axonal expression of acetylcholinesterase, the principal enzyme responsible for degrading acetylcholine, within the hippocampus increases through early to mid-adolescence (P21–P35) (Armstrong et al., 1987; Gould et al., 1991). Similarly, levels of choline acetyltransferase (ChAT), the enzyme responsible for acetylcholine synthesis, peak in hippocampus during early adolescence (about P28) and remain relatively stable until approximately P65, whereas activity of the high-affinity choline transporter was found to increase sharply during mid-adolescence (P40) and return to baseline levels at about P50 (Zahalka et al., 1993). Thus, basal forebrain cholinergic neurons have a developmental trajectory beginning in embryonic development that extends to dynamic maturational synaptic refinement during adolescence.
The NADIA Consortium has repeatedly found that basal forebrain cholinergic neurons are vulnerable to AIE exposure (Fig. 4). AIE causes a loss of ChAT-immunopositive neurons in the basal forebrain of both rats and mice that persists well into adulthood (Coleman et al., 2011; Ehlers et al., 2011; Vetreno et al., 2014). This effect appears to be somewhat selective for cholinergic neurons, as mouse basal forebrain parvalbumin-positive GABAergic neurons were not affected by AIE exposure (Coleman et al., 2011). AIE also reduces expression of the vesicular acetylcholine transporter, which transports cytosolic acetylcholine into synaptic vesicles for storage until release, in the adult basal forebrain (Vetreno et al., 2014), consistent with the loss of cholinergic neurons. In binge ethanol–exposed adolescent mice, the reduction in cholinergic expression in the basal forebrain is accompanied by downregulation of multiple muscarinic and nicotinic receptors (Coleman et al., 2011) (see Fig. 5). AIE-induced loss of ChAT expression is adolescent-specific because CIE treatment of adults (P70–P90) did not reduce ChAT (Vetreno et al., 2014). AIE exposure resulted in fewer ChAT plus immunoreactive (IR) neurons at late adolescence (P56) that persisted at similarly reduced levels into young adulthood (P80) and to older ages (P220) (Vetreno et al., 2014). Interestingly, exposure to endotoxin, a known neuroimmune activator, induced a similar decrease in ChAT plus IR, supporting the hypothesis that persistent AIE-induced neuroimmune activation (Vetreno and Crews, 2012, 2015; Vetreno et al., 2013) contributes to the loss of ChAT plus IR. Assessments of ChAT expression in postmortem alcoholic brain found a loss of both ChAT and the vesicular acetylcholine transporter, both markers of cholinergic neurons (Fig. 4) (Vetreno et al., 2014). Additional studies are needed to understand the role of cholinergic loss in alcoholism; however, given that human alcoholics tend to start drinking early in adolescence and adult rats exposed to AIE show similar deficits in ChAT expression, it is an intriguing possibility that these two phenomena may be related.

Alcohol disrupts the basal forebrain cholinergic system in rats and humans. Top: Simplified schematic of the cholinergic system of the brain. Animal studies have implicated the cholinergic system as important in a host of functions, including cognition and executive function, behavioral control, reward processes, and sleep. AIE exposure causes a reduction of ChAT + IR neurons, which synthesize acetylcholine, throughout cholinergic nuclei of the brain (Vetreno et al., 2014); this loss might contribute to persistent cognitive and emotive dysfunction in adulthood. Bottom left: Multi-site analysis of data from the NADIA Consortium reveals that AIE exposure causes a 35% reduction of ChAT + IR neurons in the adult basal forebrain. Bottom right: ChAT protein expression is reduced by 51% in the postmortem human alcoholic basal forebrain, relative to moderate drinking controls. Furthermore, protein expression of vesicular acetylcholine transporter, which packages acetylcholine into synaptic vesicles, is reduced by 30% in the postmortem human alcoholic basal forebrain, relative to moderate drinking controls. Multisite analysis was calculated by Dr. Margaret Burchinal from five independent data sets (unpublished data from Crews’ laboratory; Ehlers et al., 2011; Boutros et al., 2014; Vetreno et al., 2014). Data are presented as a mean ± S.E.M. *P < 0.05, **P < 0.01, relative to CON.

Adolescent binge ethanol exposure reduces cholinergic marker expression in the whole mouse brain. Adolescent mice received either water (CON) or ethanol (EtOH; 5.0 g/kg, i.g.) once per day for 10 consecutive days from P28 to P37. Alcohol treatment ended on P37. Shown in (A–D) are expression levels (mRNA) 1 day after the last AIE dose of ethanol and 50 days after the last dose in AIE animals (Coleman et al., 2011). Changes in controls represent maturation from adolescence to adulthood. (A) mRNA levels of ChAT, the acetylcholine-synthesizing enzyme, were reduced by 55% in adolescent mouse whole brain samples 24 hours after the conclusion of EtOH exposure (P38) as well as by 58% in adulthood (P88) compared with CON. (B) Comparison of ChAT immunohistochemistry revealed an 8% reduction of ChAT–immunopositive cells in the posterior basal forebrain of EtOH-treated adult mice, relative to CON. (C) mRNA expression of muscarinic acetylcholine receptor subtypes R1 and R5 was reduced 24 hours after the conclusion of EtOH exposure by 62% and 54%, respectively, which persisted into adulthood (R1: ↓45%; R5: ↓50%). (D) Similarly, mRNA expression of nicotinic acetylcholine receptor subtypes α4 and α7 was reduced by 30% and 56% at the conclusion of EtOH exposure, respectively, that persisted into adulthood (α4: ↓48%; α7: ↓54%). These data reveal that adolescent binge ethanol exposure leads to long-term alterations in the cholinergic system that might contribute to cognitive dysfunction in adulthood. Data are presented as mean ± S.E.M. *p < 0.05, relative to CON, and are adapted from Coleman et al. (2011).
XIII. Monoamine System Development and Effects of AIE
A. Dopamine
Adolescent behavior is characterized by impulsive and risky decision making, which can contribute to alcohol use. These behavioral characteristics are often attributed to specific maturational processes in the brain (Varlinskaya et al., 2013). A circuit of interest for these behaviors includes the ventral tegmental area, nucleus accumbens, and PFC, which are anatomically connected and play key roles in motivated behaviors (Berridge and Robinson, 1998; Schultz, 1998; Miller and Cohen, 2001; Wise, 2004; Goto and Grace, 2005; Watanabe and Sakagami, 2007). Notably, the flow of information through this circuit is clearly multidirectional, involves specific subregions of the PFC and accumbens, and is not completely understood. Studies suggest that signals of motivational significance first enter this circuit at the ventral tegmental area, which sends dopamine projections to the PFC and accumbens to trigger orienting and reward-seeking behavior (Bromberg-Martin et al., 2010). The PFC and nucleus accumbens also project to the ventral tegmental area (Sesack and Pickel, 1992; Williams and Goldman-Rakic, 1998; Frankle et al., 2006); for example, PFC stimulation can modulate dopamine neuron firing (Gariano and Groves, 1988; Svensson and Tung, 1989; Gao et al., 2007; Jo et al., 2013). The PFC additionally sends glutamatergic projections to the accumbens, where inputs are integrated into the direct and indirect pathways of the basal ganglia to produce motor output (such as reward seeking). Importantly, both the mesolimbic and mesocortical dopamine systems are changing during adolescence, but in different ways. Electrophysiology and microdialysis studies indicate that mesolimbic dopamine activity peaks during mid-to-late adolescence (approximately P45), whereas mesocortical dopamine activity appears to increase into adulthood. Specifically, the mesolimbic dopamine system, which is critical for reward-seeking and approach behaviors, exhibits peak activity during adolescence, with higher tonic dopamine levels and greater receptor expression during adolescence as compared with juvenile or adult stages (Andersen et al., 1997; Badanich et al., 2006; McCutcheon and Marinelli, 2009; Philpot et al., 2009). PFC regions involved in executive control (Blakemore and Robbins, 2012) that would moderate reward-seeking and approach behavior develop more slowly. During youth and adolescence, frontal lobe maturation begins with the primary motor cortex, whereas the PFC develops last (Gogtay et al., 2004). At the same time, adolescence is characterized by gradual increases in dopaminergic innervation to the PFC (Rosenberg and Lewis, 1995; Spear, 2000; Wahlstrom et al., 2010; Naneix et al., 2012) as well as changes in dopamine receptor expression in the PFC (Andersen et al., 2000; Naneix et al., 2012).
Many studies demonstrate alcohol-induced alteration of dopamine neurotransmission in adulthood, as acute alcohol increases the firing rate of dopamine neurons in the ventral tegmental area (e.g., Gessa et al., 1985) and increases both tonic and phasic release of dopamine in the accumbens (e.g., Imperato and Di Chiara, 1986; Robinson et al., 2009). Less is known about alcohol effects on dopamine in the medial PFC, although alcohol challenge can increase cortical dopamine concentrations (Schier et al., 2013) and alcohol-preferring P rats exhibit lower levels of medial PFC dopamine than Wistar rats (Engleman et al., 2006). Most of this research has been done in adults, with few studies measuring the effects of alcohol on dopamine during adolescence. Of note are microdialysis studies by Philpot and Kirstein showing that adolescent rats have higher basal dopamine levels in the accumbens and a greater dopamine increase to alcohol challenge than adults (Philpot and Kirstein, 2004; Philpot et al., 2009).
Emerging data also suggest that AIE has long-term consequences on dopamine function. In adulthood, tyrosine hydroxylase immunoreactivity was reduced in the prelimbic PFC after an extended AIE (P28–P53), and these rats also displayed a preference for risky choice (Boutros et al., 2014). In one study, microdialysis measurements of tonic dopamine in the accumbens demonstrated that repeated alcohol exposure during preadolescence and early adolescence decreased the ability of acute alcohol challenge to induce dopamine release in the nucleus accumbens (Philpot et al., 2009), whereas another study reported no difference in the effect of ethanol challenge after AIE, but an elevation in basal dopamine levels in the accumbens (Pascual et al., 2009). We recently reported that AIE during early to mid-adolescence (P25–P45) blunted the effect of an alcohol challenge to reduce the concentration of dopamine released per impulse in adulthood compared with controls (Shnitko et al., 2016). This finding suggests that AIE exposure results in larger phasic dopamine signals after an alcohol challenge, at least those phasic signals arising from burst firing of dopamine neurons. Consistent with this interpretation, rats that consumed alcohol during adolescence exhibited high-risk preference as adults as well as higher phasic dopamine release in the accumbens to the risky choice (Nasrallah et al., 2011). Moreover, this effect was specific to AIE, as a comparable adult ethanol exposure regimen did not alter risk preference (Schindler et al., 2014). Another dopamine-associated behavior that is altered by AIE is anhedonia, measured with intracranial self-stimulation. AIE-exposed rats did not differ from controls in reward current threshold at baseline, but were less likely to exhibit reward deficits (increased reward current thresholds) after a single or repeated alcohol challenge (Boutros et al., 2014).
Less is known about consequences of AIE on mesocortical dopamine systems. AIE induced downregulation of dopamine receptor expression in the medial PFC (Pascual et al., 2009), and preliminary data suggest that AIE impairs function of dopamine D1, but not D2-type, receptors in the same region (Trantham-Davidson et al., 2015). AIE impacts on mesocortical dopamine may be postsynaptic rather than presynaptic, as one study found that early to mid-adolescent ethanol exposure (P25–P45) did not alter the response of electrically-evoked dopamine release to an alcohol challenge (Shnitko et al., 2014). However, negative data can be difficult to interpret—it is possible that a later AIE exposure targeting the mid-to-late adolescent period during which the medial PFC matures might have a greater impact on mesocortical dopamine release, or it is possible that AIE alters some aspects of cortical dopamine release (e.g., tonic levels) other than impulse-dependent release. Indeed, there is much unknown about AIE alterations in both striatal and cortical dopamine function, including local regulation of dopamine release by D2 autoreceptors, cholinergic receptors, and glutamatergic receptors at dopamine terminals and in microcircuits involving interneurons.
In summary, AIE produces effects on dopamine-associated behavior and neurophysiology that persist into adulthood and may contribute to behavioral phenotypes such as risky choice and sensitivity to alcohol reward that can lead to excessive alcohol intake in adulthood.
B. 5-HT
5-HT is an important neuromodulatory neurotransmitter synthesized in the raphe nucleus. It is one of the first systems to develop in the mammalian brain (Rubenstein, 1998), as 5-HT–immunopositive neurons are generated during early embryonic development (Wallace and Lauder, 1983). Although studies describing serotonergic system development during adolescence are limited, the existent data suggest that this system continues to mature during adolescence, similar to other neurotransmitter systems. Levels of 5-HT within the central nervous system are at approximately 68% of adult values by P32 in Wistar rats (Loizou, 1972). Furthermore, cortical serotonergic synapses increase from birth into adolescence (P35) in the Wistar rat (Dori et al., 1996). The hippocampus, a target of serotonergic innervation from the raphe nucleus (Hensler, 2006), undergoes a modest developmental peak in expression of 5-HT terminals at approximately P50 (late adolescence) in rats (Xu et al., 2001). In humans, there is an age-associated decline in 5-HT1A autoreceptor expression in the human dorsal raphe from approximately age 18 through adulthood (Dillon et al., 1991). Thus, although the existing literature reports that serotonergic projections and receptors undergo maturational refinement across mammalian adolescence similar to other neurotransmitter systems, more detailed studies are needed.
The continued maturational refinement of the serotonergic system most likely increases the vulnerability of this system to the effects of adolescent binge drinking. Recent work from the Crews laboratory found that intragastric AIE exposure (single 5 g/kg dose on a 2-day-on/2-day-off schedule from P25 to P55, producing mean BECs of 0.18 g/dL) reduced 5-HT–immunoreactive cells in the dorsal, but not median, raphe nucleus of adult male Wistar rats. AIE also reduced 5-HT–immunoreactive terminal field densities by 38% in the hypothalamus and 20% in the amygdala (see Fig. 6). Another study in male Wistar rats, using low-dose, sole-source ethanol exposure (6.6% ethanol continuously for 6 weeks from ∼P45 to P87, producing mean BECs of 0.02 g/dL), found transient reductions of 5-HT immunoreactivity (∼30%) in the dorsal, but not median, raphe nucleus, that were no longer evident following a 10-week recovery period (Evrard et al., 2006). One interpretation of these findings is that a persistent loss of 5-HT neuronal markers requires binge alcohol exposure as modeled by AIE. In any case, these data indicate an earlier age of ethanol exposure and/or binge doses of ethanol, which model human adolescent binge drinking, is detrimental to the developing adolescent serotonergic system.

AIE reduces 5-HT neurons in dorsal raphe nucleus, leading to alterations in terminal field projection densities. Top: Simplified schematic of the serotonergic system of the brain. The serotonergic system innervates the entire brain and plays a neuromodulatory role in mood regulation, memory, behavioral control, and reward processes. Dysregulation of this system has been identified as an etiological factor underlying several psychiatric disorders, including depression, impulsivity, and alcohol dependence (Michelsen et al., 2007; Donaldson et al., 2013; Muller and Homberg, 2015; Nautiyal et al., 2015). Bottom: Adult rats with a history of AIE exposure (5.0 g/kg, i.g., 2 days on/2 days off from P25 to P55) exhibit a 20% reduction of 5-HT–immunoreactive neurons in the adult (P80) dorsal raphe nucleus (DRN), whereas those in the median raphe nucleus (MRN) are spared. Quantification of serotonergic terminal field densities revealed reductions of 20% and 38% from control (CON) in both the amygdala and hypothalamus, respectively. The loss of 5-HT + IR neurons might contribute to AIE-induced cognitive and emotive dysfunction as well as increased alcohol self-administration in adulthood. Data are presented as a mean ± S.E.M. *p < 0.05, **p < 0.01, relative to CON.
XIV. Hippocampal Development and Effects of AIE
The hippocampus is among the brain regions whose development has been studied across mammalian species, relating morphologic and physiologic maturation during adolescence (Gogtay et al., 2006; Hunsaker et al., 2014). In addition, hippocampal neurogenesis, wherein newborn neurons are formed and functionally integrated into the hippocampal circuits, is a unique process that continues into adulthood (Zhao et al., 2006, 2008) and has been implicated in hippocampal-mediated cognitive and emotive function (Madsen et al., 2003; McHugh et al., 2004). Relative to adults, adolescents have greater levels of hippocampal neurogenesis (He and Crews, 2007; Vetreno and Crews, 2015) that is associated with increased levels of neuroplasticity. In addition, there is concomitant refinement of hippocampal neurotransmitter innervation and receptor expression across adolescence. Early in adolescence, hippocampal expression of dopaminergic D1, D2, and D4 receptors increases several-fold until approximately P35, when levels stabilize and persist into adulthood in the rat (Tarazi and Baldessarini, 2000). Expression of the inhibitory GABAA receptor γ2 subunit undergoes a maturational decline beginning on P30 and progressing into adulthood in the rat (Yu et al., 2006; Centanni et al., 2014), whereas maturation of synaptic activity of the GABAB receptor occurs during mid-adolescence (i.e., P35–P45) (Nurse and Lacaille, 1999). In parallel, expression of the excitatory glutamatergic receptor NMDA undergoes substantial pruning during adolescence, as indicated by an approximate 25% reduction of NMDA receptors between P28 and P60 in rats (Insel et al., 1990). The hippocampus is also a target of serotonergic innervation from the raphe nucleus (Hensler, 2006), which is the principal source of 5-HT synthesis, and undergoes a modest late-adolescent peak (about P50) in the expression of 5-HT terminals in rats (Xu et al., 2001). Activity of ChAT, the enzyme responsible for acetylcholine synthesis, increases dramatically to P18 in the hippocampus, followed by general stability through adolescence, whereas activity of the high-affinity choline transporter increases sharply at approximately P40 to levels observed at birth, followed by a return to baseline levels at about P50 (Zahalka et al., 1993). In addition to hippocampal neurotransmitter systems, other neuromodulatory proteins undergo maturation during adolescence. Phosphorylated cAMP-response element-binding protein, which is the transcriptionally active form of the protein and critical for the induction of BDNF and other trophic factors, is expressed at high levels in the hippocampus early in postnatal development (P7) and at progressively lower levels during adolescence and adulthood (Toscano et al., 2003). Studies of human hippocampus GABAergic and synaptophysin, an integral synaptic vesicle protein, find increases between adolescence and adulthood that are consistent with changes in rat hippocampus (Eastwood et al., 2006; Hyde et al., 2011). Thus, the mammalian hippocampus undergoes extensive maturation during adolescence that is particularly sensitive to adolescent alcohol abuse or exposure-induced pathology.
The hippocampal dentate gyrus is one brain region known to form new neurons long into adulthood. Multiple studies have found that both acute and chronic alcohol inhibits hippocampal neurogenesis (e.g., Crews and Nixon, 2009). Multiple NADIA Consortium laboratories have found that AIE exposure, whether administered intragastrically, i.p., or through vapor inhalation, diminishes hippocampal neurogenesis, as discussed in detail below. AIE exposure also alters maturational refinement of neurotransmitter systems in the hippocampus. Indeed, adolescent (P30–P40), but not adult, binge ethanol exposure reduced tonic GABAA receptor-mediated inhibition in the adult rat hippocampus (Fleming et al., 2013). Similarly, Guerri and colleagues found that AIE exposure (P25–P38) in rats reduced expression of the dopamine D2 receptor and phosphorylated NR2B protein expression in the hippocampus 24 hours after the last ethanol administration (Pascual et al., 2009). In contrast, ethanol vapor exposure during postweaning and early adolescence (P23–P37) led to an increase in protein expression of the NR1 and NR2A subunit of the NMDA receptor 2 weeks following the conclusion of exposure in rats (Pian et al., 2010). In addition, AIE exposure leads to an adult reduction of BDNF in the hippocampus that was specific to the BDNF IV promoter (Sakharkar et al., 2016). Although the mechanism underlying the persistent loss of hippocampal neurogenesis remains to be fully elucidated, the data implicate a shift in the innate immune-neurotrophic balance. Binge-like AIE exposure upregulates innate immune genes in the adult hippocampus (Vetreno and Crews, 2015) while reducing expression of BDNF (Sakharkar et al., 2016). Interestingly, treatment with the histone deacetylase inhibitor trichostatin A reversed the AIE-induced reduction of BDNF and recovered the loss of neurogenesis (Sakharkar et al., 2016). Together, these data show that the maturing adolescent hippocampus is vulnerable to developmental modifications as a consequence of binge ethanol exposure.
A. Neurogenesis in Development and Adulthood
Neurogenesis is a conserved process observed in mammals (Kuhn et al., 1996; Gould et al., 1999), including humans (Eriksson et al., 1998). Although neurogenesis is primarily associated with prenatal and early postnatal development, this process continues to occur in the subventricular zone of the lateral ventricles and the hippocampus of adults (Altman and Das, 1965; Eriksson et al., 1998). Hippocampal neurogenesis is restricted to the mitotically active subgranular zone of the dentate gyrus (Abrous et al., 2005), and is modulated by internal and external factors, such as neurotransmitter systems (e.g., acetylcholine) (Cooper-Kuhn et al., 2004), enriched environments (Cotman and Berchtold, 2002), traumatic brain injuries and other pathologies (Richardson et al., 2007), and drugs of abuse (He et al., 2005). The generation and integration of nascent neurons into established hippocampal neural circuitry are thought to underlie adaptation to novelty (Kempermann, 2002) and contribute to both cognitive processes (e.g., Shors et al., 2001) and affective states (e.g., Malberg et al., 2000).
Although the role of neurogenesis in adolescent brain refinement and behavior has not been fully elucidated, neurogenesis is more pronounced in the adolescent hippocampus relative to adults (He and Crews, 2007; Vetreno and Crews, 2015) and is highly vulnerable to dysregulation by alcohol (Crews et al., 2006b). The increase in neurogenesis in the adolescent hippocampal dentate gyrus could reflect increased neuroplastic processes. Whereas the link between hippocampal neurogenesis and learning has not been fully explained (Leuner et al., 2006), data support the hypothesis that neurogenesis contributes to hippocampal-dependent cognitive processes (Shors et al., 2001; Kempermann, 2002). Animals exposed to environmental enrichment exhibit both increased learning skills and hippocampal neurogenesis (Kempermann et al., 1997), whereas neurogenesis inhibition impairs associative memory (Shors et al., 2001). An age-associated decline in hippocampal neurogenesis has been observed in humans (Spalding et al., 2013) and rodents (Kuhn et al., 1996; Broadwater et al., 2014b), which might contribute to age-associated cognitive decline (van Praag et al., 2005). The Crews laboratory observed that expression of the immature neuron marker doublecortin (Brown et al., 2003) was significantly decreased from the end of adolescence (P56) into adulthood (P220) throughout the dentate gyrus (Vetreno and Crews, 2015). Similarly, Broadwater et al. (2014b) found a similar age-associated reduction of neurogenesis from P74 to P116 in the hippocampal dentate gyrus. The reduction in neurogenesis with age could contribute to maturation of cognitive and emotive factors as well as the deficits observed in senescence. Models of depression in mice find reduced neurogenesis related to neuroimmune gene induction. These models show that antidepressants reverse both stress-induced inhibition of neurogenesis and depression-like behavior, linking antidepressant mechanisms to increased adult neurogenesis (Banasr and Duman, 2007; Iwata, et al., 2013). Indeed, reductions of plasticity and neurogenesis early in life may manifest as vulnerability to psychopathologies such as depression later in life (Klempin and Kempermann, 2007). Although the precise role of adult neurogenesis is complex and poorly understood, studies of hippocampal neurogenesis provide insight into hippocampal plasticity, neuronal health, and growth as well as psychologic and cognitive functions.
B. Long-Lasting Loss of Neurogenesis after AIE
As mentioned above, new neurons derived from neural stem cells are continuously produced in the hippocampal dentate gyrus (Altman and Das, 1965; Eriksson et al., 1998; Alvarez-Buylla and Garcia-Verdugo, 2002). As described above, adolescent hippocampal neurogenesis is far greater than adult neurogenesis, with neurogenesis declining with age across mammalian species (He and Crews, 2007; Chesnokova et al., 2016). The Crews laboratory and many others have found that both acute and chronic ethanol exposure reduces neurogenesis in the adult hippocampus (Jang et al., 2002a,b; Nixon and Crews, 2002; Herrera et al., 2003; He et al., 2005). Furthermore, ethanol exposure during adulthood blunts the growth of the progenitor’s dendritic arbor (He et al., 2005). Interestingly, adult hippocampal neurogenesis is resilient, recovering over a 30-day period from the 4-day binge alcohol model (Nixon and Crews, 2002) and a 7-week chronic, relapsing model of alcohol dependence (Hansson et al., 2010). In contrast to adult recovery from chronic ethanol inhibition of hippocampal neurogenesis during withdrawal and abstinence, AIE exposure causes a persistent loss of hippocampal neurogenesis (Ehlers et al., 2013b; Broadwater et al., 2014b; Sakharkar et al., 2016; Vetreno et al., 2016b). For example, Vetreno et al. (2015) found that AIE exposure in rats led to reduced neurogenesis (i.e., loss of doublecortin-immunoreactive neurons) in late adolescence (P56) that persisted through to adulthood (P220) in both dorsal and ventral dentate gyrus of the hippocampus (Fig. 7). AIE-induced loss of neurogenesis most likely occurs due to both reduced neuroprogenitor proliferation and increases in cell death. AIE in rats results in loss of cells immunopositive for Ki-67, a marker of proliferating cells (Ehlers et al., 2013b; Broadwater et al., 2014b; Sakharkar et al., 2016; Vetreno et al., 2015>), and similar decreases are reported in nonhuman primate models of AIE (Taffe et al., 2010). Furthermore, AIE in rodents increases in hippocampal dentate gyrus expression of the cell death markers cleaved caspase-3 (Broadwater et al., 2014b; Vetreno and Crews, 2015) and FluoroJade (Ehlers et al., 2013b), consistent with loss of neurogenesis due to decreased progenitor proliferation and increased death. The rodent subventricular zone has a neurogenic region along the lateral ventricles that also forms oligodendroglia progenitors, and preliminary studies suggest that AIE reduces subventricular zone progenitors (unpublished data). Broadwater et al. (2014b) found that the persistent loss of neurogenesis was specific to binge ethanol exposure during adolescence and not adulthood, indicative of a unique vulnerability of the adolescent hippocampal dentate gyrus to the neurotoxic effects of alcohol. AIE exposure in nonhuman primates also causes persistent reductions of neuroprogenitor markers NeuroD and polysialylated neural cell adhesion molecule in the hippocampal dentate gyrus as well as increased FluoroJade markers of cell death (Taffe et al., 2010). Thus, binge levels of alcohol exposure in adolescents and adults reduce new neuron formation in hippocampal dentate gyrus. Whereas adult hippocampal neurogenesis recovers from alcohol with weeks of abstinence, adolescent intermittent binge-drinking models repeatedly find a loss of neurogenesis that persists long into adulthood. As the adolescent brain is uniquely sensitive to alcohol neurotoxicity (Crews et al., 2007), decreased adult neurogenesis could contribute to increased risks of adult psychopathology and cognitive dysfunction.
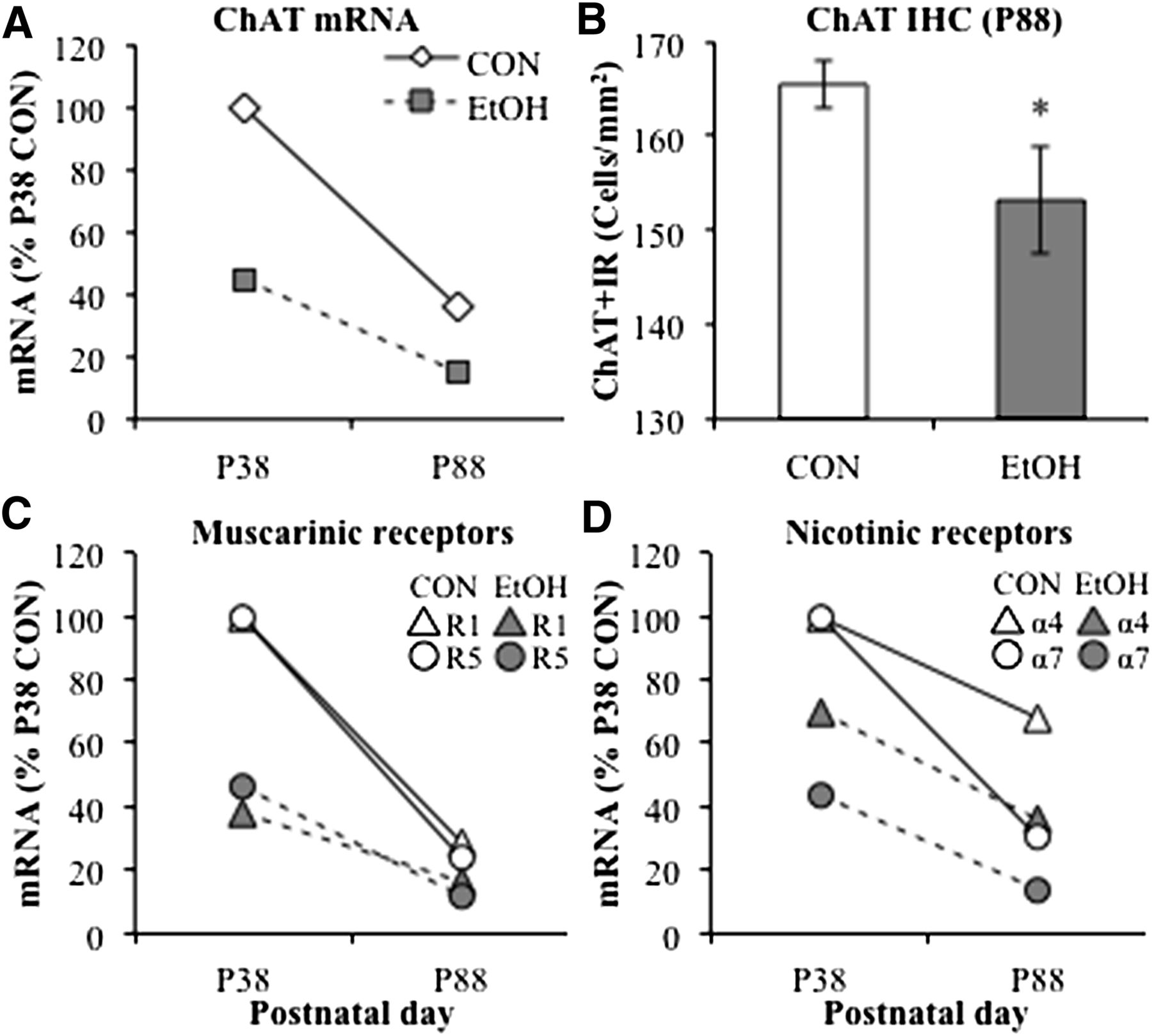
Hippocampal neurogenesis is highly vulnerable to the neurodegenerative effects of adolescent binge ethanol exposure. Representative photomicrographs of doublecortin (DCX) immunoreactivity, a neuroprogenitor microtubule-associated protein expressed by immature neurons, in the adult dorsal and ventral hippocampal dentate gyrus following control (CON) and AIE (5.0 g.kg, i.g., 2 days on/2 days off from P25 to P55). Scale bars, 100 μm. The middle bar graph depicts multisite analyses of data from the NADIA Consortium. In adulthood (e.g., P80) following AIE, DCX + immunoreactive (+IR) cells are reduced by 36%, which is accompanied by a concomitant 25% reduction in Ki-67 + IR, which is an endogenous marker of progenitor cells (Vetreno and Crews, 2015). In parallel, cleaved caspase-3, which is a marker of cell death, was increased by 31% in the adult hippocampal dentate gyrus of AIE-exposed animals. These data reveal that AIE leads to long-term reductions of hippocampal neurogenesis that could contribute to cognitive deficits in adulthood. Multisite analysis was calculated by Dr. Margaret Burchinal from four independent data sets (Ehlers et al., 2013b; Broadwater et al., 2014b; Swartzwelder et al., 2015; Vetreno et al., 2015). Data are presented as a mean ± S.E.M. **p < 0.01, relative to CON.
Although animal models have consistently found that AIE reduces neurogenesis within the adult hippocampal dentate gyrus, the underlying mechanism remains to be illuminated. However, studies from the Crews laboratory using hippocampal-entorhinal cortex (HEC) slice culture suggest that upregulation of the innate immune system contributes to the detrimental effects of ethanol on neurogenesis. Specifically, 100 mM ethanol applied to HEC slices for 4 days diminished expression of neurogenesis markers, including 5-bromo-2-deoxyuridine (an S-phase cell cycle mitotic marker), Ki-67, and doublecortin, that was paralleled by elevated expression of cytokines and the inflammasome IL-1β/IL-18 complex (Zou and Crews, 2012). In addition, HEC slices treated with either the proinflammatory cytokine IL-1β or anti-inflammatory drugs decreased and increased doublecortin expression, respectively. In the AIE model, Vetreno and Crews (2015) found a long-term upregulation of proinflammatory cytokines (i.e., TNF-α, MCP-1, and HMGB1), TLR4, the TLR4 adaptor protein CD14, and nuclear factor κ-light-chain enhancer of activated B cells (NF-κB) p65. NF-κB p65 mediates nuclear translocation of NF-κB, where it modulates the generation of proinflammatory cytokines. The subsequent release of proinflammatory cytokines induces further synthesis and activation of NF-κB, thereby providing support for the generation of positive-feedback loops of innate immune activation (Crews et al., 2011). Although the mechanism of innate immune-induced reductions of hippocampal neurogenesis in the AIE model remains to be fully understood, Rolls et al. (2007) reported that neurogenesis levels are increased in TLR4 transgenic knockout mice. Furthermore, reduced levels of neurogenesis were found in hippocampal precursor cell cultures treated with either recombinant IL-6 or TNF-α (Monje et al., 2003). Treatment with the TLR4 agonist lipopolysaccharide, which causes proinflammatory innate immune gene induction, reduced hippocampal neurogenesis comparable to adult animals exposed to AIE (Vetreno and Crews, 2015). In vitro studies of neurogenesis in hippocampal slice cultures found inhibition of ethanol induction of IL-1β and inflammasome proteins; for example, NLRP, a protein involved in IL-1β synthesis, reduced ethanol inhibition of neurogenesis (Zou and Crews, 2014). Increased inflammasome proteins and IL-1B were also found in the postmortem hippocampus of individuals with alcohol-use disorder (Zou and Crews, 2014). Although a direct mechanistic relationship between innate immune genes and loss of neurogenesis remains to be established in the AIE model, findings from the NADIA Consortium of persistent upregulation of hippocampal innate immune markers, coupled with data implicating innate immune involvement in the loss of neurogenesis (Monje et al., 2003; Crews et al., 2006b; Rolls et al., 2007; Zou and Crews, 2012), support the hypothesis that the innate immune system contributes to the AIE-induced loss of neurogenesis.
Although induction of innate immunity in adolescent binge drinking models most likely contributes to the observed loss of neurogenesis, AIE-induced loss of cholinergic inputs to the hippocampus might also contribute to the reductions of hippocampal neurogenesis. Basal forebrain cholinergic neurons regulate hippocampal neurogenesis through direct projections to neural progenitor cells. Indeed, specific lesions of cholinergic neurons and cholinergic agonists inhibit and facilitate neurogenesis, respectively (Kaneko et al., 2006; Van Kampen and Eckman, 2010). The NADIA Consortium has consistently reported reductions of cholinergic cell markers in the rodent basal forebrain (Coleman et al., 2011; Ehlers et al., 2011; Vetreno et al., 2014), an effect that was replicated in the postmortem basal forebrain of individuals with alcohol-use disorder (Vetreno et al., 2014). Intriguingly, cholinergic neurons of the basal forebrain also regulate the innate immune system (Su et al., 2010; Zhou et al., 2011; Sitapara et al., 2014), as studies employing specific lesions of cholinergic neurons find increased innate immune gene induction and microglial activation in hippocampus, whereas administration of cholinergic agonists reduces proinflammatory innate immune gene induction (Su et al., 2007, 2010; Lim et al., 2011; Field et al., 2012). Although further research is needed to fully elucidate the anti-inflammatory effects of the basal forebrain cholinergic system, animal studies suggest that these are mediated, in part, through activation of the α-7 nicotine receptor on microglia that reduce HMGB1 release (Sitapara et al., 2014). Thus, it is likely that both reduced cholinergic innervation and induction of innate immunity in the hippocampus play mechanistic roles in the long-lasting AIE-induced reduction of neurogenesis.
As outlined above, neurogenesis in the adolescent hippocampus is especially vulnerable to the deleterious effects of alcohol (Crews et al., 2006a). Multiple models of AIE have produced reductions in hippocampal neurogenesis (Crews et al., 2006b; Ehlers et al., 2013b; Broadwater et al., 2014b), which is consistent with reports of enhanced sensitivity of hippocampal-dependent behaviors to ethanol during adolescence (White and Swartzwelder, 2004). Reductions in neurogenesis were found to be associated with more disinhibitory behavior in the open-field conflict test at 2 and 8 weeks following termination of AIE vapor exposure (Ehlers et al., 2013b). Vetreno and Crews (2015) found that AIE-induced loss of doublecortin-immunopositive cells in the dorsal hippocampal dentate gyrus was positively correlated with performance on the novel object recognition task, such that lower expression levels of doublecortin were associated with diminished object recognition memory. Thus, the AIE-induced loss of hippocampal neurogenesis persists into adulthood and is unique to adolescent exposure. A variety of adult cognitive and affective changes could result from the AIE-induced loss of neurogenesis.
XV. Adolescent Alcohol-Induced Epigenetic Alterations in Gene Expression
Epigenetics is an emerging area in neuroscience focused on processes such as methylation and acetylation that change gene transcription without changing the DNA sequence, leading to modifications that can be transmitted to daughter cells. Recent findings indicate that epigenetic mechanisms contribute to alcohol and other addictions (Renthal and Nestler, 2008; Krishnan et al., 2014). For example, recent studies indicate epigenetic inheritance through the male germline during alcohol exposure (Rachdaoui and Sarkar, 2014). Epigenetic modifications, such as histone and DNA methylation and acetylation, contribute to both neurodevelopment directly and the influence of environment on neurodevelopment (Kofink et al., 2013; Szyf, 2013). Gene expression can be dynamically changed by histone acetylation through histone acetyltransferase and histone deacetylase (Kalkhoven, 2004; Lilja et al., 2013; Valor et al., 2013; Sheikh, 2014; Swaminathan et al., 2014). Acetylation of lysine by histone H3 alters transcription and has been implicated in ethanol-induced alterations to synaptic plasticity that contribute to anxiety and alcohol self-administration (Pascual et al., 2012; Moonat et al., 2013; Krishnan et al., 2014; Sakharkar et al., 2014). Notably, epigenetic mechanisms can impact both neurons and glia.
Using various adult animal models, the Pandey laboratory found that ethanol alters expression of the neurotrophic factor BDNF and neuropeptide Y, whose expression is mediated by histone H3 acetylation through histone deacetylase and cAMP-response element-binding protein in the amygdala (Pandey et al., 2008; Sakharkar et al., 2012, 2014; Moonat et al., 2013). In adult rats, chronic ethanol exposure also alters expression of BDNF in the hippocampus (Tapia-Arancibia et al., 2001; Hauser et al., 2011). Adolescent binge-like exposure to ethanol has also been found to reduce hippocampal expression of BDNF and neurogenesis markers that is reversed by a BDNF agonist (Briones and Woods, 2013). Hippocampal BDNF is thought to play a regulatory role in synaptic plasticity and neurogenic processes, and aberrations in these biologic processes have been implicated in neurologic disorders (Duman, 2004; Duman and Monteggia, 2006; Taliaz et al., 2010; Andero et al., 2014; Schoenfeld and Cameron, 2015). The decrease in BDNF expression is complex, but appears to be due to epigenetic mechanisms that reduce BDNF expression in neurons. AIE exposure reduces phosphorylated cAMP-response element-binding protein plus IR, consistent with reduced BDNF transcription (Pandey et al., 2015) and with studies finding that alcohol reduces cAMP response element-binding protein and increases NF-κB through complex signaling mechanisms (Zou and Crews, 2006). AIE also reduced protein levels of the epigenetic marker H3-K9Ac in hippocampus in association with decreased BDNF in CA1, CA2, and CA3 regions, but not in the dentate gyrus (Sakharkar et al., 2016). Thus, the loss of dentate neurogenesis may be associated with the AIE-induced increased neuroimmune signals in the dentate gyrus rather than reduced BDNF. However, neuroimmune signaling and BDNF expression interact in a reciprocal manner, such that epigenetic reductions in BDNF might contribute to increased neuroimmune gene expression, or more likely, increased neuroimmune signaling might contribute to epigenetic decreases in BDNF expression (Fig. 8). The complex effects of AIE on neuroimmune signaling and trophic factor expression require additional research to define how these signaling mechanisms contribute to the lasting effects of adolescent binge-like alcohol exposure.

Duality of stressors and enrichment on neuronal–glial signaling. Neuronal–glial communication lies along a continuum with devitalization-malaise on one end and vigor-endurance on the other end. Stimuli such as alcohol, stress, and endotoxins activate neurons and glia to release proinflammatory signals. As a consequence of innate immunity-system activation, neurotrophic support is reduced, leading to neurotransmitter system disruption as well as increased anxiety and cognitive dysfunction. In contrast, stimuli such as exercise and enrichment induce neuronal–glial communication to increase neurotrophin expression, such as BDNF, that blunts the innate immune system, creating an environment that facilitates neurotransmitter system survival and increases mood and cognition.
XVI. Conclusions
Accumulating evidence indicates that adolescence is a unique developmental period of malleable brain and body maturation that includes puberty, socialization, improvements in abilities, and the transition to independence. Neurodevelopmental programs in early life and adolescence are uniquely responsive to activities, both enriching and dampening. Across species, adolescent emergence of sexual identity and puberty are developmental processes controlled by brain maturation. Also across species, adolescent characteristic risk taking, thrill seeking, and peer social motivations subside with brain frontal cortical development, myelin growth, and increased functional connectivity. Frontal cortical maturation coincides with the emergence of prefrontal executive functions that underlie the maturation of adult self-control and reflection on the future consequences of actions. Cortical maturation includes both within-cortical refinements as well as increased frontal cortical connectivity to neuronal networks across the brain. Increasing myelination and brain regional connectivity during adolescence most likely contribute to long-lasting adult motivation, planning, reflecting on consequences of actions, and successful independence within society.
The immature adolescent brain has unique responses to alcohol and possibly other drugs of abuse. Adolescents have a low sensitivity to alcohol-induced motor incoordination and sedative/hypnotic responses. This low physical sensitivity coupled with thrill seeking and peer/social factors can promote extreme binge drinking and very high blood alcohol levels. Adolescents are more sensitive to alcohol disruption of cognition than adults, further increasing risks of accidents, heavy binge drinking, and unwanted consequences. Blackouts are common among adolescents, consistent with heavy binge drinking. Compared with adults, the developing adolescent cortex is more sensitive to alcohol-induced neuronal death, and adolescent hippocampal neurogenesis is more sensitive to alcohol toxicity. Immature brain connections and synapses most likely contribute to the adolescent low-sedative sensitivity, increased neurotoxic sensitivity, and cognitive disruption to ethanol.
The association of an early age of drinking onset with lifelong risks of alcoholism and alcohol-related violence and trauma could be due to neurodevelopmental insult due to high levels of alcohol exposure. Alternatively, it could represent individuals with emerging mental disorders, genetic, and/or other factors leading to early onset drinking as an identifier of those of high innate risks of developing alcohol dependence. The relative contributions of these intertwined factors are confounded in human studies. For example, it is difficult to untangle inherited genetic factors in families with a history of alcohol dependence from the increased availability of alcohol, expectations of positive responses to alcohol drinking, poor parental interaction, and sibling–peer factors in families of alcoholics that encourage and facilitate drinking alcohol. Studies of adopted twins were needed to clearly establish a genetic component that is now accepted to be between 40 and 60% of the risks of alcohol dependence. Likely individual responses vary due to a diversity of innate predisposing and protective factors that are influenced by familial–environmental circumstances that combine in a dynamic manner to influence the trajectory of maturation of brain and behavior to adult abilities and character. A clear understanding of the impact of adolescent alcohol abuse on brain development and function also requires preclinical studies that focus on the impact of alcohol while keeping genetic and other influencing factors constant. The nature of adolescent drinking (e.g., periodic extreme binge drinking, followed by periods of abstinence) differs from drinking patterns of most adult drinkers and alcohol-dependent individuals. AIE models of adolescent drinking support the hypothesis that adolescent binge drinking changes adult brain structure and function. Preclinical controlled studies provide insight to human investigators in the hope that focused specific hypotheses based on the preclinical discoveries will be more easily confirmed than elucidated de novo in human studies.
AIE models have found long-lasting changes in adult brain (Fig. 9). Two persistent molecular changes in adult brain following AIE are increased neuroimmune gene expression and reduced expression of the neuronal trophic factor BDNF. A reciprocal, complex relationship between BDNF and neuroimmune genes most likely involves neuronal–glial signaling. Current studies of neuroimmune gene induction have focused on PFC and hippocampus, whereas BDNF has been investigated primarily in amygdala, but more recently in hippocampus. In hippocampus, parallel but distinct studies find that reversing the AIE-induced loss of BDNF or prevention of the AIE induction of neuroimmune genes can prevent and/or reverse the AIE-induced loss of adult neurogenesis. The AIE-induced reduction in adult BDNF is linked to epigenetic acetylation–methylation changes in neuronal BDNF, whereas microglial priming is linked to persistent increases in brain neuroimmune gene expression. Microglial and neuronal signaling changes lead to a loss of trophic resilience combined with increasing innate immune signals that can be toxic. Microglia are involved in innate immune gene induction by alcohol that includes cytokines, chemokines, HMGB1, TLRs, and other signaling molecules. Microglia respond to and regulate synapses, particularly during development. Innate immune signaling clearly contributes to brain development; however, it is poorly understood. Microglial signaling between neurons, astrocytes, oligodendrocytes, and other microglia is unique to brain, although the signaling molecules are common to other tissues and cells. Induction of innate immune-signaling molecules by AIE persists long into abstinent adulthood, perhaps for life. Interestingly, multiple innate immune-signaling proteins are increased in postmortem human alcoholic PFC and positively correlate with age of drinking onset. Supporting a role of innate immune gene activation are studies finding that endotoxin treatment of rats, a known activator of innate immune signaling, mimics AIE-induced changes in brain. Additional studies are needed to understand how trophic signals or microglial sensitization mechanisms vary across brain regions; however, emerging studies suggest that anti-inflammatory agents and reversal of epigenetic suppression of BDNF can also reverse AIE-induced neuropathology. Thus, neuroimmune-trophic balance mechanisms appear to underlie AIE-induced adult molecular neuropathology.
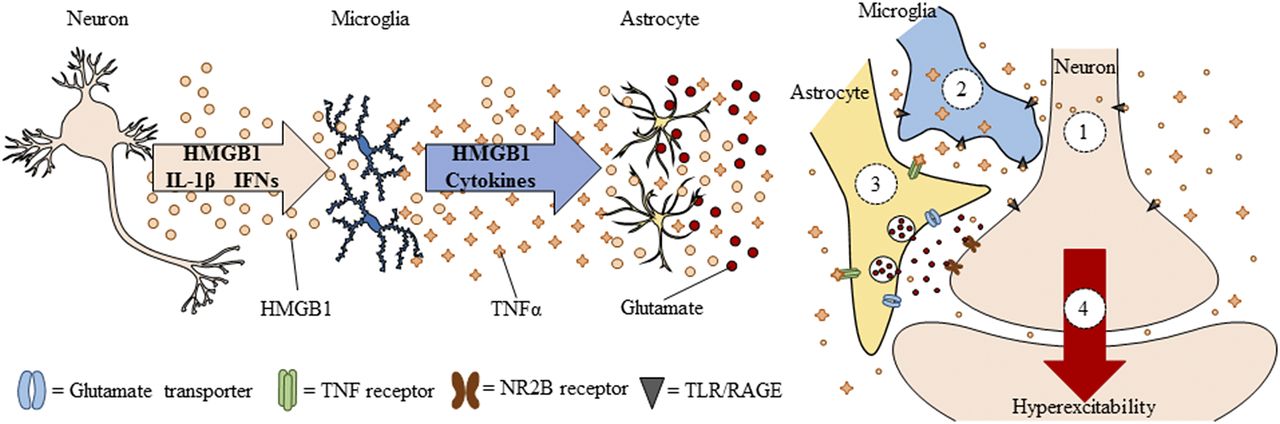
Schematic summary of preclinical findings on the lasting consequences of adolescent binge drinking in adulthood. Shown are the summarized, consensus findings of persistent adult pathologies from across the NADIA Consortium as well as other investigators, as described in this review. Multiple studies find that AIE leads to adult impairments in cognitive and executive functioning, increases depressive- and anxiety-like behaviors as well impulsivity, and increases alcohol self-administration in adulthood. Potential mechanisms of AIE disruption of maturation include increased expression of brain cytokines and other innate immune genes, loss of cholinergic and serotonergic neurons, and epigenetic changes that continue into adulthood following AIE treatment. Many of these changes are not found after similar treatment of adults, as outlined in the review. Additional studies are needed to clearly link mechanisms to adult behavioral pathologies; however, the persistent changes found are consistent with human studies, indicating that adolescent binge drinking is associated with lifelong risks for alcohol-related problems.
Evidence indicates that brain structure continues to change across adolescence in parallel with maturation of adult characteristics. AIE changes adult global brain morphology, causing subtle and diffuse degeneration-like changes that are consistent with the diffuse neurodegeneration found in adult alcoholism. More robust evidence indicates that AIE alters specific neuronal subtypes and that these effects can last into adulthood. In some cases, AIE-induced structural changes have been linked to neuroimmune gene induction or reduced BDNF. Hippocampal neurogenesis and forebrain cholinergic neurons are persistently decreased by AIE, but not by comparable adult treatment, consistent with high adolescent sensitivity to binge alcohol exposure. Serotonergic neurons and dopaminergic neurons are also sensitive to AIE, and alterations in these broadly projecting regulatory neurotransmitter systems would be expected to influence many adult brain functions. AIE also changes adult synaptic inhibitory and excitatory synaptic responses and synaptic spine morphology, microscopic changes that would alter local and regional neuronal connectivity and function. These and other to-be-discovered AIE-induced structural changes in adult brain are likely to underlie the impact of AIE on adult characteristics and alcohol responses.
Adolescent alcohol exposure also causes long-lasting changes in adult brain function and responses to alcohol, many of which appear to be retention of certain adolescent-like characteristics (Fig. 9). Indeed, physiologic findings from a large number of studies and multiple endpoints support the hypothesis that AIE causes a lock-in or retention of some adolescent characteristics into adulthood. Both hippocampal slice and whole rat brain electrophysiological studies suggest that AIE causes a persistence of adolescent-like physiology in adults as well as a retention of adolescent-like behavioral responses to an ethanol challenge. AIE-induced changes in adult P3 ERP, waking EEG, and sleep EEG are consistent with a persistence of adolescent-like brain information processing and brain regional connectivity. AIE-induced changes in hippocampal tonic current are consistent with alterations in inhibitory synaptic maturation and altered adult excitatory/inhibitory synaptic balance. Persistent adolescent-like behavioral characteristics found in adult rodents after AIE include increased risk taking, increased reward seeking, and enhanced disinhibition. AIE-induced changes in adult responses to an alcohol challenge also suggest a persistence of adolescent-like responses, including lessened sensitivity to aversive response, anxiolytic response, and withdrawal from ethanol as well as increased sensitivity to ethanol-induced social facilitation, rewarding effects, and memory impairment. Another adolescent-like characteristic is high alcohol consumption, and AIE increases adult alcohol self-administration and preference for alcohol. Although there are many adolescent-like characteristics that have not been tested after AIE exposure, the adolescent-like characteristics discussed above that persist in adulthood after AIE exposure converge as risk factors for the development of adult alcoholism, consistent with the hypothesis that an early age of drinking onset blunts maturational mechanisms and increases risks.
The maturation of adult characteristics parallels frontal cortical development of executive functions. Executive functions reduce impulsivity and are needed to adjust and/or extinguish habitual responses when rewards or circumstances change. Multiple studies of various endpoints find no effect of AIE on initial learning in adults, but deficits in behavioral flexibility when reward contexts are altered, suggestive of a loss of frontal cortical control. Together, these studies find that AIE can cause molecular, structural, and functional changes in brain that increase risks for the development of adult alcohol dependence and a persistence of adolescent-like characteristics in adulthood. These findings support the proposal that eliminating adolescent alcohol abuse could significantly improve adult brain health as well as possibly decrease risks for adult alcoholism and alcohol dependence. Continuing investigations into the mechanisms of the persistent changes associated with adolescent binge drinking will allow better prevention and treatment of risks that contribute to adult neuropathology and the development of alcoholism.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Crews, Vetreno, Broadwater, Robinson.
Footnotes
This work was supported in part by the National Institutes of Health, National Institute on Alcoholism and Alcohol Abuse [Grants AA019767, AA11605, AA007573, and AA021040], Neurobiology of Adolescent Drinking in Adulthood Consortium [Grants AA020023 and AA020024], and National Institute on Alcoholism and Alcohol Abuse Training Grant AA007573.
Abbreviations
- AIE
- adolescent intermittent ethanol
- BDNF
- brain-derived neurotrophic factor
- BEC
- blood ethanol concentration
- ChAT
- choline acetyltransferase
- CIE
- chronic intermittent ethanol
- EEG
- electroencephalography
- EPM
- elevated plus maze
- ERO
- event-related oscillation
- ERP
- event-related potential
- GABA
- γ-aminobutyric acid
- HEC
- hippocampal-entorhinal cortex
- HMGB1
- high-mobility group protein B1
- 5-HT
- serotonin
- IL
- interleukin
- IR
- immunoreactive
- LTP
- long-term potentiation
- MCP-1
- monocyte chemoattractant protein-1
- NADIA
- Neurobiology of Adolescent Drinking in Adulthood
- NF-κB
- nuclear factor κ-light-chain enhancer of activated B cells
- NMDA
- N-methyl-D-aspartate
- NREM
- nonrapid eye movement
- OFC
- orbitofrontal cortex
- P
- postnatal day
- PFC
- prefrontal cortex
- RAGE
- receptor for advanced glycation end products
- REM
- rapid eye movement
- SWS
- slow-wave sleep
- TLR
- Toll-like receptor
- TNF
- tumor necrosis factor
- Copyright © 2016 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
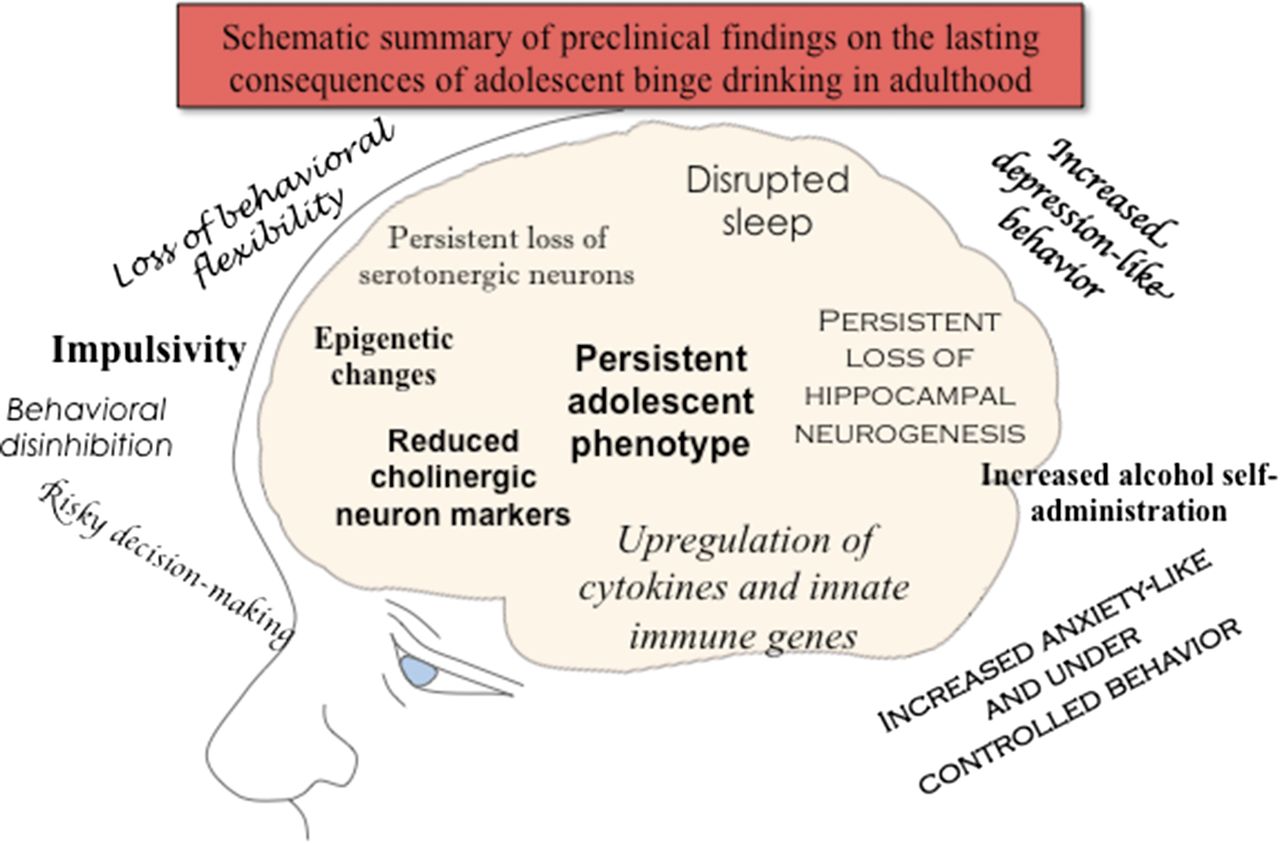{kind=link}
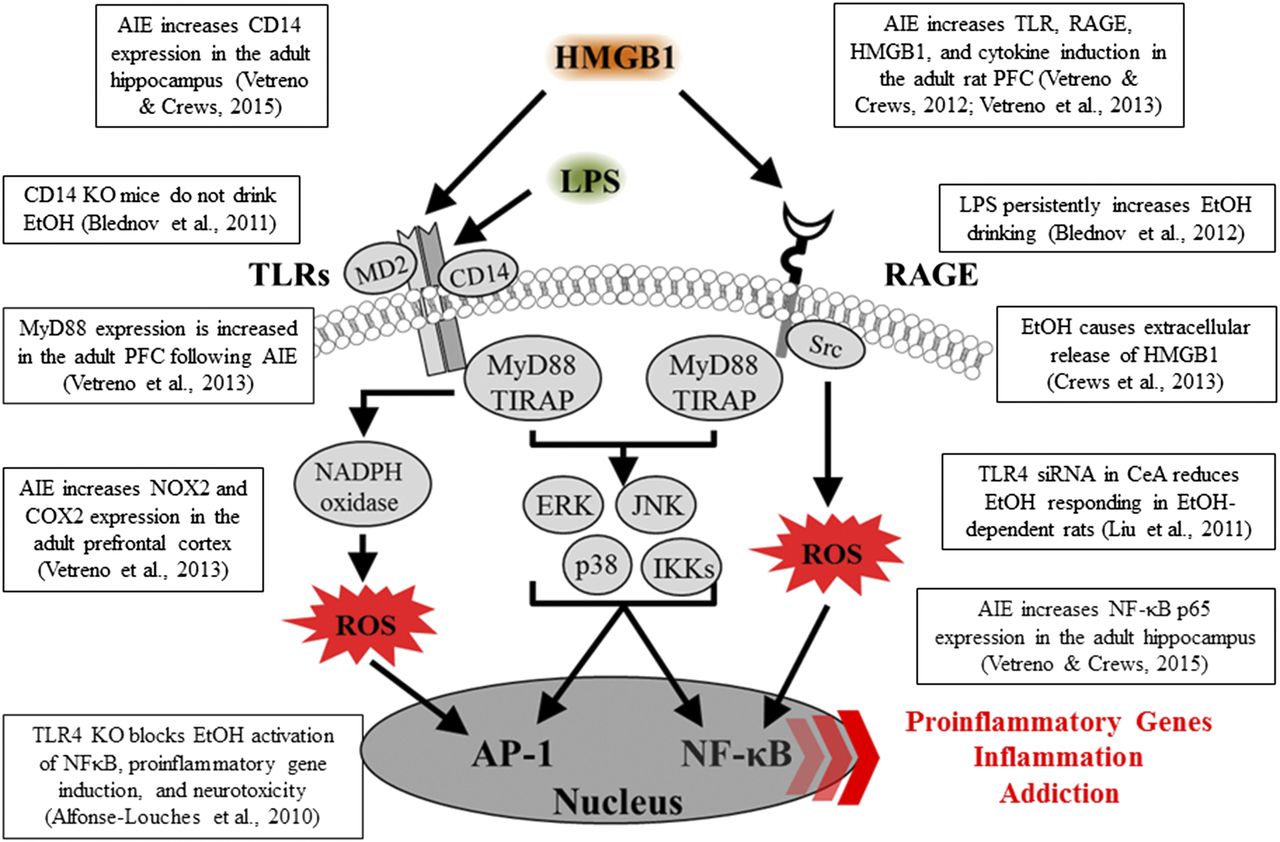{kind=link}
{kind=link}
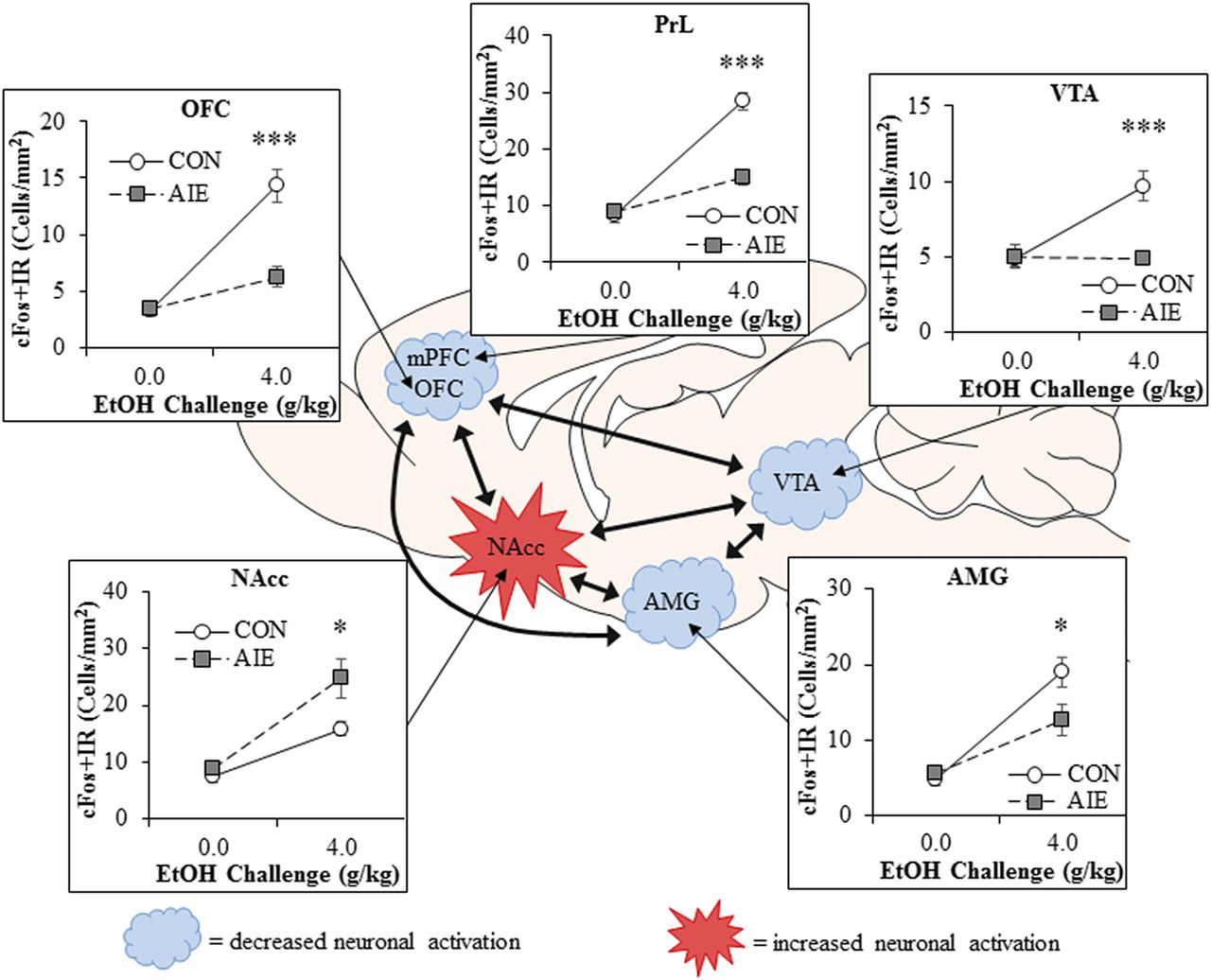{kind=link}
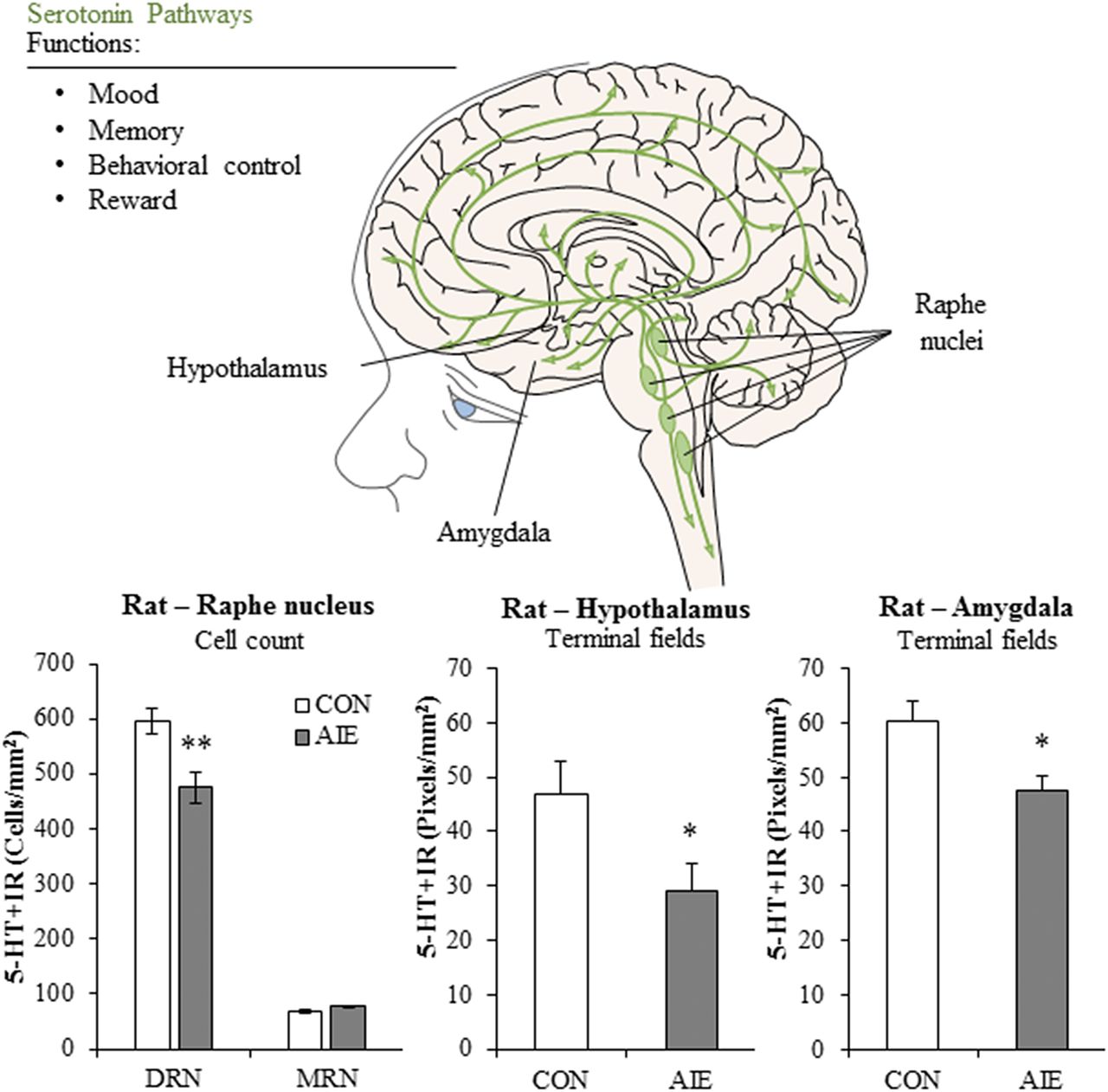{kind=link}
{kind=link}
{kind=link}
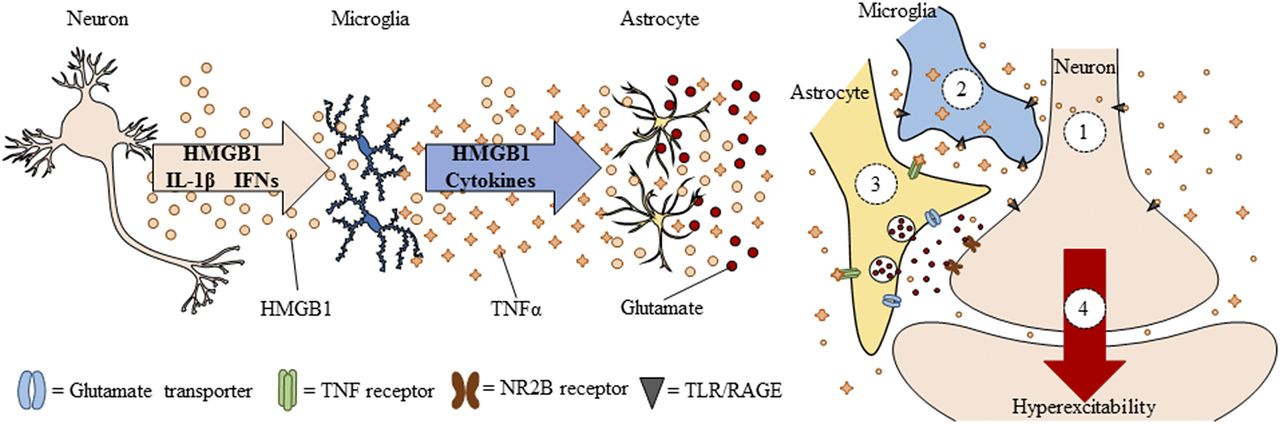{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. Brain Maturation and Adolescence
- III. Adolescent Alcohol Sensitivity
- IV. Adolescents Binge Drink
- V. Modeling Adolescent Alcohol Drinking
- VI. Lock-In—Persistence of an Adolescent Phenotype in Adulthood, Including an Adolescent-Typical Response to Ethanol
- VII. AIE Increases Ethanol Self-Administration in Adulthood
- VIII. AIE Results in Decreased Behavioral Flexibility
- IX. Adolescent Alcohol Effects on Anxiety and Negative Affective Behavior
- X. Adolescent Alcohol-Induced Neuroimmune Gene Induction
- XI. Brain Electroencephalography and Sleep
- XII. Cholinergic System Development and Effects of AIE
- XIII. Monoamine System Development and Effects of AIE
- XIV. Hippocampal Development and Effects of AIE
- XV. Adolescent Alcohol-Induced Epigenetic Alterations in Gene Expression
- XVI. Conclusions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters