


Visual Overview

Abstract
α-Synuclein (α-Syn) aggregation in Lewy bodies and Lewy neurites has emerged as a key pathogenetic feature in Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Various factors, including posttranslational modifications (PTMs), can influence the propensity of α-Syn to misfold and aggregate. PTMs are biochemical modifications of a protein that occur during or after translation and are typically mediated by enzymes. PTMs modulate several characteristics of proteins including their structure, activity, localization, and stability. α-Syn undergoes various posttranslational modifications, including phosphorylation, ubiquitination, SUMOylation, acetylation, glycation, O-GlcNAcylation, nitration, oxidation, polyamination, arginylation, and truncation. Different PTMs of a protein can physically interact with one another or work together to influence a particular physiological or pathological feature in a process known as PTMs crosstalk. The development of detection techniques for the cooccurrence of PTMs in recent years has uncovered previously unappreciated mechanisms of their crosstalk. This has led to the emergence of evidence supporting an association between α-Syn PTMs crosstalk and synucleinopathies. In this review, we provide a comprehensive evaluation of α-Syn PTMs, their impact on misfolding and pathogenicity, the pharmacological means of targeting them, and their potential as biomarkers of disease. We also highlight the importance of the crosstalk between these PTMs in α-Syn function and aggregation. Insight into these PTMS and the complexities of their crosstalk can improve our understanding of the pathogenesis of synucleinopathies and identify novel targets of therapeutic potential.
Significance Statement α-Synuclein is a key pathogenic protein in Parkinson’s disease and other synucleinopathies, making it a leading therapeutic target for disease modification. Multiple posttranslational modifications occur at various sites in α-Synuclein and alter its biophysical and pathological properties, some interacting with one another to add to the complexity of the pathogenicity of this protein. This review details these modifications, their implications in disease, and potential therapeutic opportunities.
I. Introduction
Pathological aggregation of α-synuclein (α-Syn) is a characteristic feature of a group of neurodegenerative disorders including Parkinson's disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA), collectively known as synucleinopathies (Goedert et al., 2017). α-Syn is a 140 amino acid protein that is intrinsically disordered with remarkable conformational plasticity, as it can adopt a broad range of structural conformations including oligomers, protofibrils, and mature fibrils (Deleersnijder et al., 2013).
Like other proteins, α-Syn undergoes a number of posttranslational modifications (PTMs) including phosphorylation, ubiquitination, SUMOylation, acetylation, glycosylation, glycation, nitration, oxidation, arginylation, polyamination, truncation, and methylation (Vicente Miranda et al., 2017a; Zhang et al., 2019b). PTMs modulate the structure and function of proteins, and dysregulated PTMs may alter the propensity for protein misfolding and aggregation (Schaffert and Carter, 2020).
In addition to the impact of PTMs on the fundamental biology of α-Syn, they have diagnostic and therapeutic implications. As a biomarker of disease pathology, phosphorylated α-Syn detected by immunohistochemistry on skin biopsy specimens is positive in 94% of patients with clinical PD, 96% of those with DLB, and 98% of MSA cases, compared with 3.3% of control individuals (Gibbons et al., 2024). This test is now available for healthcare providers to order for their patients. In terms of treatment, understanding the specific PTMs involved in disease pathways and their interplay among them have the potential to lead to targeted therapies. Strategies to mimic or inhibit specific PTMs can restore normal cellular functions in various disorders. Therefore, the study and manipulation of protein PTMs hold substantial promise for advancing the diagnosis and treatment of diseases.
This review provides a comprehensive analysis of α-Syn PTMs and their interplay in influencing α-Syn structure, function, and misfolding. Understanding the crosstalk between PTMs can provide insight into potential novel therapeutic targets to impact the pathogenesis of synucleinopathies and their progression.
A. Posttranslational Modifications
Posttranslational modifications fill a unique niche among the many multilayered regulatory mechanisms that control the physiology of eukaryotic cells because they are extremely dynamic and largely reversible. By attaching a modifying chemical group or another protein to one or more of a protein's amino acid residues, PTMs affect a variety of protein properties, such as structure (Macek et al., 2019; Zecha et al., 2022), enzymatic activity (Deribe et al., 2010), interaction with other proteins (Li et al., 2013; Duan and Walther, 2015), and subcellular localization (Karve and Cheema, 2011). Thus, these modifications are crucial for controlling how proteins function in health and disease.
More than 200 distinct types of PTMs are already recognized (Minguez et al., 2012), ranging from modest chemical alterations, such as phosphorylation and acetylation, to the incorporation of whole proteins, e.g., ubiquitination. The majority of these modifications are added following translation (synthesis of the polypeptide chain), so the phrase “posttranslational modifications” is frequently used to describe them. However, some of these alterations, such as amino-terminal (N-terminal) protein acetylation (Ree et al., 2018) or N-glycosylation (Latousakis and Juge, 2018), take place concurrently with translation. In addition, PTMs can occur at any stage of the protein life cycle, altering protein folding, subcellular localization, and activity in time and space (Didonna et al., 2016).
PTMs that involve covalent attachment of functional groups include phosphorylation, acetylation, glycosylation, acylation, ubiquitination, SUMOylation, and oxidation. Some PTMs are added enzymatically, such as phosphorylation, acetylation, glycosylation, methylation, ubiquitination, SUMOylation, palmitoylation, biotinylation, chlorination, polyamination, and arginylation (Folk et al., 1980; Saha and Kashina, 2011; Santos and Lindner, 2017), while others, such as glycation, nitration, and oxidation (Kakizawa, 2013; Nedić et al., 2015; Greifenhagen et al., 2016), do not require an enzyme. Other unusual PTMs, including glypiation, neddylation, siderophorylation, AMPylation, and cholesteroylation, are also known to affect the structure and function of proteins (Basak et al., 2016).
In the complex processes occurring within cells, proteins are subject to various PTMs that regulate their function and cellular activities. Crosstalk between PTMs establishes a dynamic and intricately regulated network of modifications, wherein one PTM can affect the occurrence, function, or removal of another. This interplay is vital for fine-tuning cellular processes and responses to various stimuli, enabling a more adaptable and responsive regulatory system (Hunter, 2007; Yang and Seto, 2008). Such interactions can profoundly impact the overall cellular environment, influencing everything from signal transduction pathways to gene expression and protein stability (Beltrao et al., 2013). Understanding PTM crosstalk is essential for decoding the complexities of cellular signaling pathways. It involves mapping out how different modifications interact, compete, or cooperate to modulate protein activity and cellular outcomes. This knowledge has significant implications for developing therapeutic strategies targeting diseases associated with PTM dysregulation, such as cancer, neurodegenerative disorders, and metabolic diseases (Choudhary and Mann, 2010; Deribe et al., 2010).
B. 2 α-Synuclein Protein Structure and Function
α-Syn is encoded by the SNCA gene located at position 21 on the long arm of chromosome 4 (Shibasaki et al., 1995). This 14 kDa protein is abundantly expressed in neurons, and its primary amino acid sequence can be divided into three main domains: the N-terminal domain (1–60), the central region (61–95), and the C-terminal domain (96–140). The N-terminal domain contains 11-amino acid repeats with an imperfectly conserved core motif KTKEGV and has the propensity to form an α-helical structure. The central region, which was first purified from amyloid plaques in brains affected with Alzheimer's disease, contains a highly hydrophobic motif that comprises amino acid residues 65–90 known as the nonamyloid component (NAC) (Uéda et al., 1993). The crystal structures of residues 68–78 (termed NACore) and residues 47–56 (termed PreNAC) using Micro-Electron Diffraction have shown that the strands in this region stack into β-sheets that are typical of amyloid assemblies (Rodriguez et al., 2015). In a drosophila model of PD, the aggregation and neurotoxicity of α-Syn are both reduced when residues 71–82 are deleted (Periquet et al., 2007). In addition, when isolated from the remainder of α-Syn, this segment is very cytotoxic and induces apoptotic cell death (El-Agnaf et al., 1998). Also, the C-terminal domain of α-Syn is enriched in negatively charged residues and provides flexibility to the polypeptide (Villar-Piqué et al., 2016). This domain, which contains 10 glutamate and 5 aspartate residues, was initially thought to be essential for protein solubility. The presence of five proline residues, which are also recognized to induce turns and disrupt secondary protein structure, suggested that this region lacks secondary structure (Mor et al., 2016). Several lines of evidence indicate that the C-terminus is crucial for the interaction of α-Syn with other proteins, lipids and small molecules including metal ions (Burré et al., 2010, 2012; Lautenschläger et al., 2018; Moons et al., 2020). Negative charges in the C-terminal region of α-Syn have been found to be important in modulating fibril formation (Izawa et al., 2012). In vitro studies have revealed that decreasing the pH, which neutralizes these negative charges, can induce α-Syn aggregation (Hoyer et al., 2002). Little aggregation was observed when full-length wild-type (WT) α-Syn was kept at 37°C without shaking, while C-terminally truncated α-Syn (residues 1–120 and 1–110) formed long filaments (Crowther et al., 1998). Moreover, C-terminal truncation has been shown to enhance in vitro fibril formation even faster than the PD-linked familial A53T mutant form of α-Syn (Murray et al., 2003).
Under physiological conditions, the secondary structure of α-Syn is dynamically balanced between a soluble state and a membrane-bound form depending on the cellular environment. The interaction between α-Syn and lipid surfaces is hypothesized to contribute to its biological activity. Soluble cytosolic α-Syn is naturally unstructured and acts like a natively unfolded protein. When human α-Syn is expressed in mouse and rat brains as well as in mammalian cell lines, similar patterns are observed (Fauvet et al., 2012). However, in disease states, α-Syn forms oligomers and eventually mature fibrils (Li et al., 2022); the oligomers are believed to be the most toxic species (Ingelsson, 2016).
Since its discovery, α-Syn has been recognized as a presynaptic protein with relatively little expression in the cell body, dendrites, or extrasynaptic sites along the axon (Maroteaux et al., 1988; Iwai et al., 1995). Various cellular and animal models have been employed to elucidate the physiological function of α-Syn. It is implicated in the compartmentalization, storage, and recycling of neurotransmitters under physiological conditions (Zhang et al., 2019b; Miquel-Rio et al., 2023). Our current understanding suggests that it plays a regulatory role in maintaining synaptic homeostasis as well as a role in exo- and endocytosis mechanisms (Gureviciene et al., 2007; Ben Gedalya et al., 2009; Nemani et al., 2010; Cheng et al., 2011; Janezic et al., 2013; Kisos et al., 2014; Lautenschläger et al., 2017).
Numerous synaptic processes have been linked to α-Syn including membrane remodeling, modulation of dopamine transporter and vesicular monoamine transporter, clustering of synaptic vesicles, maintenance of synaptic vesicle pools, stimulating soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-complex assembly necessary for neurotransmitter release (Gerst, 1999), and regulating synaptic vesicle recycling (Burré, 2015; Sharma and Burré, 2023). The overall impact of α-Syn on synaptic release is believed to be an equilibrium between its inhibitory effect through synaptic vesicle clustering and a release-promoting effect through SNARE-complex chaperoning and fusion pore opening (Sharma and Burré, 2023). Direct interactions between α-Syn and the SNARE protein synaptobrevin-2 facilitate the development of SNARE complexes (Burré et al., 2010). α-Syn has also been shown to regulate the number of vesicles docked at synapses during neurotransmitter release by participating in the dynamics of synaptic vesicle trafficking (Burré, 2015). Collectively, these findings indicate that α-Syn plays a role in synaptic homeostasis and neurotransmitter release.
II. α-Synuclein Posttranslational Modifications
α-Syn undergoes many PTMs as illustrated in Fig. 1. Under physiological conditions, α-Syn, like many proteins, is subjected to multiple PTMs at various sites, many of which occur at the same residue (Fig. 2). These modifications influence the protein’s structure and conformation and consequently its localization, function, and eventual fate within the cell. However, in some pathological conditions, the regulation of these PTMs is disrupted. This dysregulation can lead to changes in α-Syn structure or conformation, affecting its folding patterns and increasing its tendency to form seeds. The latter serve as nuclei for the further aggregation of α-Syn monomers, a process that is closely linked to the onset and progression of synucleinopathies. This sequence of events highlights the delicate balance maintained by various PTMs in the normal functioning of α-Syn and how a disturbance in this balance can contribute to pathological aggregation.

α-Synuclein posttranslational modifications. Summary of the various posttranslational modifications of α-Syn categorized by the type of modification including addition of chemical groups (red), a polypeptide (green), complex molecules (black), and modifications involving amino acids (blue). 4-Hydroxynonenal is another reported posttranslational modification not depicted in this figure.

α-Synuclein structure with posttranslational modification sites. Created with BioRender.com pursuant to its Academic License Terms.
The role of PTMs as potential biomarkers for diagnosis and tracking disease progression in PD and related synucleinopathies, as well as possible targets for treatment, are increasingly recognized (Brembati et al., 2023). To gain insight into the potential therapeutic implications of these modifications and the crosstalk between them, we first review the impact of all PTMs on the structure, function, and aggregation propensity of α-Syn (Table 1).
α-Synuclein posttranslational modifications
A. Phosphorylation
Phosphorylation is a reversible PTM that regulates the physiological and pathological functions of proteins involved in processes such as cell cycle progression, intercellular communication, cellular metabolism, gene expression, differentiation, and migration (Manning et al., 2002a,b).
α-Syn is phosphorylated at serine (S42, S87, S129), tyrosine (Y39, Y125, Y133, Y136) (Okochi et al., 2000; Nakamura et al., 2001; Chen et al., 2009; Xu et al., 2015; Kleinknecht et al., 2016; Manzanza et al., 2021; Zhang et al., 2023), and threonine (T64, T72, T75, T81) (Matsui et al., 2023) residues. Detection of pathologic α-Syn aggregates in postmortem human tissues and experimental cellular and animal models typically utilizes phospho-S129-α-Syn antibodies (Chen and Feany, 2005; Gorbatyuk et al., 2008; Muntané et al., 2012; Awa et al., 2022). Except for pS87 and pY39, the majority of phosphorylated residues are concentrated in the C-terminal domain (Figs. 2 and 3), which is believed to be involved in α-Syn pathogenicity (Nakamura et al., 2001; Chen et al., 2009; Xu et al., 2015).

Schematic diagrams of the impact of posttranslational modifications on α-Syn. The three domains of α-Syn are depicted: the N-terminal domain (1–60), the central region (61–95), and the C-terminal domain (96–140). PTMs can a) promote (red circles) or inhibit (green circles) α-Syn aggregation, b) promote (red circles) or inhibit (green circles) toxicity, and c) inhibit (red) or promote (green) clearance. Polyamination that targets acidic and negatively charged residues promotes α-Syn aggregation (not shown in the diagram). Created with BioRender.com pursuant to its Academic License Terms.
1. α-Synuclein Phosphorylation at Serine Residues
α-Synuclein phosphorylation occurs at three serine sites: S129, S87, and S42. Among these, serine 129 (pS129) is the most extensively studied PTM due to its significant role and as a key marker of pathological α-Syn in PD and related synucleinopathies. In addition to the brain (Kim et al., 2019; Schaser et al., 2019; Zhang et al., 2020; Manzanza et al., 2021; Gibbons et al., 2024), pS129 α-Syn has been detected in various biological fluids and tissues including serum (Cariulo et al., 2019; Chatterjee et al., 2020), red blood cells (Tian et al., 2019; Li et al., 2020, 2021b), cerebrospinal fluid (El Turk et al., 2018; Schmitz et al., 2019), plasma exosomes (Zheng et al., 2021), gut (Chen et al., 2018; Li et al., 2018; Kishimoto et al., 2019; Beck et al., 2020; Bu et al., 2020; Harapan et al., 2020; Izco et al., 2021), retina (Ortuño-Lizarán et al., 2018), salivary glands (Fernández-Arcos et al., 2018; Iranzo et al., 2018), extracellular vesicles from saliva (Cao et al., 2020), cutaneous nerve fibers (Donadio et al., 2018a,b, 2019; Kuzkina et al., 2019; Liu et al., 2020; Giannoccaro et al., 2022; Gibbons et al., 2024), peripheral sensory nerves (sural nerve) (Zhang et al., 2019a; Rong et al., 2021), Schwann cells of sciatic nerves (Sun et al., 2021), and external urethral sphincter (Ding et al., 2020).
Despite extensive investigations on this particular PTM, questions remain regarding whether S129 phosphorylation contributes to α-Syn toxicity or is protective. Several lines of evidence suggest that phosphorylation of α-Syn at S129 increases its tendency to aggregate and is associated with the production of toxic α-Syn species and neurotoxicity in various in vitro and in vivo models of PD (Kahle et al., 2000; Okochi et al., 2000; Fujiwara et al., 2002; Chen and Feany, 2005; Anderson et al., 2006; Lee et al., 2011) (Fig. 3). In Drosophila, mutation of Ser129 to alanine to avoid phosphorylation fully suppressed the dopaminergic neuronal loss caused by overexpression of human α-Syn. In contrast, the toxicity of α-Syn is markedly increased in dopaminergic neurons when Ser129 is substituted with the phosphomimetic aspartate (Chen and Feany, 2005). These findings have been supported by studies in rodents as well. Compared with wild-type α-Syn preformed fibrils (PFF), injection of pS129 α-Syn fibrils into the mouse striatum induced the formation of more α-Syn aggregates in the substantia nigra, worsened pathology in the cerebral cortex, caused greater dopaminergic neuronal loss, and impaired fine motor activity as early as 2 months postinjection (Karampetsou et al., 2017). In addition, pS129 α-Syn accumulates in association with pathological lesions in the cerebellar cortex of transgenic PLP-α-Syn mice, in which human α-Syn was expressed under the control of the PLP promoter (Kahle et al., 2002), and in several brain regions of transgenic mice expressing pathogenic A30P mutant human α-Syn under the control of the neuronal Thy1 promoter (Freichel et al., 2007). Conversely, using a pharmacological approach to promote the dephosphorylation of pS129 α-Syn, we observed a significant decrease in α-Syn aggregation, preserved neuritic processes, and improved behavioral outcome of Thy1 promoter-driven human wild-type α-Syn transgenic mice (Lee et al., 2011). Additional support for the pathogenic role of S129 phosphorylation comes from a human postmortem study showing that ∼90% of α-Syn in the urea-soluble fraction of cerebral cortex in DLB-affected brains is phosphorylated at Ser 129. Similar results were observed in LBs isolated from individuals with DLB, suggesting that pS129 α-Syn is the most abundant modified form of α-Syn in LBs and that only a small amount is present in the soluble fraction of both control and DLB brains (Anderson et al., 2006). On the other hand, only 4% of the total normal adult rat brain α-Syn is phosphorylated at S129, highlighting the importance of α-Syn phosphorylation in the pathogenesis of synucleinopathies (Fujiwara et al., 2002). Quantification of pS129 α-Syn levels in human cerebral spinal fluid (CSF) was found to have a significant correlation with the severity of clinical manifestations in individuals diagnosed with PD, suggesting that pS129 could function as a prognostic biomarker for disease progression (Wang et al., 2012; Stewart et al., 2015).
In contrast, other lines of evidence suggest that phosphorylation of α-Syn at S129 reduces its tendency to misfold or has no effect on inclusion formation or toxicity (Chen and Feany, 2005; Gorbatyuk et al., 2008; Paleologou et al., 2008; Tenreiro et al., 2014; Weston et al., 2021a). Expression of wild-type or mutant α-Syn isoforms that prevent S129 phosphorylation (S129A and S129G) in yeast cells has shown that blocking α-Syn phosphorylation accelerates inclusion formation and exacerbates its toxicity. Moreover, cells expressing S129A α-Syn fail to activate the autophagy pathway, suggesting involvement of phosphorylation at S129 in the clearance of α-Syn through autophagy (Tenreiro et al., 2014) (Fig. 3). Additionally, GFP-tagged human α-Syn expressed in zebrafish could be phosphorylated by endogenous Polo-like kinase. In this model, α-Syn aggregation was not affected either by administering a Polo-like kinase inhibitor or expressing S129A or S129D α-Syn (Weston et al., 2021a). Furthermore, injections of adeno-associated viral vector serotype 2 expressing human WT or S129A α-Syn into the substantia nigra of rats resulted in greater toxicity with the S129A isoform 4 weeks postinjection. This was demonstrated by a significant decrease in nigral dopaminergic neurons and striatal dopamine and tyrosine hydroxylase content (Gorbatyuk et al., 2008). A more recent study showed that following α-Syn PFF injection into the dorsal striatum of mice, a small amount of pS129 α-Syn was detected 1 to 2 weeks postinjection, but this became more apparent by week 4 postinjection, suggesting that phosphorylation occurs after the initial seeding and protein aggregation (Ghanem et al., 2022). Despite the aforementioned contradictory findings about the disease relevance of pS129 α-Syn, it is widely observed as a disease-associated PTM, which needs additional research to determine its full significance in synucleinopathies.
Various biochemical techniques have confirmed the presence of α-Syn tetramers in healthy cells and brain tissue (Fonseca-Ornelas et al., 2022). Certain mutations in α-Syn reduce the tetramer-to-monomer (T:M) ratio, resulting in the formation of round, cytoplasmic inclusions that react to α-Syn immunostaining and are linked to neurotoxicity (Dettmer et al., 2015). Moreover, there is a marked inverse relationship between the α-Syn T:M ratio and the levels of Ser129 phosphorylation. Specifically, a lower T:M ratio in human neurons correspond to increased phosphorylation of α-Syn (Fonseca-Ornelas et al., 2022), suggesting a potential mechanism by which phosphorylation could drive α-Syn to undergo conformational changes.
a. α-Synuclein Phosphorylation at Serine Residues in the gut
In people with Parkinson's disease, the presence of pS129 α-Syn has been detected in the upper gastrointestinal tract, specifically in the submandibular glands and distal esophagus. In the rostrocaudal axis, which extends from the stomach through the small and large intestine to the rectum, the occurrence of p-α-Syn gradually diminishes (Gelpi et al., 2014; Beck et al., 2020). In A53T mutant human α-Syn transgenic mice, pS129 α-Syn has been found to accumulate in the enteric nervous system before motor symptoms appear (Bencsik et al., 2014). The salivary glands, particularly the submandibular glands, have been identified as another possible site for early identification of PD pathology. Notably, samples from PD patients have shown substantial positivity for pS129 α-Syn (Adler et al., 2014, 2019). Research conducted on the colon has identified the presence of pS129 α-Syn in a substantial number of people with PD but not in healthy individuals. This suggests that pS129 α-Syn analysis may have diagnostic significance (Lebouvier et al., 2010; Clairembault et al., 2015). However, certain authors present contrasting findings regarding the identification of pS129 α-Syn in colon biopsies, primarily highlighting the prevalence of high positivity in persons without health issues, thus diminishing the diagnostic utility of this marker for PD (Visanji et al., 2015; Antunes et al., 2016).
2. α-Synuclein Phosphorylation at Tyrosine Residues
α-Syn is known to undergo phosphorylation at several tyrosine residues including Y125, Y39, Y133, and Y136. α-Syn that is phosphorylated at tyrosine 125 (Y125) has been found in human brains (Chen et al., 2009) and is reported in LBs in a case of familial PD with the G51D mutation (Kiely et al., 2013). While a study reported higher levels of pY125 in control brains compared with DLB brains (Chen et al., 2009), another study found comparable levels between control and PD brains using immunoblotting analysis (Mahul-Mellier et al., 2014). However, other reports have suggested that pY125 is not a prominent component of LB pathology in murine models or in human PD and DLB (Anderson et al., 2006; Fayyad et al., 2020).
Y39 phosphorylation affects the structure and function of α-Syn, and pY39 α-Syn PFFs are more cytotoxic than wild-type fibrils in rat primary cortical neurons (Zhao et al., 2020). This PTM could also regulate differential binding of the helix-2 region of the N-terminal domain of α-Syn to lipid membranes, hence influencing the interaction with docked synaptic vesicles and the plasma membrane (Dikiy et al., 2016). While tyrosine phosphorylation at Y39 has been reported to decrease α-Syn fibril formation in vitro (Dikiy et al., 2016), it has been suggested to increase α-Syn aggregation in vivo (Brahmachari et al., 2016) (Fig. 3). Quantification of pY39 spanning peptide EGVLpYVGSK, which is shared between α-Syn and β-Syn (αβ-Syn), in CSF has been studied as a potential biomarker for the diagnosis and prognosis of PD. Although the absolute levels of endogenous pY39 αβ-Syn did not show a statistically significant difference between PD patients and control subjects, the ratio of phosphorylated Y39 to nonphosphorylated Y39 αβ-Syn was markedly higher in the PD group (Na et al., 2020).
Few studies have addressed the physiological function(s) and relevance of pY133 and pY136 α-Syn in the pathogenesis of LB disease in the human brain. The level of pY133 α-Syn is similar in DLB, PD, and control brains (Fayyad et al., 2020), suggesting that this PTM may not be an essential pathogenetic factor. However, in yeast cells, expression of human α-Syn has shown that Y133 is necessary for a protective S129 phosphorylation through autophagy (Fig. 3). In fact, Y133 mutation led to a loss of phosphorylation at S129 and prevented aggregate clearance by autophagy. α-Syn has also been shown to be highly phosphorylated at Y136 in Lewy body disease brains (Sano et al., 2021, 2). The extent to which phosphorylation at these tyrosine residues impacts α-Syn function and aggregation remains unclear.
In a recent study, significant amounts of soluble α-Syn phosphorylated at Y39 and S87 were detected in the brains of patients with Lewy body disease and MSA as well as from normal control individuals. The impact of phosphorylation of soluble α-Syn at these sites and at Y125 and Y133 on the seeding ability of pathological α-Syn sourced from these disease brains (LB-α-Syn) and glial cytoplasmic inclusions (GCI-α-Syn), and synthetic α-Syn PFF were examined in HEK293 cells made to express glutamic acid substitutions to mimic phosphorylation. Y39 phosphorylation blocked seeding by LB-α-Syn and to a lesser extent GCI-α-Syn but not by synthetic α-Syn PFF. On the other hand, S87 phosphorylation slightly increased the seeding ability of LB-α-Syn but markedly blocked the seeding ability of GCI-α-Syn. Similar observations were made in primary neurons. Additionally, in in vitro experiments, synthetic α-Syn PFF could seed pY39 α-Syn and pS87 α-Syn equally, but LB-α-Syn preferentially seeded pS87 α-Syn, while GCI-α-Syn preferentially seeded pY39 α-Syn. These findings suggest that the phosphorylation of soluble α-Syn influences the seeding potential of pathological α-Syn in a conformation- and phosphorylation site-specific manner (Zhang et al., 2023).
In addition to phosphorylation at serine and tyrosine residues, a novel phosphorylation site on α-Syn at Threonine 64 (T64) has recently been identified. This particular PTM was found to be increased in SH-SY5Y cells treated with α-Syn fibrils, in transgenic zebrafish expressing human α-Syn, in α-Syn PFF-injected mouse brains, and in postmortem human PD brains. Moreover, T64D phosphomimetic mutation resulted in the formation of oligomers with a structure similar to that of pathogenic A53T mutant α-Syn, exhibited high degree of toxicity in both cell culture and zebrafish, leading to swimming movement disorder and neurodegeneration (Matsui et al., 2023) (Fig. 3).
3. Regulation of α-Synuclein Phosphorylation
Phosphorylation is regulated by kinases and phosphatases. Several kinases are known to phosphorylate α-Syn. These include Casein kinases (CK1, CK2) (Takahashi et al., 2007; Waxman and Giasson, 2008), Glycogen synthase kinase-3β (GSK-3β) (Hu et al., 2020; Takaichi et al., 2020), Polo like kinase 2 (PLK2) (Inglis et al., 2009), death-associated protein kinase 1 (Shin and Chung, 2020), inflammation-associated serine-threonine kinase, PKR (Reimer et al., 2018), and G protein-coupled receptor kinases including G protein-coupled receptor kinase (GRK)2 (Pronin et al., 2000), GRK3 (Sakamoto et al., 2009), GRK5 (Pronin et al., 2000; Arawaka et al., 2006), GRK6 (Sakamoto et al., 2009), and nonreceptor Abelson tyrosine kinase (c-Abl) (Mahul-Mellier et al., 2014). Colocalization of GRK5 (Arawaka et al., 2006) and Casein kinase II beta subunit (Ryu et al., 2008) with α-Syn in LBs in postmortem brains of PD patients has been reported. However, it remains unclear whether all kinases work in concert, operate independently under different conditions, or function in different cell types. Further investigations are needed to understand the precise coordination and regulation of these kinases in the phosphorylation of α-Syn, particularly in pathological states.
Although all these kinases promote α-Syn phosphorylation, it is believed that their effects on α-Syn aggregation are distinct. For example, GRK2 (Chen and Feany, 2005) and GRK6 (Sato et al., 2011) promote the aggregation and toxicity of α-Syn. In contrast, PLK2, which is a key factor in α-Syn phosphorylation (Inglis et al., 2009), has been reported to either reduce α-Syn accumulation by inducing its autophagic degradation (Oueslati et al., 2013) or have no effect on α-Syn aggregation or toxicity (Kofoed et al., 2017; Elfarrash et al., 2021; Weston et al., 2021a,b). Mechanisms other than phosphorylation of α-Syn at Ser129 are likely involved in PLK2 effect. In cellular models and mouse brains, overexpression of PLK2 reportedly decreases α-Syn levels without S129 phosphorylation, while a highly selective PLK2 inhibitor raises α-Syn mRNA transcription and protein levels in brain tissue and primary neurons, suggesting that PLK2 targets other proteins that are important in regulating α-Syn levels (Kofoed et al., 2017).
Studies using neuroblastoma cell lines and primary cultures of mouse cortical neurons have demonstrated that Y39 in α-Syn, and to a lesser extent Y125, are phosphorylated by c-Abl. Phosphorylation at Y39 can be effectively inhibited by specific c-Abl inhibitors (imatinib, nilotinib, and GNF-2) or increased by activating c-Abl with (5-[3-(4-fluorophenyl)-1-phenyl-1H-pyrazol-4-yl]-2,4-imidazolidinedione. Phosphorylation of α-Syn by c-Abl protects it from degradation by both the autophagy and proteasome pathways in cortical neurons (Mahul-Mellier et al., 2014). Yet, the specific molecular process underlying the pathology associated with pY39 α-Syn remains unclear.
Phosphatases are equally important in regulating the steady-state phosphorylation of proteins. This is true for α-Syn S129 phosphorylation (Braithwaite et al., 2012). A particular isoform of protein phosphatase 2 A (PP2A), namely B55alpha subunit, is responsible for dephosphorylating pS129 α-Syn (Lee et al., 2011). Carboxyl methylation of the C-subunit of this isoform is critical for the assembly of the functional trimer and the regulation of its phosphatase activity with substrate specificity (Bryant et al., 1999; Leulliot et al., 2004; Park et al., 2018). This methylation, in turn, is controlled by two opposing enzymes: the specific methylating enzyme leucine carboxyl methyltransferase-1 (LCMT-1) and the specific demethylating enzyme protein phosphatase methylesterase-1 (PME-1) (Lee et al., 1996; Ogris et al., 1999). In postmortem brains from PD and DLB patients, LCMT-1 levels are significantly lower in the frontal cortex and substantia nigra compared with age-matched controls. On the other hand, PME-1 levels are higher in the PD nigra. These changes in the regulating enzymes are associated with marked reduction in the ratio of methylated PP2A to demethylated PP2A in PD and DLB brains (Park et al., 2016). Interestingly, evidence has been presented that α-Syn itself negatively controls PP2A methylation. Overexpression of α-Syn inhibits PP2A activity by increasing demethylation at L309 in SK-N-SH cells and primary cortical neurons from Thy1-α-Syn transgenic mice. This was associated with downregulation of LCMT-1 and upregulation of PME-1 (Tian et al., 2018), which mirrors the profile seen in postmortem brains from PD and DLB brains (Park et al., 2016). Thus, there appears to be a feedback loop between α-Syn and PP2A dysregulation. Exploiting the therapeutic potential of this mechanism has proven to be promising and is discussed in greater detail in Section 3.1.
B. Ubiquitination
Ubiquitin is a 76 amino acid protein with an approximate molecular weight of 8.5 kDa. It attaches covalently to target proteins through the formation of a peptide bond between the carboxyl group of the C-terminal residue of ubiquitin and the side chain amino group of a lysine residue of the protein substrate (Hershko and Ciechanover, 1998). Three types of enzymes, including activating (E1), conjugating (E2), and ligating (E3) act sequentially for a successful ubiquitination process. First, E1 activates ubiquitin before transferring it to an E2-conjugating enzyme. The E3 ubiquitin ligase then binds simultaneously with a ubiquitin-loaded E2 and the substrate protein to promote the formation of an isopeptide bond between its C-terminal residue and a substrate lysine residue (Hershko and Ciechanover, 1998; Guo and Tadi, 2022). Various forms of ubiquitination have been identified. These include monoubiquitination (one ubiquitin molecule attaches to a target protein), multimono-ubiquitination (attachment of a single ubiquitin molecule to multiple lysine residues in the substrate), and poly-ubiquitination (ubiquitin chains attached end-to-end to a single lysine residue) (Ronai, 2016). Advances in the field of proteomic mass spectrometry, along with the development of specific antibodies against ubiquitin chains attached to substrates, have enabled researchers to trace ubiquitination precisely and comprehensively (Popovic et al., 2014).
Ubiquitination is essential for a variety of physiological functions such as cell survival (Chen and Qiu, 2013), differentiation (Suresh et al., 2016), innate and adaptive immunity (Hu and Sun, 2016), and many more. Ubiquitin plays a crucial regulatory role in the dynamic and complex process of eukaryotic protein degradation by the proteasome and lysosome (Tai and Schuman, 2008). The ubiquitin-proteasome system (UPS) degrades most soluble intracellular proteins (Ciechanover, 2005), but it can also break transmembrane proteins if they are released into the cytosol (Nakatsukasa et al., 2008). Despite the fact that UPS function and lysosomal degradation differ in various ways, ubiquitin might act as a general recognition signal for selective autophagy (Kraft et al., 2010).
Given the variety of functions and substrates targeted by the ubiquitin pathway, it is not surprising that abnormalities of ubiquitination directly or indirectly contribute to the etiology of numerous diseases. The pathological conditions linked to the ubiquitin system can be divided into two categories: 1) those caused by loss of function due to mutations in ubiquitin system enzymes or in the target substrate's recognition motif, which result in the stabilization of specific proteins, and 2) those caused by an increase in ubiquitin system activity, which leads to abnormal or accelerated degradation of the protein target (Ciechanover and Schwartz, 2004). Dysregulated expression of genes that control protein turnover and degradation, including that of ubiquitin, contribute to a number of neurodegenerative disorders (Schmidt et al., 2021). In addition, in cellular models, pathogenic proteins including α-Syn can form ubiquitin-containing aggresomes that have features of Lewy bodies (Lam et al., 2000; Waelter et al., 2001; Junn et al., 2002; Tanaka et al., 2004; Hara et al., 2006).
α-Syn has been shown to be mostly mono- or diubiquitinated at several lysine residues in vitro and in vivo by a number of E3 ubiquitin ligases including parkin (Shimura et al., 2001; Conway et al., 2022), seven in absentia homolog (SIAH) (Tofaris et al., 2003; Liani et al., 2004; Anderson et al., 2006; Lee et al., 2008), or Nedd4 ubiquitin ligases (Tofaris et al., 2011). Parkin, which was identified as the initial E3 ubiquitin-protein ligase to ubiquitinate α-Syn in vitro, requires the E2 ubiquitin-conjugating enzyme UbcH7 for this activity. However, parkin is able to ubiquitinate only a 22-kilodalton O-glycosylated version of α-Syn, which has also been observed in the brains of patients with PD and DLB (Shimura et al., 2001). Mutations in parkin associated with autosomal recessive PD (Kitada et al., 1998) inhibit its ubiquitination function (Dawson and Dawson, 2010).
The proteasome degrades monoubiquitinated α-Syn (Rott et al., 2011; Abeywardana et al., 2013), whereas polyubiquitination via Nedd4 results in lysosomal degradation (Tofaris et al., 2011). Monoubiquitination appears to promote α-Syn aggregation and enhance the formation of toxic α-Syn inclusion bodies and neurotoxicity in different cell lines including SH-SY5Y, PC12, and HeLa cells (Lee et al., 2008; Rott et al., 2008). Lewy bodies isolated from MSA (Hasegawa et al., 2002), PD, and DLB (Tofaris et al., 2003; Anderson et al., 2006) brains have been demonstrated to be mono- or diubiquitinated. Additionally, the finding of SIAH immunoreactivity in Lewy bodies of PD patients provides evidence that SIAH proteins may contribute to inclusion formation (Liani et al., 2004).
The effect of ubiquitination on α-Syn structure and aggregation is site-specific based on in vitro studies. Depending on the particular lysine residue that is modified, ubiquitination can either substantially hinder or enhance fibril formation (Meier et al., 2012). For example, ubiquitination at K6, K23, and K96 prevents the development of amyloid fibers but does not completely block α-Syn aggregation. However, fibers that form when α-Syn is ubiquitinated at K6 or K23 are structurally comparable to those created by unmodified α-Syn. In contrast, K96 ubiquitination results in fibers that are shorter than unmodified α-Syn (Moon et al., 2020). Other in vitro evidence supports the hypothesis that ubiquitination of α-Syn at K6 stabilizes the monomeric form of the protein and, therefore, inhibits its oligomerization and fibrillogenesis (Hejjaoui et al., 2011).
Proteasome activity has been found to be reduced in the nigra of PD patients (McNaught and Jenner, 2001; Rott et al., 2008), suggesting that dysfunction of the ubiquitin-proteasome system may contribute to the disease. In an early study on the role of ubiquitination on α-Syn degradation, we showed a slower rate of degradation for both wild-type and disease-causing A53T mutant α-Syn in transiently transfected human neuroblastoma SH-SY5Y cells under proteasome suppression, indicating regulation of α-Syn levels by the ubiquitin proteasome system (Bennett et al., 1999). The latter study also showed that mutant α-Syn is degraded slower than the wild-type protein, which supported the hypothesis that the pathogenic mutant protein tends to accumulate in neurons. In an animal model of PD using adeno-associated viral vectors-mediated overexpression of mutant α-Syn in dopaminergic neurons, UPS dysfunction is reportedly associated with pS129 α-Syn accumulation before dopaminergic neurodegeneration and behavioral deficits (McKinnon et al., 2020) (Fig. 3).
The protein de-ubiquitinase enzyme, USP13, has been shown to be upregulated in postmortem PD brains (Moussa, 2016; Liu et al., 2019), whereas knockdown of USP13 by injecting a lentiviral vector expressing USP13 shRNA in the striatum of A53T α-Syn transgenic mice resulted in increased α-Syn ubiquitination and clearance (Liu et al., 2019). α-Syn is the target of other de-ubiquitinase enzymes as well, such as USP9X, which reduces SIAH-dependent α-Syn proteasomal degradation (Rott et al., 2011), and USP8, which removes K63-linked ubiquitin chains from α-Syn and inhibits it from being degraded through the lysosome (Alexopoulou et al., 2016). These findings suggest that modulating UPS is a plausible strategy to lower the risk associated with protein aggregates and neuronal damage.
C. SUMOylation
SUMOylation is a posttranslational modification that involves the covalent conjugation of the small ubiquitin-like modifier (SUMO) to target proteins. Similar to ubiquitination, SUMOylation needs a series of enzymatic processes involving an E1 activating enzyme, an E2 conjugating enzyme (Ubc9), and an E3 SUMO ligase (Geiss-Friedlander and Melchior, 2007). Mammalian cells express five SUMO isoforms: SUMO1, SUMO2, SUMO3, and the less well-studied SUMO4 and SUMO5 (Guo et al., 2004; Liang et al., 2016; Celen and Sahin, 2020). SUMOylation regulates numerous cellular activities including protein stability (Seeler and Dejean, 2001), nucleo-cytoplasmic transport (Pichler and Melchior, 2002), transcriptional control (Gill, 2003), stress response, and apoptosis (Li et al., 2021a). SUMO binds the lysine side chains of target proteins through an ATP-dependent mechanism, and it can be released from the target protein by proteases/isopeptidases. Therefore, SUMOylation is a dynamic and reversible process, and proteins undergo cycles of SUMOylation and SUMO deconjugation (Melchior et al., 2003).
α-Syn undergoes SUMOylation, which may be crucial for its intracellular targeting. Among its 15 lysine residues that are potential SUMOylation sites, K96 and K102 are the primary sites (Dorval and Fraser, 2006; Krumova et al., 2011). Mutations at these two sites reduce α-Syn SUMOylation (Krumova et al., 2011). SUMOylation can control certain aspects of α-Syn including its interaction with proteins and membranes, degradation, aggregation, and toxicity (Savyon and Engelender, 2020) (Fig. 3). Studies in primary cortical neurons and HEK293 cells have shown that SUMOylation of α-Syn enhances its release from cells within extracellular vesicles by promoting α-Syn binding to membranes. In fact, SUMO-deficient α-Syn mutations significantly reduce the ability of α-Syn to attach to membranes compared with wild-type α-Syn (Kunadt et al., 2015). Blood level of SUMOylated α-Syn has been proposed as a potential biomarker for PD. A study measuring SUMO-1-ylated α-Syn levels in PD patients has revealed a decrease, with a notable association between the extent of this decrease and disease severity, as quantified by the United Parkinson’s Disease Rating Scale III motor score (Vicente Miranda et al., 2017a). Larger cross-sectional and longitudinal studies are needed to verify the utility of this measure as a biomarker of synucleinopathy.
Whether α-Syn SUMOylation promotes or inhibits its aggregation is debatable. Induction of SUMOylation by the SUMO E3 ligase human Polycomb protein 2 in HEK293 cells promotes α-Syn aggregation (Oh et al., 2011). Accumulation of SUMO-positive aggresome-like α-Syn inclusions in various cell lines may further contribute to the elevation of α-Syn SUMOylation in a positive-feedback loop (Kim et al., 2011; Oh et al., 2011). In an animal model of PD, injection of the mitochondrial complex I inhibitor rotenone into the medial forebrain bundle of mice resulted in an increase of SUMO1 and high molecular weight α-Syn species (Weetman et al., 2013). In PD and DLB affected brains as well as in COS-7 cells, we have observed that α-Syn and SUMO1 colocalize in Lewy bodies and aggresome-like structures, respectively (Kim et al., 2011). Consistent with these findings, the level of SUMOylated α-Syn immunoprecipitated from the cerebral cortex of PD patients with dementia is reportedly increased compared with age-matched controls (Rott et al., 2017). Moreover, in postmortem brain tissue from MSA-affected subjects, pathogenic oligodendroglial cytoplasmic α-Syn positive aggregates display significant punctate SUMO-1 immunostaining (Pountney et al., 2005).
On the other hand, SUMOylation can prevent α-Syn from forming mature fibrils. Consistent with reports suggesting that SUMO conjugation increases the solubility of target proteins (Marblestone et al., 2006; Guerra de Souza et al., 2016), double mutations of K96 and K102 residues of α-Syn increased its aggregation and toxicity in both HEK293T cells and in the rat substantia nigra injected with an adeno-associated viral vector serotype 2 vector expressing α-Syn (Krumova et al., 2011) (Fig. 3). A recent in vitro study showed that SUMO1, which targets the N-terminus of α-Syn, binds to it transiently in a noncovalent manner. This binding leads to compaction within α-Syn, which in turn slows down fibrillization (Panigrahi et al., 2023). Furthermore, another in vitro study suggests that, compared with SUMOylation of K96, reduction in fibrillization is more evident with SUMOylation of K102, particularly with SUMO1 relative to SUMO3 (Abeywardana and Pratt, 2015). We recently found that rotenone treatment of SH-SY5Y cells reduces global SUMOylation and autophagy. Boosting the SUMOylation machinery by overexpressing SUMO-1 prevented α-Syn aggregation and phosphorylation and restored autophagy function (Hassanzadeh et al., 2023). α-Syn has also been shown to be SUMOylated in yeast cells, and impairment of this process leads to increased inclusion formation and impaired autophagy-mediated aggregate clearance, suggesting that this PTM reduces α-Syn toxicity and serves as a protective mechanism (Shahpasandzadeh et al., 2014). Mutations in the main SUMOylation acceptor sites of α-Syn at K96 and K102 accelerate α-Syn aggregation in the mouse striatum and inhibit α-Syn degradation via the ubiquitin-proteasome system and the autophagy-lysosome pathway (Zhu et al., 2018) (Fig. 3). Overexpressing the SUMO-conjugating enzyme Ubc9 in N27 cells prevented PFF-induced toxicity. In addition, compared with wild-type mice, transgenic mice overexpressing Ubc9 treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) showed less damage to nigrostriatal dopaminergic neurons (Verma et al., 2020).
The mechanism through which SUMOylation may contribute to synucleinopathies is complex. It is either by altering the propensity of α-Syn to aggregate, or influencing the cell's degradation machinery and clearance of protein aggregates, or a combination of both (Vijayakumaran et al., 2015). In addition, SUMOylation may hinder α-Syn ubiquitination, hence preventing its degradation and causing accumulation (Rott et al., 2017). Altogether, it is believed that SUMO conjugation induces protein aggregation through the following mechanisms: 1) noncovalent interactions between SUMOylated proteins may act as molecular “glue” (Matunis et al., 2006); and 2) SUMOylation at multiple lysine residues changes the conformation of the target protein (Wilkinson and Henley, 2010), alters protein–protein interactions (Song et al., 2004), eventually leading to protein aggregation in neurodegenerative disorders.
Altogether, this evidence highlights the significance and complexity of the SUMOylation machinery in modulating α-Syn aggregation and toxicity in the pathophysiology of synucleinopathies. However, there are still many unknowns to elucidate the mechanisms behind the role of SUMOylation in α-Syn function and aggregation.
D. Acetylation
Acetylation is the addition of an acetyl group (CH3CO) to a molecule. The acetyl group on a target molecule can react with a wide range of atoms or functional groups. Protein acetylation is one of the most prevalent posttranslational modifications in eukaryotes, in which an acetyl group is transferred from acetyl coenzyme A to a specific site on a polypeptide chain (Verdin and Ott, 2015). Although acetylation has been detected on serine, threonine (Mukherjee et al., 2006, 2007), arginine, and histidine (Jedlicka et al., 2018) residues of proteins, amino group (nitrogen) acetylation has been investigated the most. Lysine acetylation (Nε-acetylation) and N-terminal protein acetylation (Nα-acetylation) are two separate processes for the acetylation of protein amino groups (Christensen et al., 2019).
α-Syn is constitutively acetylated under physiological conditions (Bartels et al., 2010). Lysines, which are known to play a role in the creation of an α-helical structure upon lipid contact, are acetylated (Plotegher and Bubacco, 2016). In addition, the N-terminus of α-Syn extracted from brain tissues (healthy subjects, PD or DLB patients) (Ohrfelt et al., 2011), erythrocytes (Bartels et al., 2011), or mammalian cell lines (Fauvet et al., 2012) is acetylated. N-terminal acetylation increases the membrane affinity of α-Syn by regulating its binding affinity for lipid vesicles (Dikiy and Eliezer, 2014) and synaptic vesicles (Runfola et al., 2020). However, by using solid-state nuclear magnetic resonance spectroscopy, N-acetylation was found to have no effect on the membrane-bound conformational properties of α-Syn (Dikiy and Eliezer, 2014; Runfola et al., 2020).
N-terminal acetylation is important for the formation of α-helical oligomers of α-Syn. Native α-Syn has been described as a monomeric protein (George et al., 1995) or α-helical tetramer (Bartels et al., 2011). Acetylation of the N-terminal region of α-Syn leads to the removal of its charge, resulting in increased hydrophobicity. This change enhances the protein’s ability to engage in hydrophobic interactions, which are crucial for its folding into the native tetrameric structure or for the aggregation process associated with PD (Trexler and Rhoades, 2012).
Additionally, N-terminal acetylation of α-Syn in vitro leads to the formation of fibrils with an identical morphology to those generated by the nonacetylated variant (Kang et al., 2012). However, acetylated α-Syn fibrils grow at a slower rate and are more resistant to aggregation (Kang et al., 2013; Bartels et al., 2014). This is believed to be due to an increase in α-helical folding propensity (Bartels et al., 2014). Furthermore, in an in vitro model using a solid-state nanopore system, N-terminal acetylation reduced α-Syn oligomerization. Molecular dynamics simulations suggest that the addition of an acetyl group at the N-terminus disrupts intermolecular hydrogen bonds, slowing the initial α-Syn oligomerization (Bu et al., 2017). However, with PD-causing mutations, A30P, E46K, and A53T, the protective effect of N-terminal acetylation against α-Syn aggregation is impaired (Ruzafa et al., 2017), suggesting a potential link between the N-terminus and the region of these mutations that may be crucial for α-Syn aggregation (Fig. 3). In addition, N-terminal acetylation modifies the conformation of monomeric α-Syn species in a region known to be essential for metal binding (Kang et al., 2012). For example, α-Syn acetylation has been suggested to affect Cu2+–α-Syn interactions in vitro. When compared with nonacetylated α-Syn, N-terminal acetylation abolishes Cu2+ binding at the high-affinity site and alters the Cu2+ interaction site, potentially resulting in significantly decreased α-Syn fibrillization (Moriarty et al., 2014).
In addition to N-terminal acetylation, lysine residues in α-Syn may potentially be acetylated (Struhl, 1998; Strahl and Allis, 2000; Zhao et al., 2010). Mass spectrometry analysis of endogenous α-Syn from wild-type mouse brains showed that K6 and K10 can be acetylated (de Oliveira et al., 2017). Furthermore, acetylation patterns in rat and human skeletal muscle biopsies revealed that acetylation occurs on α-Syn lysines 6, 34, 45, and 96 (Lundby et al., 2012). Acetylation at K6 and K10 sites reduces α-Syn aggregation in vitro as well as toxicity in vivo. In fact, mutations at these two sites that inhibit acetylation exacerbate α-Syn toxicity in the substantia nigra of rats. Assessing the impact of soluble α-Syn acetylation on the seeding potential of various pathological α-Syn forms (LB-α-Syn, GCI-α-Syn, or PFF) in HEK293 cells revealed that acetylation at positions K21, K43, and K45 significantly diminished the seeding potential of LB-α-Syn. Yet, acetylation at only K43 and K45 effectively decreased the seeding potential of GCI-α-Syn. Interestingly, only K43 acetylation impacted the seeding potential of PFFs. This indicates that acetylation of soluble α-Syn influences the propagation of pathological α-Syn in a site and strain-dependent manner (Zhang et al., 2023).
Genetic inhibition of sirtuin 2, a protein that removes α-Syn acetyl groups, alleviates the deleterious effects of α-Syn in two animal models of PD, including adeno-associated viral vectors-mediated α-Syn expression in the substantia nigra and chronic MPTP mouse model (de Oliveira et al., 2017), suggesting an important regulatory role for α-Syn acetylation in its aggregation tendency and toxicity (de Oliveira et al., 2017; González et al., 2019).
E. Glycosylation
Glycosylation is the most abundant and diverse form of PTM of proteins (Schjoldager et al., 2020) and is critical for physiological and pathological cellular functions (Reily et al., 2019). The two major types of protein glycosylation are N-glycosylation and O-glycosylation. N-glycosylation is the most common (Spiro, 2002), involving an N-glycosidic bond that links the nitrogen of an asparagine residue amide group to the N-acetylglucosamine (GlcNAc) of a glycan (Nalivaeva and Turner, 2001). O-glycosylation in humans often occurs via an N-acetylgalactosamine attached to the hydroxyl group of serine or threonine residues [3]. O-GlcNAcylation is highly abundant in the mammalian brain (Khidekel et al., 2004; Lee et al., 2020), in which the monosaccharide GlcNAc is attached to serine or threonine residues of various nuclear, cytosolic and mitochondrial proteins (Holt and Hart, 1986; Love et al., 2003). This modification is important for regulating cellular processes such as signal transduction and protein homeostasis (Hart et al., 2011; Balana and Pratt, 2021).
Similar to several aggregation-prone proteins that directly contribute to neurodegeneration and are modified by O-GlcNAcylation, several proteomics investigations have revealed that α-Syn can be O-GlcNAcylated (Wang et al., 2009, 2010, 2017b; Alfaro et al., 2012; Morris et al., 2015). Interestingly, O-glycosylated α-Syn is a substrate for parkin’s E3 ubiquitin ligase activity in the normal human brain, and there is an accumulation of GlcNAcylated α-Syn in parkin-linked PD-affected brains (Shimura et al., 2001).
At least nine different Ser/Thr residues with O-GlcNAcylation modification on α-Syn have been identified in vivo in mouse and human tissues (Levine et al., 2019). Initial studies on O-GlcNAcylated α-Syn at threonine 72 (T72) demonstrated that this modification has a substoichiometric inhibitory effect on α-Syn aggregation and inhibits the toxicity of α-Syn (Marotta et al., 2015). Subsequently, O-GlcNAcylation at Serine 87 (S87) was shown to have a similar effect on α-Syn aggregation, but to a lesser extent than that for the same modification at T72 (Lewis et al., 2017). An in vitro study using synthetic α-Syn with O-GlcNAcylation at S87 or T72 showed that O-GlcNAcylation at these two sites prevents α-Syn aggregation and enhances soluble and Thioflavin T negative oligomers (Zhang et al., 2017a) (Fig. 3). More recently, O-GlcNAcylated α-syn(gS87) PFF injected in the striatum of wild-type mice was found to result in a milder pathology than that caused by unmodified α-syn PFF, with no significant loss of nigral TH-positive neurons and fewer pS129-positive inclusions, highlighting the reduced potential of α-syn(gS87) PFF to induce neuronal pathology (Balana et al., 2024). Consistent with these in vivo results, a glycoside hydrolase O-GlcNAcase inhibitor that significantly increased O-GlcNAcylated α-Syn was able to improve motor performance and decrease astrogliosis and pS129 immunoreactivity in Thy1-Syn transgenic mice (Permanne et al., 2022). Furthermore, O-GlcNAcylation at both S87 or T72 residues inhibits α-Syn cleavage by the protease calpain (Levine et al., 2017). The precise effect of calpain proteolysis in PD is unknown. Calpain-derived α-Syn fragments have been identified in aggregates from human PD and DLB brains (Dufty et al., 2007). However, in vitro data reveal that calpain-mediated cleavage of α-Syn near and within its middle region yields fragments that do not aggregate (Mishizen-Eberz et al., 2005).
In vitro characterization of six out of nine sites of O-GlcNAcylation on α-Syn demonstrates that this PTM in general has largely inhibitory but site-specific effects on the aggregation and cellular toxicity of α-Syn. For example, O-GlcNAcylation at T75, T81, or S87 prevents the extension of PFFs, whereas the same modification at T72 does not (Levine et al., 2019). Interestingly, many of the O-GlcNAcylation modified Ser/Thr residues are also known phosphorylation sites, leading to a reciprocal relationship with often opposite downstream effects (Whelan et al., 2008; Hart et al., 2011; van der Laarse et al., 2018; Schwein and Woo, 2020), suggesting a regulatory crosstalk between O-GlcNAcylation and phosphorylation, which is further discussed later.
F. Glycation
Glycation differs from glycosylation and other PTMs in that it is spontaneous, nonenzymatic, and typically irreversible. It is the covalent attachment of a reducing sugar to the free amino groups of a protein, lipid, or DNA that forms advanced glycation end products (AGEs) (Fu et al., 1996). AGEs may cause cell damage via various pathways that have implications in several diseases (Jomova et al., 2010; Vicente Miranda and Outeiro, 2010). Glycation is an age-dependent PTM that can alter the structure and function of multiple proteins. In PD-affected brains, glycation can be detected at the periphery of Lewy bodies (Vicente Miranda et al., 2016). This has been reported in the substantia nigra and locus coeruleus of PD patients (Castellani et al., 1996). It has also been observed in the cerebral cortex, amygdala, and substantia nigra of healthy older individuals, but the levels are higher in PD, implying a potential pathogenetic role for glycation in this disease (Dalfó et al., 2005).
α-Syn is one of the most abundantly glycated proteins in PD (Vicente Miranda et al., 2017b; Videira and Castro-Caldas, 2018). It has 15 lysine residues that can potentially be glycated at multiple sites (Fig. 2), leading to the formation of a variety of early glycation products and AGEs (Vicente Miranda and Outeiro, 2010; Guerrero et al., 2013). In a comprehensive analysis focused on identifying PTMs of soluble α-Syn purified from synucleinopathies as well as control brains, glycation in the form of carboxymethylation and carboxyethylation have been identified on lysine residues (Zhang et al., 2023).
AGEs have been found to be colocalized with α-Syn where they have been linked to accelerated protein aggregation (Padmaraju et al., 2011). In vitro studies have shown that the dicarbonyl compound methylglyoxal (MGO) and the sugar ribose are the most effective agents for inducing α-Syn glycation and preventing fibril formation (Lee et al., 2009; Farzadfard et al., 2022) by decreasing the conformational flexibility of the protein (Lee et al., 2009) during the fibril elongation step rather than nucleation (Farzadfard et al., 2022). However, there is also evidence that glycation of α-Syn affects the nucleation of protein aggregates and that glycated α-Syn is more prone to oligomerization in both human cell lines and animal models (Padmaraju et al., 2011; Vicente Miranda et al., 2017b) (Fig. 3). MGO injection in the substantia nigra of wild-type and Thy1-α-Syn transgenic mice induced α-Syn glycation and aggregation as well as loss of TH-positive neurons (Vicente Miranda et al., 2017b). In addition, intracerebroventricular injection of MGO in the same transgenic animals increased glycation of glutamatergic-associated proteins, such as N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid, glutaminase, vesicular glutamate transporters, and excitatory amino acid transporter 1; induced glutamatergic hyperactivity in the midbrain; and exacerbated motor and nonmotor behavioral performance, indicating that glycation can also modulate glutamatergic signaling (Chegão et al., 2022).
Glycated α-Syn oligomers may exert toxicity in neuronal cells through multiple mechanisms including increased oxidative stress, proteasome dysfunction, activated microglia, and neuroinflammation (Guerrero et al., 2013) (Fig. 3). Furthermore, glycation of α-Syn can result in inactivation of glyceraldehyde 3-phosphate dehydrogenase, a glycolytic enzyme linked to neurodegenerative diseases (Semenyuk et al., 2019). Inactivation of glyceraldehyde 3-phosphate dehydrogenase in turn results in redirection of the glucose flux and potentially increased dicarbonyl formation rate (König et al., 2018). Glycation of α-Syn also disrupts its homeostasis through impaired lipid membrane binding, altered aggregation behavior, localization, and clearance, which might be due to reduced α-Syn ubiquitination and SUMOylation, the two other PTMs that also target lysine residues (Plotegher and Bubacco, 2016; Vicente Miranda et al., 2017b; Farzadfard et al., 2022). Altogether, characterization of the glycosylation and glycation patterns of α-Syn can shed light on the significance of these sugar-mediated PTMs in α-Syn homeostasis in physiological and pathological conditions.
G. Nitration
Protein nitration is a nitrosative stress-related PTM that affects tyrosine (Y) residues. Excess nitric oxide (NO), oxidants, and transition metal centers interact to cause protein tyrosine nitration, which mostly happens through free radical routes (Radi, 2004).
α-Syn is nitrated at its C-terminal residues Y125, Y133, and Y136, as well as at its N terminal residue Y39 (Benoit I. Giasson et al., 2000). Nitrated α-Syn is abundant in the brains of patients with neurodegenerative disorders (Duda et al., 2000) including PD, DLB, the Lewy body form of Alzheimer's disease, and MSA (Giasson et al., 2000; He et al., 2019). In addition to the brain (Giasson et al., 2000; Reynolds et al., 2008; Sonustun et al., 2022), nitrated α-Syn has been detected in the gastrointestinal tract (Xuan et al., 2016), salivary gland (Ma et al., 2019), and blood cells of PD patients (Prigione et al., 2010). Systemic administration of MPTP to mice increased nitrated and phosphorylated α-Syn levels in the enteric glial cells of the gastric myenteric plexus. This increase in nitrated α-Syn was directly correlated with the number of MPTP administrations, highlighting the dose-dependent nature of this toxin’s effects on the enteric nervous system (Heng et al., 2022). Compared with healthy age-matched individuals, blood samples from PD patients showed a significant increase in n-Y39-Syn levels. Notably, this elevation was even more significant in those diagnosed with the disease for 10 years or more (Vicente Miranda et al., 2017a). This finding suggests that the nitration of α-Syn at this site might serve as a potential biomarker for the disease.
In human postmortem brains of PD patients, evaluation of the differential distribution and abundance of p-S87 α-Syn, p-S129 α-Syn, and n-Y39 α-Syn revealed that p-S129 α-Syn is the most dominant and earliest PTM, followed by nitrated Y39 α-Syn, while p-S87 α-Syn was detected in fewer LBs in PD brains and appeared later in the disease course (Sonustun et al., 2022). In addition, in the MPTP mouse model of PD, α-Syn was found to be nitrated in the ventral midbrain and striatum (Przedborski et al., 2001).
α-Syn is nitrated through inducible nitric oxide synthase (iNOS), and overexpression of iNOS in vitro leads to reduced α-Syn monomers while increasing high molecular weight species (>30 kDa) (Stone et al., 2012). Furthermore, nitrated α-Syn induces cell death through activation of iNOS and inhibition of phosphorylated focal adhesion kinase in SH-SY5Y cells, suggesting that the cytotoxicity of nitrated α-Syn might be mediated through an integrin-iNOS/-focal adhesion kinase signaling pathway and that α-Syn nitration contributes to neuronal degeneration (Liu et al., 2011).
The effects of α-Syn tyrosine nitration on aggregation (Norris et al., 2003; Yamin et al., 2003; Hodara et al., 2004; Uversky et al., 2005), vesicle binding, and proteolytic degradation (Hodara et al., 2004) have been assessed in several studies. Nitration of the tyrosine Y39 impairs the affinity of α-Syn for lipid membranes through electrostatic repulsion. In fact, membrane binding is mediated by α-Syn (aa: 1–95), whereas Y39 nitration within this region interferes with binding (Hodara et al., 2004; Sevcsik et al., 2011). The nitration of Y39 resulted in disruption of the alpha helical shape of nitrated α-Syn in the presence of vesicles, which was accompanied by a reduction in the affinity of α-Syn monomers to vesicles (Hodara et al., 2004). In addition, nitration of C-terminal residues Y125, Y133, and Y136 decreases α-Syn membrane-binding affinity by changing the ensemble of conformational states and eliminating those capable of membrane binding (Sevcsik et al., 2011). The interaction of α-Syn with membranes is believed to protect it from oxidation and nitration and eventually reduces the number of protein molecules that are accessible to form aggregates (Trostchansky et al., 2005).
Nitrated α-Syn oligomers produced from exposure of human recombinant α-Syn to nitrating agents such as peroxynitrite/CO2 or myeloperoxidase/H2O2/nitrite are highly stabilized due to the covalent crosslinking that occurs when tyrosine is oxidized to form o,o′-dityrosine (Souza et al., 2000). Although substantial oligomerization occurs upon nitration of Y39, it is believed that Y125 is more important for α-Syn dimer formation (Takahashi et al., 2002). Moreover, coincubation of low concentrations of monomers and dimers of nitrated α-Syn with unmodified α-Syn has been shown to enhance the rate of fibril formation. Nitrated α-Syn monomer alone, on the other hand, was incapable of forming fibrils (Hodara et al., 2004). This finding is supported by some evidence suggesting that nitration effectively inhibits α-Syn fibrillization (Yamin et al., 2003; Uversky et al., 2005; Barrett and Timothy Greenamyre, 2015).
Nitrated α-Syn is significantly more toxic to dopaminergic neurons than nonnitrated α-Syn (Yu et al., 2010; He et al., 2019) (Fig. 3). In vivo, nitrated α-Syn injection into the substantia nigra of rats caused motor dysfunction as well as a significant decrease in the number of dopaminergic neurons (Yu et al., 2010). Additionally, compared with unmodified α-Syn, nitrated α-Syn has the ability to elicit an immune response (Benner et al., 2008), activate a neurotoxic microglial phenotype, and result in a significant decrease in viable dopaminergic MES23.5 cells (Reynolds et al., 2008). On the other hand, an aging-related hyperinflammatory response in the nigrostriatal system elicited by intrapallidal lipopolysaccaride injection in 16-month-old rats increased α-Syn nitration, which further exacerbated brain inflammation (Choi et al., 2010).
S-nitrosylation is another PTM that occurs when a nitrogen monoxide group is attached to a thiol side chain of cysteine (Hess et al., 2005). S-nitrosylation differs from nitration, and α-Syn has not been observed to undergo this modification.
Given the role of nitration in driving the aggregation and toxicity of α-Syn, further investigations of this target using pharmacological tools can have the potential to expand our understanding of how this PTM contributes to α-Syn-related pathology and open new avenues for therapeutic interventions.
H. Oxidation
All amino acid residues in proteins can be oxidized. Methionine, cysteine, tyrosine, and tryptophan residues are especially susceptible to oxidation by almost all reactive oxygen species (ROS) (Andrés et al., 2022). Cysteine residues are transformed into disulfides, and methionine residues are changed into methionine sulfoxide (MetO) residues, even under mild oxidative circumstances (Berlett and Stadtman, 1997). Two enantiomers, known as Met-R(O) and Met-S(O), are produced as a result of the formation of an asymmetric center at the sulfur atom (Glaser et al., 2005). Under physiological conditions, methionine undergoes reversible oxidation to methionine sulfoxide, and under certain conditions, it can be reduced back to the original amino acid. The irreversible oxidation of methionine to methionine sulfone is uncommon and only occurs in the presence of powerful oxidants (Hoshi and Heinemann, 2001). The MetO content of proteins rises with aging (Stadtman et al., 2005), and its level is controlled by a number of mechanisms, including the rate of ROS production, antioxidant capacity, and proteolytic activities that degrade oxidized proteins, and changes to the ability to convert MetO residues back to Met residues (Stadtman et al., 2005).
Like other proteins, α-Syn is a target for oxidation, but it lacks a cysteine residue, making methionines and tyrosines the primary amino acids susceptible to oxidation (Chavarría and Souza, 2013). α-Syn has four methionines: M1 and M5 at the N-terminal and M116 and M127 at the C-terminal of the protein. M1, M116, and M127 are more resistant to oxidation than M5, most likely because of the structure of the natively unfolded α-Syn. Electrostatic and hydrophobic interactions in α-Syn are altered by methionine oxidation (Zhou et al., 2010). Using nuclear magnetic resonance analysis, the two N-terminal Met residues M1 and M5 were found to be the only ones that undergo oxidation upon interaction with lipid vesicles, and the C-terminal Met residues do not. The oxidation of M1 decreases the rate of oxidation of M5 and vice versa (Maltsev et al., 2013). This is a site-specific PTM crosstalk in which a PTM on one site controls the same PTM on the other site of a protein (Fig. 2).
Protein oxidation on methionine residues to produce sulfoxide is thought to influence the function of proteins through structural modification (Schildknecht et al., 2013). Such structure alteration results in a significant reduction of the hydrophobic property of the methionine side chain, which may impact secondary and tertiary structures (Uversky et al., 2002). Oxidized α-Syn has been shown to exhibit restricted secondary structure transitions in response to dehydration and modestly increased tertiary structure transitions in response to ligand binding. This variety in susceptibility to forced folding could explain the loss of fibrillization potential of oxidized α-Syn (Ponzini et al., 2019). The degree to which methionine oxidized α-Syn inhibits fibrillization is believed to be proportional to the quantity of oxidized methionines. With one methionine oxidized, the fibrillization kinetics are comparable to those with nonoxidized α-Syn, and with increasing numbers of methionine sulfoxides, the fibrillization kinetics grow increasingly slower (Hokenson et al., 2004) (Fig. 3). Methionine oxidation of α-Syn has been suggested as a mechanism for preventing lipid oxidation in one report (Zhu et al., 2006). While α-Syn is subject to oxidation, its overexpression is known to exacerbate oxidative stress in various models (Junn and Mouradian, 2002; Dias et al., 2013).
Metal-mediated oxidation, which may cause structural damage to proteins and has been linked to aging and disease (Requena et al., 2001) also occurs with α-Syn. This protein is sensitive to oxidation catalyzed by copper leading to significant oligomerization and precipitation (Paik et al., 2000; Requena et al., 2001). Copper-induced oxidation involves the reduction of Cu2+ by an electron donor and the conversion of molecular oxygen into reactive oxygen species that induce oxidative changes in the protein (Binolfi et al., 2010). Notably, prolonged exposure to manganese, which can promote α-Syn fibril formation (Xu et al., 2021), may result in parkinsonism among welders in a dose-dependent manner (Racette et al., 2017). Organotypic brain slices obtained from postnatal days 3–4 rats and treated with manganese exhibited a significant increase in α-Syn oxidation, oligomerization, as well as neurotoxicity (Xu et al., 2013).
Another mechanism for α-Syn oxidation is through cytochrome c (Hashimoto et al., 1999; Bayir et al., 2009), which is a protein that resides in the intermembrane space of mitochondria and is released into the cytosol in response to proapoptotic signals (Garrido et al., 2006). Cytochrome c can colocalize with α-Syn in SH-SY5Y cells exposed to a proapoptotic or pro-oxidant stimulus (Bayir et al., 2009) and in Lewy bodies of PD patients (Hashimoto et al., 1999). These findings collectively suggest that α-Syn oxidation and its functional consequences have an important biological impact on its aggregation and the development of synucleinopathies.
I. Arginylation
Protein arginylation is a PTM mediated by arginyltransferase, an enzyme that exists in all eukaryotic cells. Arginylation, which modifies the molecular interactions and activity of numerous proteins in vivo, is necessary for embryogenesis and controls angiogenesis, tissue morphogenesis, and heart development (Saha and Kashina, 2011).
α-Syn is known to be an effective target for arginyltransferase in vitro. Mass spectrometry identified arginylated α-Syn at glutamate residues E46 and E83 in the mouse brain (Wang et al., 2017a). Arginylated α-Syn has the same vesicle affinity as the unmodified protein (Pan et al., 2020).
Residues E46 and E83 have been considered to be important in α-Syn function and the pathology of PD (Waxman et al., 2010; Boyer et al., 2020). Mutation of residue 46 from glutamate to lysine (E46K) is one of the genetic causes of autosomal dominant synucleinopathies presenting as parkinsonism and dementia (Spillantini and Goedert, 2018). The E46K mutation modifies α-Syn fibril structure and speeds up filament assembly to the same extent as the A53T mutation (Choi et al., 2004), as well as increases the pathogenicity of α-Syn fibrils compared with the wild-type protein (Boyer et al., 2020). In addition, the presence of the highly charged E83 residue inhibits the formation of α-Syn amyloid fibrils (Waxman et al., 2010). Therefore, a double mutation at both these sites to generate an α-Syn mutant incapable of arginylation led to an even greater α-syn aggregation in the brain of mice and in cultured cells, as well as a reduction in its capacity to be eliminated via typical degradation mechanisms (Wang et al., 2017a), suggesting that these mutations likely work in concert to promote intracellular α-Syn accumulation. Moreover, in vitro data reveal that arginylation at both sites slows down the formation of fibrils. However, arginylation at E83, but not at E46, reduces α-Syn aggregation and lowers the proportion of monomer integration into fibrils in a dose-dependent manner (Pan et al., 2020) (Fig. 3). Recent findings from a human brain study showed an inverse relationship between arginylation level, total α-Syn level, and patient age, suggesting a possible causative link between a decline in arginylation and α-Syn-dependent neuropathology. α-Syn arginylation is, therefore, proposed to be a potential neuroprotective mechanism in the human brain during neurodegeneration and aging (Zhao et al., 2022).
J. Polyamination
The addition of polyamines to proteins, also known as polyamination, can be facilitated by transglutaminases (Folk et al., 1980). Polyamines, which are small cationic molecules, are found at millimolar levels in the brain (Morrison et al., 1995). The addition of positively charged polyamines to a protein surface can affect protein-protein interactions and likely other posttranslational modifications (Schuster and Bernhardt, 2011). Polyamines such as spermidine and spermine are able to regulate the tendency of α-Syn to form fibrils and may, therefore, play a role in the formation of α-Syn aggregates (Antony et al., 2003; Fernández et al., 2004; Krasnoslobodtsev et al., 2012). A nuclear magnetic resonance study revealed that polyamines accelerate the aggregation of α-Syn through a direct interaction with the C-terminus of the protein (Fernández et al., 2004). Under physiologic conditions, the addition of spermidine dramatically increases the susceptibility of both wild-type and mutant α-Syn to misfold, suggesting that elevated levels of spermidine and possibly other polyamines can contribute to the pathogenesis of synucleinopathy (Krasnoslobodtsev et al., 2012). Spermine has also been reported to enhance the rate of α-Syn aggregation by modifying protein conformation, which then proceeds to form aggregates (Grabenauer et al., 2008) (Fig. 3).
One member of the transglutaminase family of enzymes, transglutaminase 2 (TG2), which is widely expressed in the human brain (Kim et al., 1999), is a multifunctional protein with roles that include crosslinking through transamidation, GTPase, protein disulfide isomerase, cell adhesion, scaffolding, and kinase activities (Tatsukawa et al., 2016). TG2 is involved in α-Syn aggregation and the pathogenesis of PD and DLB (Citron et al., 2002; Junn et al., 2003; Andringa et al., 2004; Wilhelmus et al., 2011; Grosso and Mouradian, 2012). We have found immunohistochemical evidence for TG2-catalyzed crosslinked α-Syn in the halo of Lewy bodies in postmortem brains from these patients (Junn et al., 2003). We have also found evidence for TG2-mediated exacerbation of α-Syn pathology in mouse models. Double transgenic mice for human α-Syn and TG2 exhibit more high-molecular-weight species of α-Syn in brain lysates and develop α-Syn aggregates in the synaptic vesicle fraction associated with exacerbated pathology and behavioral deficits (Grosso et al., 2014). On the other hand, TG2 KO/α-Syn transgenic mice have fewer α-Syn aggregates in the brain and milder pathologic and behavioral deficits compared with α-SynTg mice, indicating that deletion of TG2 mitigates α-Syn mediated neurodegeneration (Zhang et al., 2020). The transamidation (Andringa et al., 2004; Schmid et al., 2009) function, rather than polyamination, is the mechanism by which TG2 affects α-Syn aggregation.
K. Truncation
Protein truncation is an irreversible modification of proteins to generate shorter proteins with new N‐ or C‐termini and is proposed to alter the protein's activity and biological properties through structural and conformational modification (Fortelny et al., 2015). Approximately 15% of α-Syn in LBs is believed to be truncated (Liu et al., 2005; Zhang et al., 2019b). The presence of various truncated forms of α-Syn with molecular weights ranging from 10–15 kDa in LBs from PD, DLB (Baba et al., 1998; Tofaris et al., 2003; Li et al., 2005; Liu et al., 2005; Grassi et al., 2018), or MSA (Anderson et al., 2006) brains raises the question of the role of truncation in α-Syn aggregation. Numerous enzymes, including neurosin (Kasai et al., 2008), calpain I (Dufty et al., 2007), cathepsin D (Sevlever et al., 2008), and matrix metalloproteinase 3 (Choi et al., 2011), have been connected to α-Syn truncation. This PTM occurs at the N-terminus (Terada et al., 2018), the NAC region (Mishizen-Eberz et al., 2005; Kasai et al., 2008), or the C-terminus (Serpell et al., 2000; Mishizen-Eberz et al., 2003; Murray et al., 2003; Mishizen-Eberz et al., 2005; Li et al., 2005; Tofaris et al., 2006; Dufty et al., 2007; Periquet et al., 2007; Kasai et al., 2008; Zhang et al., 2017b; Terada et al., 2018; van der Wateren et al., 2018; McGlinchey et al., 2019; Ni et al., 2019) of α-Syn, and modification at each site has a particular effect on α-Syn aggregation.
Evidence from in vitro studies using recombinant α-Syn variants and in vivo studies indicate that α-Syn truncation at its C-terminus tends to boost its aggregation and pathological features (Murray et al., 2003; Li et al., 2005; Tofaris et al., 2006; Periquet et al., 2007; van der Wateren et al., 2018; McGlinchey et al., 2019; Ni et al., 2019) (Fig. 3). In vitro studies have shown that the truncated α-Syn variants including 1-89, 1-102, 1-110, 1-120, and 1-130 aggregate faster than the full-length protein (Murray et al., 2003). Additionally, the truncation of the 16 (α-Syn 1-124) or 32 (α-Syn 1-109) C-terminal amino acid residues of wild-type α-Syn leads to a noticeable acceleration of aggregation (Hoyer et al., 2004). In SH-SY5Y cells, co-overexpression of truncated α-Syn with the full-length protein enhances sensitivity to oxidative stress, suggesting that truncated α-Syn has a role in the pathogenesis of synucleinopathies (Liu et al., 2005). Truncation of α-Syn in the gut could influence its aggregation and propagation to the brain. Prolonged exposure of mice to oral rotenone causes an increase in the expression of asparagine endopeptidase in the colon, which cleaves α-Syn at the N103 residue. The resulting C-terminally truncated α-Syn is prone to aggregate and form fibrillary inclusions that can then propagate to the brain through the vagus nerve (Wang et al., 2023).
In addition to protease mediated truncations, there are at least three alternative splicing isoforms of α-Syn that lack exon 3 (residues 41–54), exon 5 (residues 103–130), or both, and these are known as α-Syn 126, α-Syn 112, and α-Syn 98, respectively (Beyer et al., 2008; McLean et al., 2012; Gámez-Valero and Beyer, 2018). These isoforms, and in particular those lacking exon 5 (C-terminal residues), may exhibit traits resembling those of C-terminally truncated α-Syn, such as enhanced aggregation and toxicity (Ma et al., 2013; Hassanzadeh et al., 2023) (Fig. 3), and their expression levels are mostly higher in PD, DLB, Alzheimer’s, and mixed pathology affected brains compared with healthy subjects (Beyer et al., 2008, 2009; McLean et al., 2012; Cardo et al., 2014; Soll et al., 2020).
Compared with C terminal truncations of α-Syn, N-terminal truncations are investigated less and are limited to in vitro studies designed to assess fibril assembly. For instance, removal of 13, 35, or 40 residues from the N-terminus altered aggregation behavior, and truncated fibrils were found to be poor seeds for soluble wild-type α-Syn (McGlinchey et al., 2021). Interestingly, even though N-terminal truncation slowed fibril assembly in vitro, mice injected intrastriatally with N-terminally 10- or 30-residue-truncated human α-Syn fibrils exhibited more α-Syn pathology than mice injected with full-length WT fibrils (Terada et al., 2018) (Fig. 3).
Truncations at the NAC region inhibit α-Syn aggregation. A study in transgenic drosophila demonstrated that α-Syn with a deletion of amino acids 71–82 in the NAC domain is incapable of aggregation, whereas the C-terminally truncated protein consisting of amino acids 1–120 exhibited enhanced aggregation into large inclusion bodies and increased toxicity to dopaminergic neurons (Periquet et al., 2007) (Fig. 3). In addition, in vitro incubation of α-Syn with the serine protease neurosin showed that α-Syn cleaved in the central NAC region (after K80) generates fragments with reduced tendency to polymerize. However, compared with full-length α-Syn, deletions of the fragment after K97 that preserves the entire NAC region and truncates the C-terminal section may have a higher tendency to polymerize (Kasai et al., 2008).
α-Syn truncation, and in particular C-terminal truncation, is believed to be increased by impaired proteostasis, whereby only partial degradation of monomeric and fibrillar forms of α-Syn occurs due to reduced lysosomal autophagy and oxidative stress (Sorrentino and Giasson, 2020) (Fig. 3). Interventions aimed at maintaining proteostatic balance and reducing truncated α-Syn have shown therapeutic potential (Spencer et al., 2013; Bassil et al., 2016; Hassen et al., 2018). However, the site-dependent impact of truncation on α-Syn aggregation should be regarded as a crucial point.
L. Methylation
Protein methylation is a PTM that involves the addition of one or more methyl groups to specific amino acids in a protein. While lysine and arginine residues are its primary targets (Blanc and Richard, 2017; Luo, 2018), methylation can also modify other residues such as histidine and glutamine, as well as the N- and C-termini of proteins (Clarke, 1992; Diaz et al., 2021; Małecki et al., 2022). The addition of the methyl group to proteins is facilitated by enzymes known as methyltransferases (Schubert et al., 2003; Falnes et al., 2016). Protein methylation is an important modification that governs various cellular functions, such as signaling, gene transcription, RNA splicing, translation, and protein–protein interactions (Biggar and Li, 2015; Cornett et al., 2019; Guccione and Richard, 2019; Lorton and Shechter, 2019). Methylation has also been identified as a factor influencing protein aggregation. In an in vitro study investigating the impact of methylation on tau protein, lysine methylation was found to reduce the natural propensity of tau to form aggregates by slowing the nucleation rate, preventing the elongation rate, and destabilizing mature filaments (Funk et al., 2014).
In a recent investigation, soluble α-Syn was isolated from the brains of patients with α-synucleinopathies and normal brains using immunoprecipitation and examined using liquid chromatography-tandem mass spectrometry. This study unveiled, for the first time, novel modifications of soluble α-Syn, including methylation and 4-hydroxynonenal on lysine residues. α-Syn can be monomethylated, demethylated, or trimethylated. In total, nine methylation sites, six dimethylation sites, and one trimethylation site were identified. As of now, the effects of these PTMs on α-Syn function and aggregation seeding potential remain unknown (Zhang et al., 2023).
III. Targeting α-Synuclein Posttranslational Modifications as a Therapeutic Strategy
α-Synuclein has been a major focus of research due to its central role in the pathogenesis of PD and other synucleinopathies. Several strategies are actively being investigated, and a large number of compounds targeting various aspects of α-Syn are currently in various stages of development (Grosso Jasutkar et al., 2022). Targeting α-Syn PTMs represents an underexplored but promising approach. Each α-Syn PTM can have unique effects on the protein's function, localization, and propensity to aggregate. Some of these modifications might exacerbate the disease by promoting α-Syn pathologic aggregation. Conversely, others may offer protection by reducing the possibility of protein misfolding and accumulation (Fig. 3). Given this intricate complexity, it is crucial to understand thoroughly the molecular mechanisms behind each PTM, develop and test specific agents targeting them, and examine the impact of each pharmacological manipulation on other PTMs in preclinical models of synucleinopathies. Table 2 provides an overview of pharmacological tools that aim to target α-Syn PTMs and their effects in cellular and animal models.
Pharmacological tools targeting α-synuclein PTMs
A. Phosphorylation
Targeting α-Syn phosphorylation, the most extensively studied PTM, is a tractable therapeutic approach. The modulation of kinases and phosphatases, key regulators of this PTM, is central in this regard. For example, the small molecule PLK2 inhibitor BI 2536 has been used as a pharmacological tool to modulate α-Syn phosphorylation in transgenic mice expressing human wild-type α-Syn-green fluorescent protein. Treating these animals with BI 2536 acutely led to a significant reduction in both phosphorylated and total α-Syn levels in synaptosomal and cytosolic pools of cortical tissue lysates. PLK2 deletion in these mice reduced presynaptic terminal pS129 α-Syn level and slowed the rate of neuronal death (Weston et al., 2021b). However, there was no effect on Lewy body-like pathology in these brains, including phosphorylation, following α-Syn PFF injection. The authors of this study raised doubt whether the neuroprotective effect of PLK2 inhibition is due to pS129 α-Syn.
Considering that multiple kinases can phosphorylate α-Syn, inhibiting a single kinase may not be an effective strategy. Alternatively, enhancing the dephosphorylation step has emerged as a viable alternative. In particular, phosphoprotein phosphatase 2 A plays a crucial role in dephosphorylating α-Syn at serine 129, a process significantly amplified by the carboxyl methylation of PP2A's catalytic C subunit. We have shown that enhancing PP2A methylation and its enzymatic activity pharmacologically can ameliorate the pathological phenotype observed in both α-Syn transgenic mice and in mice injected with α-Syn PFF in the striatum (Lee et al., 2011; Yan et al., 2018). Eicosanoyl-5-hydroxytryptamide, a fatty acid derivative of serotonin found in coffee that inhibits the PP2A methylesterase PME1 to maintain PP2A in a highly active methylated state, reduces the accumulation of phosphorylated α-Syn, ameliorates neuroinflammation, mitigates the propagation of α-Syn PFF and the ensuing nigrostriatal pathology, and improves behavioral performance (Yan et al., 2018). Overall, these observations demonstrate the importance of PP2A regulation in α-Syn phosphorylation and synucleinopathies.
B. Ubiquitination
As discussed earlier, α-Syn is a substrate for various deubiquitinase enzymes, including USP13. A recent study involving mice injected with a lentiviral vector expressing α-Syn into the substantia nigra revealed that treating mice with the USP13 inhibitor BK50118-C resulted in enhanced proteasome activity, significant reduction in α-Syn levels, preservation of dopamine neurotransmission, and improved motor and behavioral outcomes (Liu et al., 2022). Additionally, spautin-1, which targets both USP10 and USP13, was identified to impact the proteasome and autophagy pathways at significantly higher concentrations than BK50118-C. However, spautin-1 does not cross the blood–brain barrier, thereby limiting its therapeutic utility for PD (Liu et al., 2021).
In a screening of approximately 300 natural compounds, canthin-6-one emerged as a potent and selective compound capable of lowering levels of three different forms of α-syn (WT, A53T, and A30P mutations) in PC12 cells. Its mechanism of action involves activating the UPS for α-syn degradation, primarily through upregulation of the PSMD1 gene. This gene encodes the 26S proteasome non-ATPase regulatory subunit 1, crucial for protein degradation. The effect of canthin-6-one is mediated by activating the protein kinase A (PKA) pathway, highlighting its potential for PD treatment by enhancing proteasome function (Yuan et al., 2019).
C. SUMOylation
Ginkgolic acid, a natural compound extracted from Ginkgo biloba leaves, is known to inhibit SUMOylation by blocking the function of Ubc9 (Fukuda et al., 2009). An in vitro study conducted on SH-SY5Y neuroblastoma cells and rat primary cortical neurons showed that both pretreatment and posttreatment with ginkgolic acid, as well as with a related compound, anacardic acid, significantly reduced the number of cells with intracytoplasmic α-Syn and SUMO-1 positive aggregates. This reduction was associated with increasing cellular levels of LC3-positive autophagosomes, suggesting that the effect on α-Syn was achieved through promoting macroautophagy and removing aggregates (Vijayakumaran et al., 2019).
D. Glycosylation
Thiamet-G, a selective inhibitor of the enzyme O-GlcNAcase (OGA) that removes O-GlcNAc from proteins, has been shown to effectively reduce the uptake of α-Syn fibrils by cells. This reduction is associated with elevated levels of proteins modified by O-linked N-acetylglucosamine (O-GlcNAc) in the nucleus and cytoplasm, is concentration and time dependent, and is evident across various cell types, including mouse primary cortical neurons. The reverse is true; when cells are treated with 5SGlcNHex, an inhibitor of O-GlcNAc transferase that catalyzes the attachment of O-GlcNAc to proteins, there is an increase in the uptake of α-Syn PFF, supporting in vivo exploration of OGA inhibitors as a potential disease-modifying approach to treat PD and other synucleinopathies (Tavassoly et al., 2021).
Another OGA inhibitor, ASN90, also known as ASN120290 or ASN-561, promotes the O-GlcNAcylation of α-Syn in the brains of transgenic mice after daily oral dosing. This is accompanied by slowed progression of motor impairment and reduced astrogliosis. Administration of ASN90 to human tauopathy mouse models also prevents the development of tau pathology (neurofibrillary tangle formation) and functional deficits in motor behavior and breathing and increases survival. These findings provide a strong rationale for the development of OGA inhibitors as disease-modifying agents in both α-synucleinopathies and tauopathies (Permanne et al., 2022). A phase I trial is underway evaluating the distribution of ASN121151, an OGA inhibitor, to the central nervous system along with its safety and pharmacokinetics in elderly individuals, both healthy and those with Alzheimer’s disease (NCT04759365). Additionally, an ongoing PET study with multiple ascending doses is examining how ASN121151 affects brain OGA occupancy and the pharmacodynamic response in peripheral blood mononuclear cells following repeated doses in healthy participants (NCT05725005).
E. Glycation
Glycation triggers the formation of α-Syn oligomers (Vicente Miranda et al., 2017b) and impedes its clearance by disrupting the ubiquitin proteasome system and hindering the autophagy–lysosome pathway (Vicente Miranda et al., 2017b). Therefore, MGO scavengers have emerged as a potential therapeutic strategy in preclinical investigations. For example, aminoguanidine and tenilsetam have been shown to reverse glycation-induced reduction of α-Syn clearance, mitigating aggregation and toxicity and ameliorating motor abnormalities in the drosophila model (Vicente Miranda et al., 2017b). In SH-SY5Y cells, morphological changes associated with MGO are prevented with the addition of aminoguanidine or tenilsetam, reducing neurite retraction compared with samples that do not receive the MGO inhibitors (Webster et al., 2005). These findings pave the way for further research into the development of MGO scavengers as pharmacological tools for mitigating the progression of neurodegeneration associated with α-Syn.
F. Nitration
NOS activation or NO donors are known to promote glutamate release (Bal-Price and Brown, 2001), and NMDA receptor activation is linked to nitric oxide neurotoxicity (Sattler et al., 1999). Both α-Syn nitration and lipopolysaccaride-induced dopaminergic neuron death were ameliorated by the NMDA receptor antagonist MK801, suggesting that glutamate excitotoxicity may be implicated in NO generation, α-Syn nitration, and neuronal death (Gao et al., 2008).
In a study using the MPTP mouse model of PD, upregulation of neuronal NO synthase activity, increased nitrative stress, and α-Syn nitration were noted within the striatum. GYY4137, an H2S slow-releasing compound, provided neuroprotection by diminishing NO production. While the overall levels of α-Syn in the striatum remained unchanged after MPTP administration, there was a significant increase in the nitrated form of α-Syn in mice challenged with MPTP. However, this increase was effectively abolished when GYY4137 was coadministered, highlighting its potential in mitigating α-Syn nitration and nitrative stress-related neurodegeneration (Hou et al., 2017).
G. Polyamination
Polyamination of α-Syn has also been observed in a mouse model with combined deficiencies in the polyamine catabolism enzymes spermine oxidase and spermidine/spermine N1-acetyltransferase. These double knockout animals exhibit ataxia and significant cerebellar injury, which might be due to polyamination and aggregation of α-Syn. Administration of transglutaminase inhibitors, cystamine, and cysteamine to these mice reduced polyamine expression and α-Syn aggregation, ameliorated the severity of cerebellar injury, and significantly delayed the onset of ataxia. These findings suggest a role for polyamination in α-Syn aggregation and pathology (Zahedi et al., 2020). In another study, the gene expression profiles of PD patients' brainstems showed a disease-related decrease in SAT1, resulting in excessive polyamine levels (Lewandowski et al., 2010). Furthermore, in α-Syn transgenic mice, DENSPM (N1, N11-diethylnorspermine), a polyamine analog that increases SAT1 activity, decreased neuronal accumulation of α-Syn in the substantia nigra and ameliorated the phenotype, while an inhibitor exacerbated it, supporting the notion that there may be a relationship between SAT1 activity and PD pathology (Lewandowski et al., 2010).
H. Truncation
Pharmacological agents have been investigated against calpain, the enzyme associated with α-Syn truncation (Dufty et al., 2007), in both animal and cellular models of PD. Inhibition of calpain by calpeptin significantly attenuated gliosis and inflammatory markers and reduced α-Syn aggregation in MPTP-lesioned mice (Haque et al., 2020). In addition, in the rat rotenone model of PD where there is increased expression of pS129 α-Syn, elevated levels of calpain-1 and calpain-2 were detected in substantia nigra dopaminergic neurons. Rotenone treatment also triggered glial activation and neuroinflammation throughout the nigrostriatal pathway. However, calpeptin promoted the differentiation of microglia, prevented astroglia/microglia activation, and prevented nigral neuron loss (Zaman et al., 2022).
IV. Crosstalk among α-Synuclein Posttranslational Modifications
Accumulating evidence suggests that multiple PTM sites within a single protein may allow different PTM types to cooperatively control the biological function and structure of the protein (Csizmok and Forman-Kay, 2018). PTMs on a protein can also interact with one another or work together to regulate downstream signals. Crosstalk among PTMs occurs when many PTMs either positively or negatively influence each other's activity. Positive crosstalk happens when one PTM acts as a signal for the addition or deletion of another PTM or as a recognition site for a binding protein that performs the second modification. Negative crosstalk is identified as either direct competition between two PTMs or indirect effects when the first PTM hides the second PTM's recognition site (Wu et al., 2019).
For reasons summarized here, our current understanding of the crosstalk among different PTMs remains incomplete. PTMs and their interactions form a biological regulation that is hard to decipher because they do not require genomic regulation or changes in protein levels. Although conventional protein detection methods such as western blots and ELISA are important for identifying and/or quantifying PTMs (Azevedo et al., 2022), they are not appropriate for identifying the interplay between PTMs. These are antibody-based techniques that are dependent on antibody availability, and antibodies might be nonspecific with possible cross-reactions. Therefore, one of the obstacles limiting progress in understanding PTM crosstalk is the lack of robust techniques that can identify concurrent PTMs.
In recent years, the development of biological methods and strategies to identify PTM co-occurrences has provided a wealth of knowledge about their interplay (Olsen and Mann, 2013; Doll and Burlingame, 2015; Zecha et al., 2022). In addition to computational methods, which have been extensively used to identify this phenomenon (Lu et al., 2011; Jahangir et al., 2014), mass spectrometry methods, particularly liquid chromatography-tandem mass spectrometry techniques (Azevedo et al., 2022) and high-field asymmetric ion mobility spectrometry (Adoni et al., 2022), allow a better understanding of the complexity of PTMs and their crosstalk. PTMs crosstalk can happen intraprotein (within one protein) or interprotein (across different proteins) (Minguez et al., 2015). Intraprotein crosstalk might happen reciprocally on the same residue or at proximal or distal locations along the protein sequence (van der Laarse et al., 2018). PTMs crosstalk between proteins (interprotein) is found to occur in close physical proximity within a complex of several proteins, as well as between proteins in a signaling pathway (Minguez et al., 2015; Huang et al., 2019). Sometimes a specific region or residue of a protein is the target of more than two PTMs crosstalk at the same time, a condition known as multiple PTMs. For example, there is an extensive crosstalk among acetylation, methylation, and ubiquitination on p53 protein lysine residues (Aggarwal et al., 2020).
Dysregulation of PTMs crosstalk is implicated in the genesis and progression of a variety of diseases including neurodegenerative disorders (Hart et al., 2011; Kontaxi et al., 2017; Rott et al., 2017; Wang et al., 2018), cardiovascular diseases (Dubois-Deruy et al., 2015; Li et al., 2019), cancers (Wu et al., 2019), and diabetes (Jahangir et al., 2014; Cao et al., 2021). As a result, learning more about the interaction of several PTMs on a protein as a key regulatory mechanism of cell activity could help us identify a PTM signature for a protein under physiological and pathological conditions and, eventually, better understand their role in disease pathogenesis.
Few studies have focused to date on α-Syn PTMs crosstalk. As shown in Table 3, all reported interactions are intraprotein, and currently there is no known interprotein PTMs crosstalk for α-Syn. Given the crucial role of phosphorylation in synucleinopathies, the majority of research on PTMs crosstalk focuses on interactions between phosphorylation and other PTMs (Table 3 and Fig. 4).
Crosstalk between α-synuclein posttranslational modifications

Crosstalk among posttranslational modifications of α-synuclein. An illustration of the complex interplay of α-Syn PTMs highlighting the dynamic relationship between various PTMs. Given that phosphorylation is the most studied PTM of α-Syn, the majority of research on PTMs crosstalk focuses on interactions between phosphorylation and other PTMs. a) Phosphorylation and ubiquitination: Increased phosphorylation of α-Syn at S129 is associated with enhanced ubiquitination at all lysine residues (K-Ub: indicated by a bracket), whereas di-ubiquitination or tetra-ubiquitination at K12 inhibits the phosphorylation of α-Syn at S129. b) Phosphorylation and SUMOylation: The impact of SUMOylation on α-Syn phosphorylation at S129 is kinase-dependent. SUMOylation of α-Syn prevents PLK2-mediated phosphorylation at S129. However, G protein-coupled receptor kinase 5 mediated phosphorylation at S129 is not impacted by the cellular SUMO machinery. c) Phosphorylation and O-GlcNAcylation: O-GlcNAcylation at T72 slightly enhances phosphorylation at S87 by CK1 but prevents phosphorylation at S129 by all three kinases. d) Phosphorylation and acetylation: Although there is presently no empirical evidence substantiating a direct correlation between the phosphorylation and acetylation of α-Syn, studies employing pharmacological methods have shown a negative association between these two PTMs, suggesting that N-terminal acetylation or lysine acetylation (indicated by a bracket) could potentially impede α-Syn phosphorylation. e) Phosphorylation and nitration: the C-terminal tyrosine residues (Y125, Y133, and Y136) of α-Syn are targets for phosphorylation and nitration. Specifically, nitration at Y133 inhibits phosphorylation at S129. f) Ubiquitination and SUMOylation: ubiquitination and SUMOylation may engage in cooperative or competitive interactions since both these PTMs aim to modify the lysine residues of α-Syn. g) Ubiquitination and glycation: the N-terminal lysine residues of α-Syn are potential targets for both glycation and ubiquitination. Evidence indicates that α-Syn glycation inhibits its ubiquitination. K-Ac, acetylation at lysine residues; K-Ub, ubiquitination at lysine residues; N,nitration; N-Ac, N terminal acetylation; O-GN, O-GlcNAcylation; P, phosphorylation; Ub, ubiquitination. Created with BioRender.com pursuant to its Academic License Terms.
A. α-Synuclein Phosphorylation Crosstalk
As mentioned, the majority of α-Syn phosphorylated residues are located in its C-terminal domain (Xu et al., 2015), which serves as a protein–protein interaction domain and a solubilizing domain and contributes to the thermal stability of α-Syn (Oueslati et al., 2010). Modifications at this domain may have a role in the regulation of α-Syn structure and physiological function as well as its pathological aggregation and propagation (Sonustun et al., 2022).
1. Phosphorylation and Ubiquitination
Few studies have explored the crosstalk between α-Syn phosphorylation and ubiquitination. In yeast cells, higher phosphorylation of α-Syn leads to greater ubiquitination and proteasome-mediated degradation (Shahpasandzadeh et al., 2014). The phosphorylated α-Syn deposited in DLB and MSA brains has been shown to be mono- or di-ubiquitinated. Additionally, ubiquitinated α-Syn is phosphorylated at S129 (Hasegawa et al., 2002). Exogenously added synthetic α-Syn PFF is ubiquitinated and phosphorylated at S129 in mammalian cell lines and following stereotaxic injection in the mouse brain (Volpicelli-Daley et al., 2011; Luk et al., 2012b). In fact, α-Syn PFFs serve as seeds and recruit endogenous α-Syn into intracellular inclusions, where they accumulate and can be ubiquitinated and phosphorylated (Luk et al., 2012a). On the other hand, in vitro and in vivo data have shown that phosphorylation at S129 had no effect on α-Syn ubiquitination (Nonaka et al., 2005). In addition, in vitro data revealed that monoubiquitination at K6 had no effect on α-Syn phosphorylation at S87 or S129 (Hejjaoui et al., 2011), while di-ubiquitination or tetra-ubiquitination at K12 prevents α-Syn phosphorylation at S129 (Haj-Yahya et al., 2013) (Fig. 4A). These findings imply an interplay between ubiquitination and α-Syn phosphorylation, which potentially impacts α-Syn aggregation and degradation.
2. Phosphorylation and SUMOylation
An interplay between phosphorylation and SUMOylation has been demonstrated in various proteins and is known to be involved in cell cycle control (Hietakangas et al., 2006; Yao et al., 2011; Khan et al., 2014; Nie et al., 2017). Both PTMs are reversible and dynamic processes that can interact directly with one another and affect each other's behavior (Yao et al., 2011). Evidence suggests that the phosphorylation of numerous substrate proteins has a negative impact on their SUMOylation. Phosphorylation of c-jun, for instance, coincides with diminished SUMO attachment (Muller et al., 2000).
As detailed earlier, α-Syn is phosphorylated as well as SUMOylated, and both modifications have been detected in Lewy bodies. Therefore, studying the crosstalk between these two PTMs is informative (Fig. 4B). In the yeast model, α-Syn phosphorylation and SUMOylation work together to regulate protein turnover. The transition between autophagic and proteasomal degradation of α-Syn is correlated with a molecular interplay between SUMOylation and phosphorylation. SUMOylation preferentially drives α-Syn aggregates into autophagy, whereas phosphorylation can reroute α-Syn toward greater ubiquitination and proteasome degradation (Shahpasandzadeh et al., 2014). In addition, the effect of SUMOylation on α-Syn phosphorylation at S129 depends on the kinase involved. Polo-like kinase 2 appears to be particularly effective against non-SUMOylated α-Syn, whereas G protein-coupled kinase 5 is less selective and can increase S129 phosphorylation regardless of the cellular SUMO machinery function (Shahpasandzadeh et al., 2014).
To date, there are no studies looking at whether SUMOylation of α-Syn directly affects S129 phosphorylation in mammalian cells. Recently, we found that enhancing total SUMOylation by overexpressing SUMO-1 in SH-SY5Y cells promotes α-Syn phosphorylation (Hassanzadeh et al., 2023). This finding does not demonstrate a direct interaction between phosphorylation and α-Syn SUMOylation, since boosting global SUMO machinery has an impact on a wide range of signaling pathways. However, there are examples of direct interplay between these two PTMs in mammalian cell models for other proteins. Overexpression of SUMO-1 in HEK293 cells with stable expression of human tau441 shows that SUMOylation of tau promotes its hyperphosphorylation at multiple Alzheimer’s disease-related sites, leading to tau accumulation and aggregation (Luo et al., 2014). In CHO-K1 cells, crosstalk between SUMOylation and phosphorylation modifies protein kinase C (PKC) activity to control oxidative stress-induced apoptotic cell death. SUMOylation of PKC increases its phosphorylation and controls H2O2-induced apoptosis through phosphorylation (Gao et al., 2021).
3. Phosphorylation and O-GlcNAcylation
α-Syn S87 is a site for phosphorylation as well as O-GlcNAcylation. The latter has been shown to inhibit the extension of PFFs formed from WT α-Syn (Levine et al., 2019). Unlike phosphorylation at this site, which is believed to alter the conformation of membrane-bound α-Syn and reduce its affinity for lipid vesicles (Paleologou et al., 2010), O-GlcNAcylation has no effect on its membrane-binding properties (Lewis et al., 2017). Therefore, the interplay between these two PTMs at S87 might influence α-Syn conformation and membrane binding characteristics. Direct evidence obtained by using semisynthetic O-GlcNAcylated proteins has shown that O-GlcNAcylation at T72 alters physiologically relevant phosphorylation of α-Syn by three different kinases: CK1, PLK1, and GRK5. Phosphorylation at S87 by CK1 was slightly increased by O-GlcNAcylation at T72, while phosphorylation at S129 by all three kinases was inhibited (Marotta et al., 2015) (Fig. 4C). A negative crosstalk between O-GlcNAcylation and phosphorylation has also been shown in other protein systems. For example, an increase in O-GlcNAcylation led to a decrease in the phosphorylation level of desmin, an intermediate filament in skeletal and some smooth muscle cells (Claeyssen et al., 2022).
4. Phosphorylation and Acetylation
An inverse correlation between protein phosphorylation and acetylation has been reported (Grimes et al., 2018) (Fig. 4D). Although there is no experimental evidence to date for a direct interplay between phosphorylation and acetylation of α-Syn, proteins such as insulin receptor substrate-1 (Barreiro et al., 2004), Bcl-2 (Choi et al., 2013), and MAPK (Liu et al., 2004) have been identified as targets of this crosstalk. As an acetyl group donor, acetylsalicylic acid (aspirin) increases acetylation while decreasing phosphorylation of total protein in wild-type C. elegans, most likely via local steric hindrance and/or allosteric impacts. The effect of aspirin on acetylation, phosphorylation, and protein aggregation suggests that acetylation may drive a negative crosstalk with phosphorylation, reducing protein aggregation and providing protection, thus lending credence to the notion that modulating PTMs could be a therapeutic approach for synucleinopathies (Ayyadevara et al., 2017).
5. Phosphorylation and Nitration
A complex crosstalk has been observed between phosphorylation at S129 and modifications of α-Syn C-terminal tyrosine residues. The former, which is abundant in Lewy bodies, has a role in autophagic clearance of protein aggregates, (Tenreiro et al., 2014). α-Syn C-terminal tyrosines (Y125, Y133, and Y136) are targets for phosphorylation (Nakamura et al., 2001; Chen et al., 2009) and nitration (Giasson et al., 2000; Kleinknecht et al., 2016). The proximity of these tyrosine residues to S129 raises the possibility that the interaction between nitration and phosphorylation at these sites may influence α-Syn degradation. In a yeast model expressing human α-Syn, mutation of tyrosine 133 to phenylalanine (Y133F) completely prevented α-Syn S129 phosphorylation, as demonstrated by immunoblotting (Kleinknecht et al., 2016). Given the suggestion in yeast cells that blocking α-Syn phosphorylation at S129 may prevent its clearance by autophagy and worsen α-Syn-induced toxicity (Tenreiro et al., 2014), nitration at Y133 exacerbates pathogenicity by preventing S129 phosphorylation (Fig. 4E) rather than through accumulation of ROS (Kleinknecht et al., 2016). In addition, phosphorylation at Y136 is associated with reduced S129 phosphorylation and α-Syn aggregation, while blocking Y136 phosphorylation promotes the formation of pS129-modified aggregates (Sano et al., 2021, 2).
B. α-Synuclein Ubiquitination Crosstalk
The ubiquitin network, which is highly incorporated with other PTMs, such as phosphorylation, acetylation, and SUMOylation, controls the activity of proteins, changing their conformation and affecting other PTMs on the protein (Grabbe et al., 2011).
Lysine residues are the primary targets for a variety of PTMs, including ubiquitination, SUMOylation, acetylation, glycation, and methylation. It is possible that one conjugation at a target lysine residue inhibits and/or modifies other PTMs at the same location (Dorval and Fraser, 2006). This scenario has been reported for different proteins under various environmental conditions (Lamoliatte et al., 2017). Therefore, there is a need to delve deeper into their interactions. Understanding the crosstalk between these PTMs in both health and disease can offer valuable insights.
1. Ubiquitination and SUMOylation
The crosstalk between ubiquitination and SUMOylation has been studied in various protein systems and can be categorized in three ways. The first is cooperative, in which SUMO comes first and facilitates ubiquitin conjugation (Denuc and Marfany, 2010). The second is competitive on the same lysine residue, where one prohibits the other and acts antagonistically (Desterro et al., 1998; Jürgen Dohmen, 2004). An example is PKC, where SUMOylation inhibits ubiquitination; hence, reducing SUMOylation results in increased PKC degradation via the ubiquitin-proteasome pathway (Gao et al., 2021). The third is differential, meaning that they can be conjugated on the same or different lysine residues in the same protein, and depending on what the cell is responding to, these modifications lead to different physiological effects (Denuc and Marfany, 2010).
For α-Syn, ubiquitination and SUMOylation may compete with one another to target the same residues as the substrate protein (Rott et al., 2017) (Fig. 4F). Accordingly, in vitro data have shown that SUMOylation of α-Syn counteracts its ubiquitination and proteasomal degradation, leading to increased α-Syn steady-state levels (Rott et al., 2017; Savyon and Engelender, 2020). Pharmacological treatment with ginkgolic acid (an SUMO E1 inhibitor) reduces steady-state levels of α-Syn in both HEK293 cells and primary neuronal cultures by promoting proteasome degradation, lending support to the role of SUMOylation in preventing α-Syn ubiquitination and degradation (Rott et al., 2017). On the contrary, it has been demonstrated that SUMOylation acts as a targeting signal for ubiquitination and ubiquitin-dependent degradation (Uzunova et al., 2007), and impaired α-Syn SUMOylation has been shown to inhibit the UPS and the autophagy–lysosome pathway and ultimately prevent α-Syn degradation. This was confirmed when non-SUMO mutants of α-Syn became more prone to aggregation (Zhu et al., 2018). However, we observed that α-Syn ubiquitination does not depend on SUMOylation. Indeed, coexpression of SUMO-1/α-Syn in COS-7 cells revealed that α-Syn SUMOylation and ubiquitination are independent processes that likely occur at different lysine residues in α-Syn (Kim et al., 2011). Altogether, the crosstalk between SUMOylation, ubiquitination, and protein degradation signaling is complex (Kim et al., 2011). Therefore, further in vivo studies are needed to elucidate the effects of this crosstalk on α-Syn aggregation and clearance.
2. Ubiquitination and Glycation
The N-terminal lysine residues of α-Syn are likely to be candidates for glycation, and several of those lysine residues are also known ubiquitination sites (Nonaka et al., 2005; Anderson et al., 2006; Vicente Miranda et al., 2017b). There is evidence suggesting that glycation competes for the ubiquitination of α-Syn (Fig. 4G). An in vitro study using MGO-induced glycation revealed that glycation inhibits α-Syn ubiquitination and impairs UPS- and autophagy-lysosome pathway-mediated α-Syn degradation (Vicente Miranda et al., 2017b).
V. Concluding Remarks
The multiple PTMs of α-Syn have been the subject of intense investigations to help elucidate their role in the normal biology of this protein and its dysfunction in disease states. Current detection and analysis of α-Syn PTMs rely primarily on antibody-based techniques and mass spectrometry (Marx, 2013). Recent advances in mass spectrometry have significantly enhanced our ability to identify PTMs (Choudhary and Mann, 2010), although challenges remain (Schmid et al., 2013) due to their low abundance, nonstoichiometric nature, and instability during analysis (Azevedo et al., 2022). In addition, environmental factors such as temperature, pH, metal ions, and shaking can affect α-Syn structure and propensity to aggregate (Candelise et al., 2020). Additionally, the complexity of biological fluids, including blood and CSF, poses significant challenges in detecting modified α-Syn forms compared with detecting its unmodified form. This increased difficulty is primarily due to the substantially lower concentrations and variations of these modified α-Syn forms in biological fluids (Schmid et al., 2013), along with technical issues associated with PTM detection protocols (Shevchenko et al., 2015). Advanced methods such as selected reaction monitoring mass spectrometry have shown promise in detecting low-abundance α-Syn in biological samples (Schmid et al., 2013), indicating the need for more sensitive detection techniques for PTMs as potential biomarkers.
With the current capabilities and limitations of the field, our understanding of the potential for PTMs to be developed as biomarkers of disease diagnosis or progression and as therapeutic targets are more advanced for some of these PTMs than others. Among these, phosphorylation, and in particular at S129, is well established as a readout of pathologic α-Syn in various experimental models and human disease tissues, with significant potential to be validated as a biomarker for synucleinopathies. Phospho-S129 α-Syn positive signal in skin biopsy specimens provides a high degree of sensitivity and specificity in synucleinopathy-affected patients (Gibbons et al., 2024). A number of pharmacological approaches aimed at mitigating this PTM have shown promising results in preclinical studies (Lee et al., 2011; Yan et al., 2018; Weston et al., 2021b). Yet, a significant gap remains in the availability of standardized and reliable detection methodologies for many of the other PTMs therefore limiting their readiness for clinical utility as biomarkers. This is compounded by the fact that individual PTMs of a protein do not function or impact their target in isolation. The complex and subtle interplay between posttranslational modifications can alter properties of the target protein, such as stability, folding, aggregation, and affinity for proteasome degradation or autophagy pathways (Fig. 3). It is clear that PTMs, which are dynamic processes, are in equilibrium under physiological conditions, whereas this homeostatic regulation and crosstalk can be impaired under pathological conditions.
Although α-Syn PTMs have received considerable attention due to their important roles in regulating the properties of this protein in health and certain neurodegenerative diseases, their crosstalk has not yet been adequately studied, and their potential as therapeutic targets is only beginning to be explored. As a result, our understanding of the effects of these modifications on the biology of α-Syn remains limited, and we are just beginning to learn how the interplay among various PTMs can orchestrate α-Syn function, misfolding, and toxicity and how pharmacological manipulation of one impacts the others (Fig. 3). This is likely due in part to the lack of a precise or specific approach to identify concurrent PTMs as well as the need for new agents that target them. In addition to the small number of studies that are focused on α-Syn PTMs crosstalk to date, their functional outcomes have been inconsistent, likely because of the varied experimental models used. Due to the substantial body of research addressing the role of phosphorylation in α-Syn aggregation and Lewy body formation, as well as ubiquitination in proteasomal degradation, most crosstalk research to date has focused on the interaction between phosphorylation or ubiquitination and other PTMs. As discussed earlier, lysine residues in α-Syn are the target for a number of PTMs, including ubiquitination, SUMOylation, glycation, acetylation, and methylation (Fig. 2). Therefore, studying the crosstalk between these PTMs provides valuable insight into the pathologies that may result from an imbalance in crosstalk. Mapping all α-Syn PTMs, and real-time analysis of simultaneous PTMs and their interactions, can yield a molecular signature for each physiological and pathological condition. This area of research paves the way for a deeper understanding of the pathobiology of α-Syn and the identification of potential innovative strategies and viable targets for therapeutic interventions to prevent or slow down the progression of synucleinopathies.
Data Availability
This review article does not contain original research data.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Hassanzadeh, Liu, Maddila, Mouradian.
Footnotes
- Received October 27, 2023.
- Revision received July 28, 2024.
- Accepted August 9, 2024.
M.M.M. is the William Dow Lovett Professor of Neurology and is supported by grants from National Institutes of Health National Institute of Neurological Disorders and Stroke and National Institute on Aging [Grants NS130702, NS101134, NS123770, NS116921, and AG075656], the Michael J. Fox Foundation for Parkinson’s Research [Grants MJFF-001006 and MJFF-022157], the American Parkinson Disease Association, and the Puri Family philanthropy.
M.M.M. is an inventor of filed and issued patents related to α-synuclein. She is a founder of MentiNova, Inc.
Abbreviations
- α-Syn
- α-synuclein
- AGE
- advanced glycation end product
- ALP
- autophagy-lysosome pathway
- c-Abl
- Ableson tyrosine kinase
- CK
- Casein kinase
- CSF
- cerebral spinal fluid
- DLB
- dementia with Lewy bodies
- GCI-α-Syn
- α-Synuclein sourced from multiple system atrophy brains with glial cytoplasmic inclusions
- GlcNAc
- N-acetylglucosamine
- GRK
- G protein-coupled receptor kinase
- iNOS
- inducible nitric oxide synthase
- LB
- Lewy body
- LB-α-Syn
- α-Synuclein sourced from Lewy body disease brains
- LCMT-1
- leucine carboxyl methyltransferase-1
- MetO
- methionine sulfoxide
- MGO
- dicarbonyl compound methylglyoxal
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MSA
- multiple system atrophy
- NMDA
- N-methyl-D-aspartate
- NO
- nitric oxide
- OGA
- O-GlcNAcase
- O-GlcNAc
- O-linked N-acetylglusosamine
- PD
- Parkinson's disease
- PFF
- α-Syn preformed fibrils
- PKC
- protein kinase C
- PLK2
- Polo like kinase 2
- PLP
- oligodendrocyte-specific proteolipid protein
- PME-1
- protein phosphatase methylesterase-1
- PP2A
- protein phosphatase 2A
- PTM
- posttranslational modification
- ROS
- reactive oxygen species
- SIAH
- seven in absentia homolog
- SNARE
- soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SUMO
- small ubiquitin-like modifier
- TG2
- transglutaminase 2
- T:M
- tetramer-to-monomer
- Ubc9
- SUMO E2 conjugating enzyme
- UPS
- ubiquitin-proteasome system
- WT
- wild-type.
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




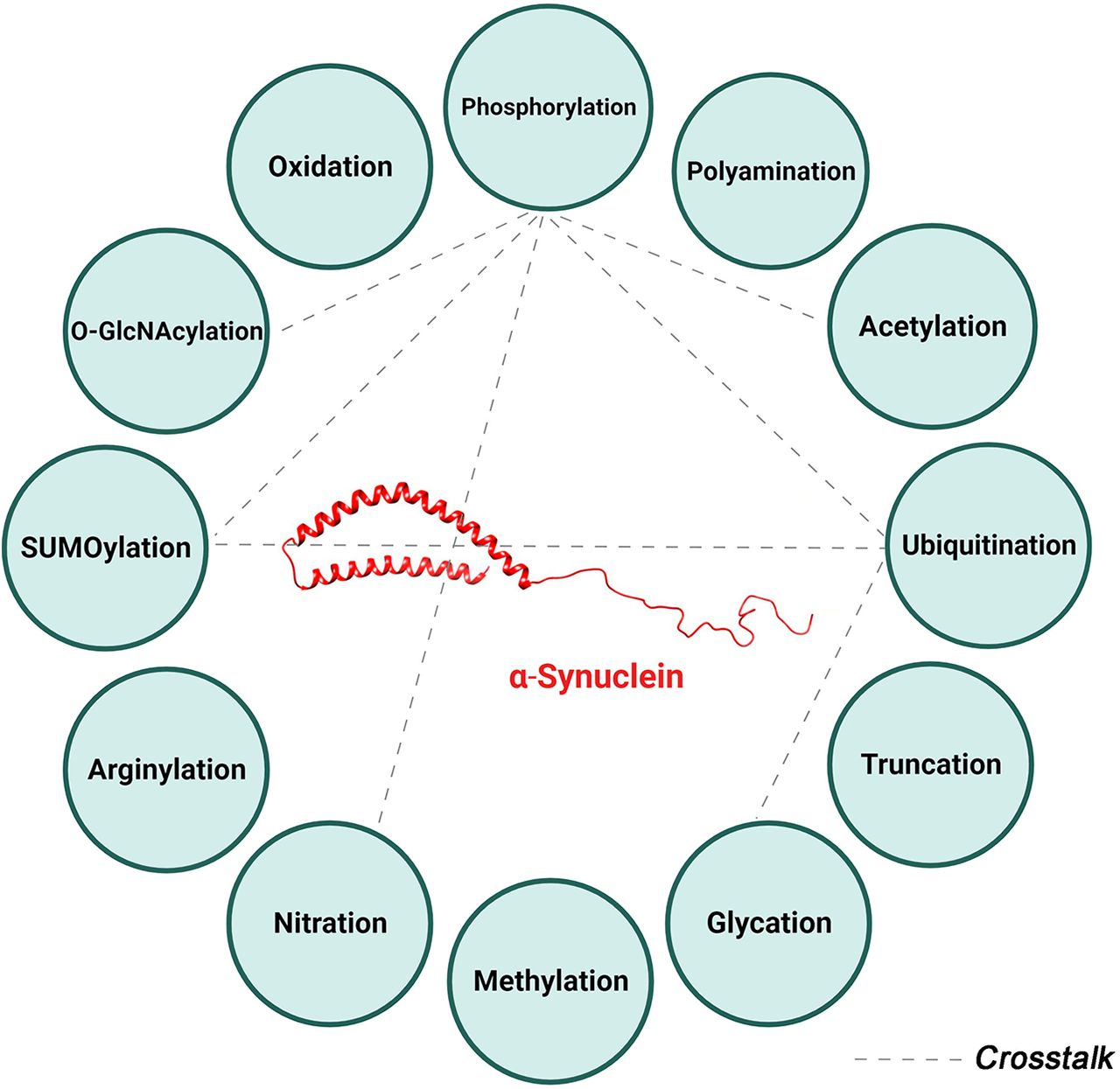{kind=link}
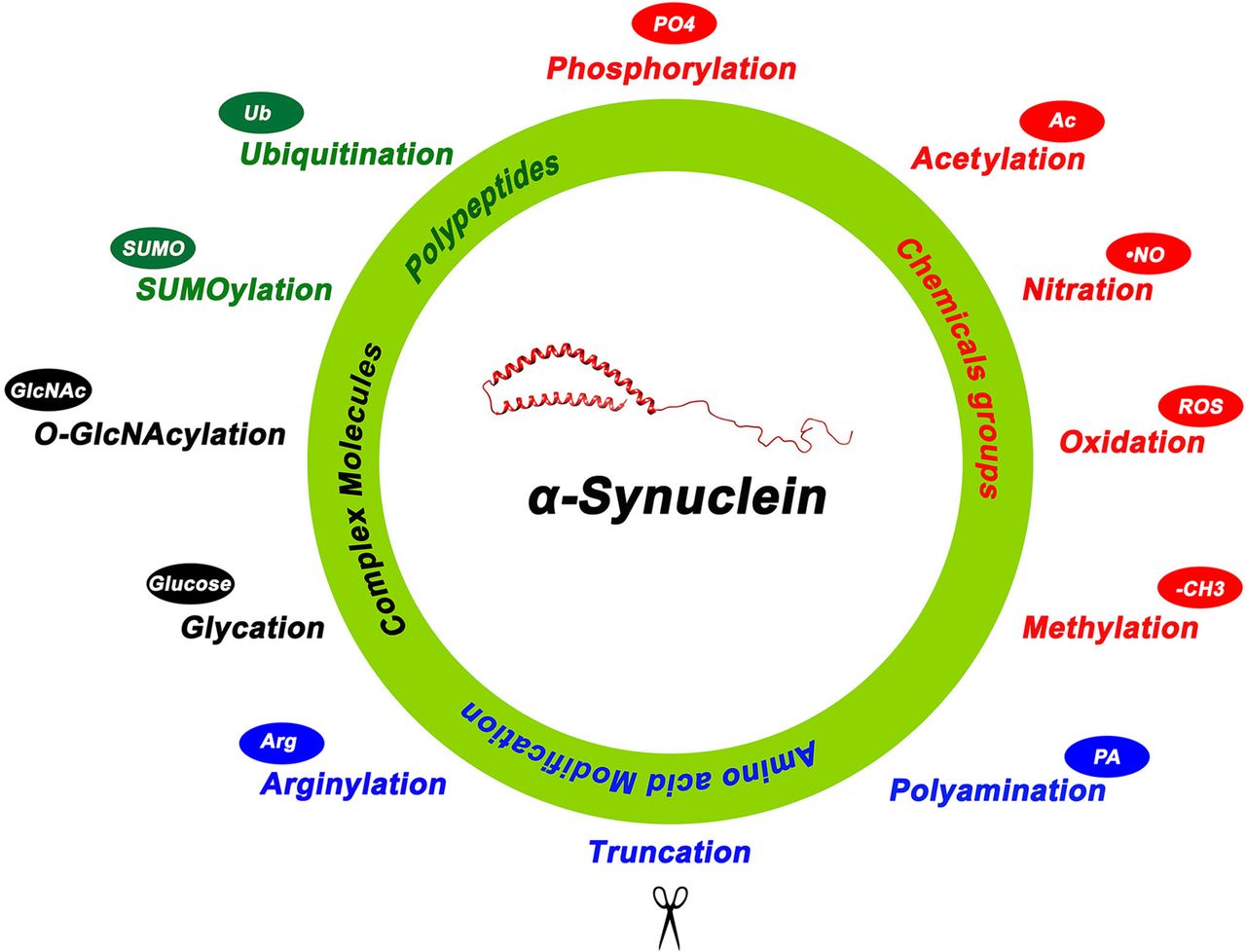{kind=link}
{kind=link}
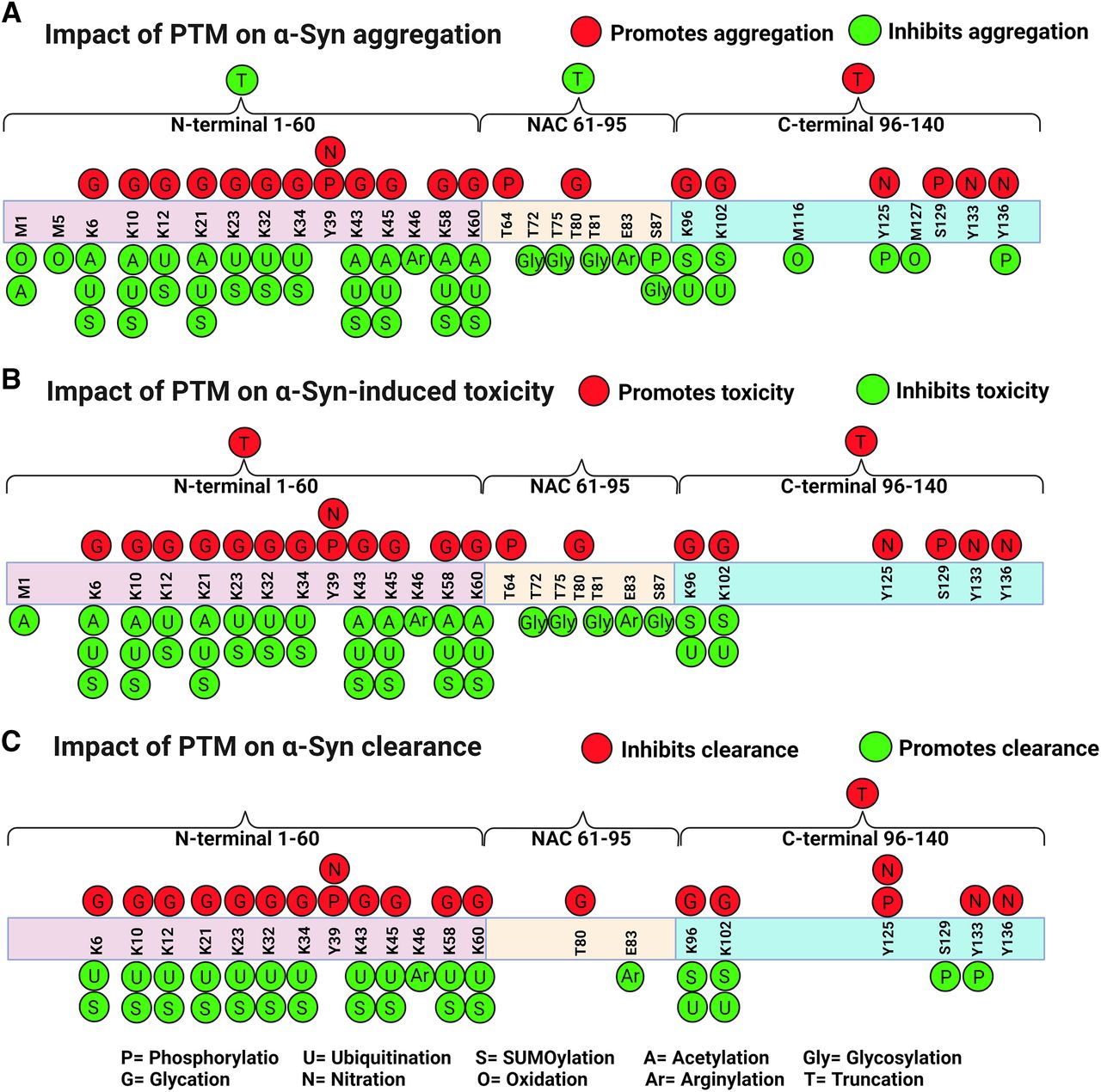{kind=link}
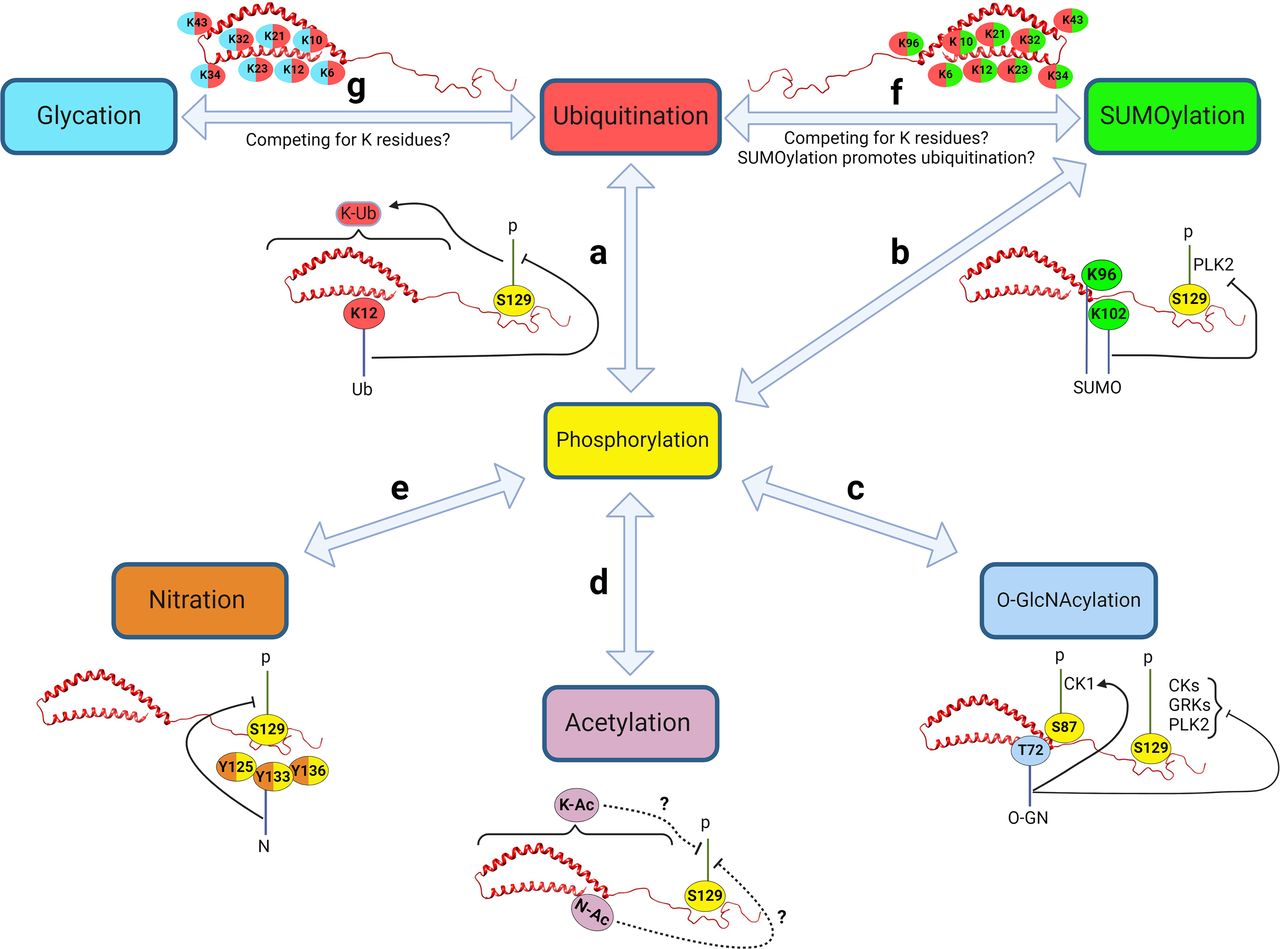{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. α-Synuclein Posttranslational Modifications
- III. Targeting α-Synuclein Posttranslational Modifications as a Therapeutic Strategy
- IV. Crosstalk among α-Synuclein Posttranslational Modifications
- V. Concluding Remarks
- Data Availability
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters