


Abstract
Over 4 decades of research support the link between Alzheimer disease (AD) and somatostatin [somatotropin-releasing inhibitory factor (SRIF)]. SRIF and SRIF-expressing neurons play an essential role in brain function, modulating hippocampal activity and memory formation. Loss of SRIF and SRIF-expressing neurons in the brain rests at the center of a series of interdependent pathological events driven by amyloid-β peptide (Aβ), culminating in cognitive decline and dementia. The connection between the SRIF and AD further extends to the neuropsychiatric symptoms, seizure activity, and inflammation, whereas preclinical AD investigations show SRIF or SRIF receptor agonist administration capable of enhancing cognition. SRIF receptor subtype-4 activation in particular presents unique attributes, with the potential to mitigate learning and memory decline, reduce comorbid symptoms, and enhance enzymatic degradation of Aβ in the brain. Here, we review the links between SRIF and AD along with the therapeutic implications.
Significance Statement Somatostatin and somatostatin-expressing neurons in the brain are extensively involved in cognition. Loss of somatostatin and somatostatin-expressing neurons in Alzheimer disease rests at the center of a series of interdependent pathological events contributing to cognitive decline and dementia. Targeting somatostatin-mediated processes has significant therapeutic potential for the treatment of Alzheimer disease.
I. Introduction
Alzheimer disease (AD) is the most common form of dementia. In the United States alone, there are an estimated 6.9 million people living with AD, which is predicted to grow to 13.8 million by 2060 (https://pubmed.ncbi.nlm.nih.gov/38689398/). AD progresses across a continuum (Jack et al., 2010; Vermunt et al., 2019). AD is preceded by a preclinical phase lasting for ∼15–20 years, associated with increasing Aβ accumulation within the brain but without cognitive impairment. A prodromal stage marks the initiation of symptoms, characterized by mild cognitive impairment (MCI), and often lasts 3–6 years. The formal dementia stage typically lasts 7–10 years and is categorized into mild, moderate, and severe AD based on the level of patient impairment. Ultimately, the capacity to speak and perform simple tasks is lost, culminating in immobility and death.
Most AD diagnoses are designated as “sporadic,” with a number of contributing genetic, environmental, and comorbidity risk factors (Samanta and Ramesh, 2022). This implicates a multifactorial etiology with potential interdependency between risk factors. Increasing age is the greatest risk factor for AD. AD diagnosis at age 65 or older is classified as late-onset AD (LOAD) and accounts for more than 95% of occurrences. AD diagnosis before age 65 is classified as early-onset AD and accounts for less than 5% of occurrences. Fewer than 1% of cases have a recognized inheritable etiology that occurs in an autosomal-dominant AD (ADAD) manner, also known as familial AD. Mutations in any of 3 specific genes [amyloid precursor protein (APP) gene, presenilin-1 (PS1) gene, and presenilin-2 (PS2) gene] results in overproduction of the amyloid-β peptide (Aβ) (Bekris et al., 2010). Inheritance of these mutations nearly guarantees AD development assuming a normal lifespan, with age of onset and rate of progression related to the severity of the mutation (Goldman et al., 2011).
The brains of individuals with AD exhibit two cardinal histopathological features: deposits of Aβ in the form of extracellular plaques and intraneuronal neurofibrillary tangles (NFTs) composed of aggregates of hyperphosphorylated tau protein (Long and Holtzman, 2019). Aβ is a primary research and drug development target due to its early-stage accumulation within the brain and genetic data supporting an Aβ-AD causal relationship (Selkoe and Hardy, 2016). Aβ is generated following the sequential cleavage of APP by β- and γ-secretase in the amyloidogenic pathway. The more hydrophobic species of Aβ (i.e., Aβ42 and longer) readily self-aggregate and are associated with greater pathologic contribution compared with less hydrophobic forms (i.e., Aβ40 and shorter). The original Aβ-cascade hypothesis posited that the deposition of Aβ in the brain is the initiating step of AD pathogenesis, with subsequent tau deposition and neuronal loss (Hardy and Higgins, 1992). This hypothesis has evolved with the increased recognition as to the role of Aβ peptide oligomers (AβOs). Soluble AβOs are now regarded as the most pathogenic and neurotoxic form of Aβ, with impairment of synaptic structure and function by AβOs preceding the formation of Aβ plaques (Gyure et al., 2001; Lacor et al., 2004; Shankar et al., 2008). There are also a number of reports identifying extensive Aβ plaque deposits in brain tissues taken from individuals who lacked definitive signs of dementia (Katzman et al., 1988; Hulette et al., 1998; Price and Morris, 1999; Aizenstein et al., 2008; Zolochevska and Taglialatela, 2016). It is hypothesized that Aβ plaques may even serve to sequester toxic AβOs (Esparza et al., 2013; Hong et al., 2014), whereas support for AβOs as the AD pathological trigger is extensive and continues to grow. AβOs directly activate N-methyl-D-aspartate (NMDA) receptors increasing neuronal hyperexcitation (Li et al., 2011; Zott et al., 2019), prevent glutamate reuptake (Li et al., 2009b; Zott et al., 2019), enhance oxidative (Sponne et al., 2003; Tabner et al., 2005; De Felice et al., 2007; Yin et al., 2021) and endoplasmic reticulum (ER) stress (Nishitsuji et al., 2009; Umeda et al., 2011; Kam et al., 2022), impair neuronal function (Heinitz et al., 2006; Chung et al., 2020), decrease trophic factors (Kitiyanant et al., 2012; Poon et al., 2013; Sen et al., 2015; Pitt et al., 2017), produce insulin resistance (Zhao et al., 2008, 2009; Ma et al., 2009), activate glial inflammatory mechanisms (Sondag et al., 2009; Maezawa et al., 2011; Ferretti et al., 2012; Yang et al., 2017), stimulate tau hyperphosphorylation (De Felice et al., 2008; Tomiyama et al., 2010; Zempel et al., 2010; Wakeman et al., 2022), produce synaptic deterioration (Lacor et al., 2004, 2007; Shankar et al., 2007, 2008), impair synaptic transport (Pigino et al., 2009; Decker et al., 2010; Poon et al., 2011; Ramser et al., 2013) and plasticity (Townsend et al., 2006; Klyubin et al., 2008; Shankar et al., 2008; Actor-Engel et al., 2021; Yan et al., 2021), and selectively induce neuronal cell death (Lambert et al., 1998; Kim et al., 2003; Salvadores et al., 2022). Notably, AβOs isolated from human AD cortical tissue impairs memory behavior when injected into the lateral ventricles of healthy adult rats (Shankar et al., 2008). Thus, although many factors contribute to AD pathogenesis, Aβ and its oligomeric forms represent the most validated therapeutic target for disease mitigation.
To date, eight drugs have been approved by the US Food and Drug Administration (FDA) for AD. Five of these drugs (donepezil, rivastigmine, galantamine, memantine, and memantine combined with donepezil) are designated as palliative therapies for symptom alleviation. Only the recently FDA-approved Aβ-directed antibodies aducanumab, lecanemab, and donanemab are specifically directed toward the underlying pathology. Nevertheless, the effectiveness of Aβ-directed antibodies in mitigating cognitive decline is greatly debated, with added concerns as to their potential to induce life-threatening brain swelling and bleeding (Shi et al., 2022; Couzin-Frankel, 2023). Given the current state of AD drug therapy, there is increasing recognition that therapeutic development needs to take into greater account the complex multifactorial nature of AD and interlinking cellular processes involved (Hampel et al., 2019). Focusing on critical neuronal networks and the targeting of key mediators involved in both neuronal health and disease progression may provide a more successful treatment approach.
Somatostatin [somatotropin-releasing inhibitory factor (SRIF)] and SRIF-expressing neurons are essential in brain function. SRIF-expressing neurons have extensive brain network interconnections, regulating hippocampal activity and memory formation (Honoré et al., 2021). The loss of SRIF and SRIF-expressing neurons in AD is a definitive pathological event (Davies et al., 1980; Rossor et al., 1980; Grouselle et al., 1998), playing a central role in a series of interdependent pathological feedback loops that drive AD progression (Fig. 1). Loss of SRIF and SRIF-expressing neurons further contributes to AD-associated neuropsychiatric symptoms, seizure activity, and inflammation. Moreover, a unique facet of SRIF is its capacity to enhance the activity of Aβ-degrading enzymes in the brain through receptor-mediated action (Saito et al., 2005; Sandoval et al., 2012; Nilsson et al., 2020). Not only does this implicate a decline in brain SRIF as a contributor to Aβ accumulation but it identifies a mechanism by which SRIF receptor (SST)-targeted therapeutics may mitigate the underlying pathology. In light of these considerations, this review aims to provide insight into the connections between SRIF and AD along with the therapeutic implications.

Flow chart of interactions between SRIF-AD. AD-associated loss of SRIF and SRIF-expressing neurons in the brain rests at the center of a series of interdependent pathological events driven by Aβ and inflammatory processes, culminating in cognitive decline and dementia. * This loss drives positive feedback loops, which feed into each other, promoting an escalating pathology (numbering is not indicative of sequence of events): (1) Loss of SRIF and SRIF-expressing neurons decreases Aβ-degrading enzyme activity, resulting in elevated Aβ brain levels and promotion of neurodegeneration. (2) Loss of SRIF and SRIF-expressing neurons results in a dysregulation of glutamate homeostasis and excitatory-inhibitory balance with corresponding neuronal hyperexcitability, which propagates further Aβ neuronal release and neuronal damage. (3) Loss of SRIF and SRIF-expressing neurons contributes to inflammation. Inflammation results in neuronal dysfunction and degeneration, perpetuating further inflammation. Inflammation-induced BBB dysregulation contributes to increased Aβ brain levels. Inflammation is linked to increased seizure activity and neuropsychiatric symptoms, which are likewise linked to the loss of SRIF and SRIF-expressing neurons as well as BBB dysregulation.
II. Somatostatin and Somatostatin Receptors
SRIF is a neuropeptide heavily involved in the regulation of endocrine, brain, and gastrointestinal processes. Brazeau et al., (1973) first isolated SRIF, identifying its ability to inhibit growth hormone release from the pituitary. Subsequent evaluations show SRIF as a primary regulator of growth hormone, prolactin, corticotrophin-releasing hormone, adrenocorticotropic hormone (ACTH), insulin, glucagon, thyroid-stimulating hormone, and vasoactive intestinal peptide, extensively reviewed elsewhere (Günther et al., 2018). Within the central nervous system (CNS), SRIF primarily acts to inhibit neuronal activity. SRIF exists in two bioactive forms: SRIF-14 and the N-terminally extended SRIF-28. Both SRIF forms have a short plasma half-life of <3 minutes (Patel and Wheatley, 1983). Although both forms are expressed at varying levels throughout the body, SRIF-14 is the predominant form in the brain (Viollet et al., 2008). Both forms are derived from the same precursor protein, prepro-SRIF, with a structure that is highly conserved across vertebrates (Conlon et al., 1997). The expression of the SRIF precursor gene is regulated by a number of growth factors, including insulin, growth hormone, leptin, brain-derived neurotrophic factor (BDNF), insulin-like growth factor (IGF)-1, and vasoactive intestinal peptide as well as steroids, inflammatory cytokines, and various neurotransmitters (Ampofo et al., 2020). The family of SRIF-related peptides also includes cortistatin (CST) and neuronostatin. CST similarly has two bioactive forms in rats (CST-14 and CST-29) and in humans (CST-17 and CST-29). Although CST is encoded by the CORT gene (Liu et al., 2010), it is highly homologous with SRIF and partially overlaps SRIF brain expression (de Lecea et al., 1997). Neuronostatin is encoded by the SRIF gene and is expressed in the pancreas, spleen, and brain (Samson et al., 2008).
SRIF produces its actions through five receptors (SST1–5), with a splice variant of SST2(B) identified in rodents (Vanetti et al., 1992). SRIF binds to all SST subtypes with high affinity, as does CST (Siehler et al., 2008). The SSTs are members of the family-A heteromeric G-protein–coupled receptors (GPCRs) of the rhodopsin-like family. All SSTs possess seven α-helical transmembrane domains, with divergence mostly occurring in the intracellular C-terminus and N-terminus domains. On basis of their phylogeny, structural homologies, and pharmacological properties, the SSTs are categorized into two families: SRIF-1 (SST2, SST3, and SST5) and SRIF-2 (SST1 and SST4). The receptors share 39%–57% homology in sequence between the subtypes, and when compared across species there is considerable sequence similarity for a given subtype (81%–99% for mouse, human, and rat homologs) (Günther et al., 2018). Although formal crystal structures are lacking, recent evaluations using cryogenic electron microscopy have characterized the structure of both SST2 and SST4 bound to ligands in different activation states (Bo et al., 2022; Robertson et al., 2022; Zhao et al., 2022). These studies show the SST ligand recognition is highly diverse, respective to each receptor, as demonstrated by ligand-induced conformational changes. The SST subtypes are further delineated by regional distribution throughout the human CNS and periphery (Consortium, 2020; Sjöstedt et al., 2020). Nevertheless, SST subtype distributions often overlap within brain regions. Immunocytochemical evaluations conducted in rat brain tissue identified that specific SST subtypes have preferential presynaptic (SST1) and postsynaptic (SST2,4,5) localization, with SST3 expressed in neuronal cilia (Schulz et al., 2000), albeit a number of exceptions exist as to the adherence of presynaptic and postsynaptic SST subtype localization (Günther et al., 2018).
All SST subtypes are Gi/Go proteins sensitive to pertussis toxin. Upon receptor activation, the Gα subunit inhibits adenylyl cyclase, which inhibits downstream formation of intracellular cAMP (Patel et al., 1994). The β/γ subunits of the GPCR can impact presynaptic and postsynaptic neuronal signaling through regulation of different ion channels. The β/γ subunits can activate G-inwardly rectifying potassium channels, resulting in K+ efflux out of the neuron and hyperpolarization. The β/γ subunits of the GPCR also bind to the α1 subunit of N-type and P/Q-type voltage-gated calcium channels, resulting in cellular inhibition (Viana and Hille, 1996; Smith et al., 2001). SSTs further modulate other pathways, including cGMP-dependent kinase, protein tyrosine phosphatase, phospholipase C, mitogen-activated protein kinase, phospholipase A2, nitric oxide synthase, and the Na+/H+ exchanger (Günther et al., 2018). The activation or inhibition of a respective pathway is dependent on the primary function of the cell acted upon, mediated through a specific SST subtype.
Receptor interactions add another layer to SST regulation. All SSTs have the capacity to dimerize (Kumar, 2013), with functional interactions between SST subtypes widely reported (Moneta et al., 2002; Cammalleri et al., 2004, 2006, 2009; Aourz et al., 2011; Prévôt et al., 2017). Homo and heterodimerization, or even oligomeric receptor complexes, can result in distinctive signal transduction responses when activated (Mores et al., 2018). Such interactions can change the receptor desensitization, internalization, postendocytic trafficking, and resensitization profiles (Grant et al., 2008; Grant and Kumar, 2010; Mores et al., 2018). The interactive arrestins also serve as scaffolding proteins for alternate intracellular signaling cascades. The recognition of these alternate pathways has led to a search for drug candidates with selective signal bias capable of favoring distinctive biochemical and physiological processes. Nevertheless, exploitation of such biased receptor-ligand complexes toward refined therapeutic targeting remains dependent on the nature of the receptor in the native tissue respective to the disorder.
III. Somatostatin-Expressing Neurons
Networks of SRIF-expressing neurons exist in the neocortex, hippocampal formation (hippocampus, dentate gyrus, entorhinal cortex, subiculum), amygdala, median eminence, preoptic area, hypothalamus, brainstem, and somatosensory cortex (Martel et al., 2012). Neuronal SRIF is colocalized with the inhibitory amino acid neurotransmitter GABA. Consequently, SRIF-expressing neurons exist as a distinct subset of GABAergic inhibitory neurons. Yet, although GABA is stored in synaptic vesicles that can be released via a single action potential, SRIF is stored in dense-core vesicles, which require high-frequency repetitive action potentials for release (Ludwig and Pittman, 2003). Moreover, unlike GABA, SRIF lacks a selective reuptake mechanism, which enhances the presence in the synaptic cleft. Thus, despite cellular coexpression and complimentary inhibitory actions, SRIF and GABA can have different release rates and receptor interaction timeframes.
A distinguishing property of SRIF-expressing neurons is the high level of spontaneous activity, enabled by intrinsic membrane conductance that can persist in the absence of synaptic input. The activity can be enhanced by other neuromodulators, including norepinephrine and acetylcholine (Paspalas and Papadopoulos, 1999; Fanselow, 2010; Chen et al., 2015). SRIF release is further regulated by GABA (Bonanno et al., 1999; Kanigowski et al., 2023) and the excitatory amino acid glutamate (Tapia-Arancibia and Astier, 1989; Fontana et al., 1996). In the hippocampus, glutamate was shown to stimulate SRIF release through the activation of ionotropic NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Fontana et al., 1996).
Upon release, SRIF acts on the SSTs of adjacent neurons in a paracrine-like manner. Presynaptically, SRIF acts to decrease neurotransmitter release. SRIF notably inhibits glutamate release, decreasing excitatory synaptic transmission in the hypothalamus, hippocampal formation, basal forebrain, and neocortex (Boehm and Betz, 1997; Tallent and Siggins, 1997; Grilli et al., 2004; Momiyama and Zaborszky, 2006; Kozhemyakin et al., 2013). This presynaptic inhibition is primarily mediated through the reduction of voltage-dependent Ca2+ currents. Other reports indicate that the modulation of presynaptic K+ channels is also involved in the inhibition of excitatory transmission via SRIF-mediated action (Tallent and Siggins, 1997). SRIF can likewise inhibit presynaptic GABA release. The application of SRIF has been shown to reduce the amplitude of the evoked GABAergic inhibitory presynaptic currents in basal forebrain cholinergic neurons (Momiyama and Zaborszky, 2006). Postsynaptically, SRIF acts to hyperpolarize neurons in the brain and spinal cord away from their firing threshold through SST-mediated enhancement of K+ efflux or a reduction of Ca2+ influx (Moore et al., 1988; Schweitzer et al., 1998; Kim et al., 2002; Qiu et al., 2008). Thus, postsynaptically, SRIF induces a slow, long-lasting inhibition.
Despite its predominately inhibitory role, SRIF can induce downstream excitatory effects. SRIF is able to increase excitation through inhibitory actions on GABA neurotransmission, contributing to long-term potentiation (LTP) (Scharfman and Schwartzkroin, 1989; Racine et al., 2021). In a study using single-unit recordings in rat hippocampus and parietal cortex, SRIF applied with acetylcholine caused a concentration-dependent increase in acetylcholine-induced excitations (Mancillas et al., 1986). In rat hippocampal slices, SRIF application enhanced acetylcholine release indirectly by a mechanism involving alterations of calcium influx during depolarization (Araujo et al., 1990). Moreover, SRIF actions can be dose dependent, with SRIF applied to cultured neurons inducing excitation at lower doses and inhibition at higher doses (Delfs and Dichter, 1983). This underscores the complex nature governing neuronal activity.
Morphologically, SRIF-expressing neurons are broadly categorized as either interneurons, which act within discrete microcircuits, or long-projecting neurons with distant projections from their cell body. Nevertheless, within these categories, SRIF-expressing neurons show substantial diversity in terms of chemical and genetic markers, intrinsic firing properties, and connectivity (Liguz-Lecznar et al., 2016; Riedemann, 2019). SRIF-expressing interneurons constitute ∼30% of all GABAergic interneurons in the brain (Rudy et al., 2011). The main SRIF-expressing interneurons in the neocortex and hippocampus are Martinotti cells. Neocortical Martinotti cells have their soma located mostly in layers 2/3 and 5 (Urban-Ciecko and Barth, 2016). Their ascending axons extensively arborize in cortical layer 1, spreading horizontally to neighboring columns. The most extensively studied SRIF-expressing interneurons of the hippocampus are those in oreins-lacunosum moleculare (O-LM). These O-LM interneurons serve as a major relay between the entorhinal cortex and the CA1 region of the hippocampus (McBain et al., 1994).
There is a significant degree of neurochemical diversity in SRIF-expressing interneurons. SRIF-expressing interneurons may coexpress a number of other mediators, including neuropeptide Y (Köhler et al., 1987; Ma et al., 2006), calbindin (Muller et al., 2007; Suzuki and Bekkers, 2010), calretinin (Gulyás et al., 2003; Xu et al., 2006), cholecystokinin (Shi et al., 2020), and neuronal nitric oxide synthase (Dun et al., 1994; Perrenoud et al., 2012). A distinct subset of SRIF-expressing neurons express nitric oxide synthase, neuropeptide Y, and the neurokinin-1 receptor for substance P, with the capacity to project to multiple brain regions (Kubota et al., 2011; Endo et al., 2016). Additional types of SRIF-expressing interneurons include those with a bitufted appearance and a subset of basket cells, which are particularly abundant in the frontal and entorhinal cortex (Kvitsiani et al., 2013; Neske et al., 2015).
Long-projecting SRIF-expressing neurons are larger in diameter and have thicker myelin layers than interneurons, which facilitate rapid inhibitory neurotransmission between brain regions (Jinno et al., 2007; Viollet et al., 2008). Although interneurons are the primary focus of learning and memory research, long-projecting SRIF-expressing neurons also play a role. The GABAergic septo-hippocampal circuit highlights this contribution. Although GABAergic neurons from the medial septum to the hippocampus are predominately parvalbumin expressing, they are reciprocally innervated by SRIF-expressing GABAergic neurons projecting from the hippocampus to the medial septum. Memory and spatial navigation are regulated, in part, through this circuit (Müller and Remy, 2018).
IV. Somatostatin in Learning and Memory
SRIF and SRIF-expressing neurons play a pivotal role in learning and memory, with recent reviews addressing the extensive cellular interactions involved (Honoré et al., 2021; Topolnik and Tamboli, 2022). At the most fundamental level, learning and memory are a function of neuronal activity. Synchronized neuronal network interactions give rise to rhythmic patterns of activity, which produce brain wave oscillations that can be measured electrophysiologically. Theta (∼4–12 Hz) and gamma (∼25–100 Hz) network oscillations help link neurons to process new information (Colgin, 2016; Nuñez and Buño, 2021). The theta rhythm is observed in multiple brain regions and is particularly robust in the hippocampal CA1. Theta oscillations, nested within gamma oscillations, are linked to the formation and retrieval of episodic memory (Griffiths and Jensen, 2023). SRIF-expressing neurons regulate both theta and gamma oscillations (Colgin, 2016; Topolnik and Tamboli, 2022). SRIF-expressing neurons terminating in the medial entorhinal cortex synchronize theta activity (Mizuseki et al., 2009; Melzer et al., 2012), mediating spatial and temporal memory coding, which underlies memory formation (Hasselmo and Stern, 2014; Siegle and Wilson, 2014). SRIF-expressing interneurons of the O-LM have shown to control theta type-2 (4–9 Hz) oscillations (Leão et al., 2012; Mikulovic et al., 2018), which are identified learning and memory behaviors associated with fear and anxiety (Castegnetti et al., 2021).
Neuronal silencing/ablation studies substantiate the role of SRIF-expressing interneurons in learning and memory. A study using the contextual fear conditioning model, widely used to study hippocampal-dependent memory behavior (Fanselow, 2010), combined with high-resolution calcium imaging and optogenetic manipulation, demonstrated that silencing SRIF-expressing interneurons in the hippocampal CA1 prevented learning behavior in mice (Lovett-Barron et al., 2014). SRIF-expressing interneuron-induced dendritic inhibition was necessary for the contextual fear learning. In another study using contextual fear conditioning, silencing of hippocampal CA1 SRIF-expressing interneurons in conditional knockin mice during the consolidation phase attenuated the increase in the fear memory (Sharma et al., 2020), confirming that hippocampal CA1 SRIF-expressing interneurons are essential for memory consolidation. The conditional fear learning notably reduced eukaryotic initiation factor 2α (eIF2α) phosphorylation in the SRIF-expressing interneurons. Phosphorylation of the α-subunit of eIF2, the central component of the integrated stress response, is associated with AD neuronal degradation and impaired memory formation (Chang et al., 2002; Oliveira and Klann, 2022), whereas a reduction in eIF2α phosphorylation in hippocampal SRIF-expressing interneurons increased general mRNA translation, bolstered synaptic plasticity, and enhanced long-term memory (Sharma et al., 2020). Lastly, a recent study in which dentate hilar SRIF-expressing interneurons were partially ablated in SRIF–internal ribosome entry site–Cre mice showed memory decline in spatial and object recognition memory behavioral tests compared with control mice (Nagarajan et al., 2023). The ablation led to increased neuronal activity in both the dentate gyrus and hippocampal CA3, consistent with age-associated learning and memory impairment (Yassa et al., 2011; Oh et al., 2016).
Pharmacological evaluations corroborate the integral role of SRIF in learning and memory processing. In an initial series of rodent behavioral studies, intracerebroventricular administration of SRIF in rats improved learned acquisition in the active avoidance foot-shock task evaluations (Bollók et al., 1983; Vécsei et al., 1983a,b) and prevented electroshock-induced amnesia in passive avoidance paradigms compared with vehicle controls (Vécsei et al., 1983c, 1984a). The antiamnesic actions show SRIF to influence both consolidation and retrieval processes as the SRIF antiamnesic effect occurred with treatment performed immediately after the shock or 1 hour prior to the retention test. Nevertheless, SRIF dosing concentration variations exist, with lower SRIF concentrations generally improving passive avoidance memory, whereas the opposite effect occurred with 10-fold higher concentrations (Vécsei et al., 1984b, 1989; Vécsei and Widerlöv, 1988). Correspondingly, use of the SRIF-depleting and antisecreting agent cysteamine (Szabo and Reichlin, 1981) induced significant memory deficits in passive avoidance and spatial discrimination tasks across a number of behavioral models and delivery routes (Haroutunian et al., 1987; Schettini et al., 1988; DeNoble et al., 1989; Fitzgerald and Dokla, 1989; Vécsei et al., 1990; Matsuoka et al., 1994), whereas treatment with SRIF or the SST2,3,5 agonist octreotide reversed the cysteamine-induced memory behavior impairments (Schettini et al., 1988; Matsuoka et al., 1994). Additionally, cysteamine administered within 4 hours following learned acquisition of a behavioral task impaired the memory behavior of the task (Haroutunian et al., 1987; Schettini et al., 1988; Vécsei et al., 1990), again supporting the role of SRIF in memory consolidation. Cysteamine administration also dose-dependently impaired mouse memory behavior in the contextual fear conditioning model (Kluge et al., 2008). The memory impairment was associated with decreased LTP in hippocampal CA1 neurons, further supporting that SRIF memory-mediated effects are hippocampal dependent.
SRIF and SRIF-expressing neuron regulation of learning and memory show an interdependency with cholinergic mechanisms. Interactions between SRIF-expressing neurons and cholinergic pathways are integral in hippocampal and neocortical information processing (Müller and Remy, 2018; Obermayer et al., 2018; Urban-Ciecko et al., 2018). Septal cholinergic input is notably involved in the hippocampal SRIF-expressing interneuron regulation of rodent learning and memory behavior, with a corresponding impact on theta oscillation generation and frequency (Leão et al., 2012; Lovett-Barron et al., 2014; Mikulovic et al., 2018; Espinosa et al., 2022). A recent study using optogenetic stimulation in anesthetized mice showed that lateral septum SRIF-expressing neurons can disinhibit the cholinergic septo-hippocampal pathway, enhancing the amplitude and synchrony of theta oscillations (Espinosa et al., 2022). Moreover, in studies assessing central cholinergic blockade in rats, through either lesioning of the nucleus basalis of Meynert (NBM) or by use of the nonspecific muscarinic antagonist scopolamine, intracerebroventricular administration of SRIF reversed memory behavior impairments in passive avoidance testing (Matsuoka et al., 1994). It is noteworthy that cognitive decline associated with cholinergic neuron loss in the NMB is a consistent finding in humans with AD and MCI (Mesulam, 1976; Whitehouse et al., 1981; Mesulam et al., 2004; Grothe et al., 2010). In evaluations of the SRIF secretagogues FK960 and FK962 in rats, both compounds produced synergistic memory behavior benefits when coadministered with the cholinesterase inhibitor (ChEI) donepezil (Tokita et al., 2002; McCarthy et al., 2011). FK962 further lessened memory deficits in passive avoidance tasks in rats treated with scopolamine or NBM lesioning (Tokita et al., 2005), whereas FK960 reduced deficits in visual discrimination memory behavior in nonhuman primates induced by scopolamine (Matsuoka and Aigner, 1997).
SST subtypes differentially regulate learning and memory. Gastambide et al., (2009, 2010) performed initial learning and memory behavior evaluations using the water-maze task and intrahippocampal injections of subtype-selective SST agonists. Subtype-selective SST1–3 agonists showed no effect on acquisition or retention of hippocampal-dependent spatial or striatum-dependent cue-based behavior when compared with vehicle controls, whereas injections of SRIF or the SST4 agonist L-803,087 impaired acquisition and retention of hippocampal-dependent spatial behavior compared with vehicle controls. Conversely, L-803,087 also showed to dose-dependently enhance cue-based memory compared with vehicle. The researchers concluded that intrahippocampal injections of an SST4 agonist was associated with switching from the use of a hippocampal-based spatial response to a dorsal striatum cue-based behavioral response (Gastambide et al., 2009, 2010). Subsequent evaluations using wild-type and Sst2-knockout mice support a hippocampal-to-dorsal striatum response switch through an interaction between SST4 and SST2 (Gastambide et al., 2010). Other reports identify a functional interaction between SST4 and SST2 within the hippocampal formation (Moneta et al., 2002; Cammalleri et al., 2006; Prévôt et al., 2017). SST4 agonist-induced enhancements of learning and memory behaviors likewise occur in mouse models of accelerated aging and AD. Administration of SST4 agonists NNC 26-9100 (intracerebroventricularly) (Sandoval et al., 2012, 2013) or KS-I-50 (intraperitoneally and orally) (Neumann et al., 2021) enhanced spatial learning and memory using the foot-shock/T-maze task in age-accelerated and AD mouse models when compared with vehicle controls. NNC 26-9100 also enhanced hippocampal-dependent memory behavior in the novel-object recognition test compared with vehicle controls (Sandoval et al., 2012). Although the parceling of SST subtype function as determined by behavioral responses is subject to numerous qualifiers, both SST2 and SST4 show to have predominant roles in learning and memory processing, which is consistent with their heightened hippocampal expression (Fehlmann et al., 2000; Sjöstedt et al., 2020).
V. Somatostatin Links to Alzheimer Disease
A. Somatostatin Decline
Declines in brain SRIF and SRIF-expressing neurons in AD coincide with progressive pathology and symptomology (Fig. 2). Although declines in brain SRIF occur as a part of natural aging (Mattson and Arumugam, 2018), the loss is accelerated in AD. Initial studies using AD post-mortem brain tissue identified that SRIF-like immunoreactivity was reduced in the cerebral cortex compared with age-matched controls (Davies et al., 1980; Rossor et al., 1980). Subsequent reports confirmed the AD-associated reductions in SRIF levels and SRIF immunoreactivity in cortical tissues (Beal et al., 1986; Dournaud et al., 1995; Bissette et al., 1998; Grouselle et al., 1998; Kumar, 2005; Saiz-Sanchez et al., 2010; Waller et al., 2020), along with decreased SRIF levels in cerebrospinal fluid (CSF) (Soininen et al., 1984; Atack et al., 1988; Davis et al., 1988; Molchan et al., 1993; Nilsson et al., 2001) compared with age-matched controls. One report found a >70% reduction in AD frontal cortex SRIF-immunoreactive neurons (Kumar, 2005), with another report showing an ∼30% reduction in AD temporal cortex SRIF-immunoreactive interneurons (Waller et al., 2020) compared with respective controls. Decreased SRIF gene expression in the AD frontal and temporal cortices parallel the loss of SRIF-expressing neurons and decline in brain SRIF (Gahete et al., 2010; Guennewig et al., 2021). A recent study generating a single-cell transcriptomic atlas covering 2.3 million cells in post-mortem tissue of the aged human prefrontal cortex from 427 individuals diagnosed with either no cognitive impairment, MCI, or AD identified selectively vulnerable SRIF neuronal subtypes in those with AD (Mathys et al., 2023). The relative abundance of the SRIF inhibitory neuron subtypes showed to be significantly higher in those with cognitive resilience to global AD pathology, neurotic plaque burden, and NFT burden. In those with a confirmed diagnosis of AD, the relative abundance of the vulnerable SRIF neurons was significantly higher in those without cognitive impairment compared to those with observed dementia. SRIF levels in the AD anterior olfactory nucleus also declined by ∼50% compared with controls (Saiz-Sanchez et al., 2010). SRIF plays a critical role in olfactory information processing, with mouse models showing that SRIF mediates olfactory detection and discrimination behaviors (Lepousez et al., 2010; Nocera et al., 2019). Notably, olfactory dysfunction is prevalent in individuals with MCI and strongly correlated with AD development (Devanand et al., 2000; Saiz-Sanchez et al., 2010), indicating early SRIF loss in this region as a risk factor for AD.

AD pathological progression. AD occurs across a continuum associated with progressive brain pathology and symptomology [adapted with permission from Long and Holtzman, (2019)].
The relationship between Aβ and SRIF loss is validated in animal models of AD. Evaluations in rats chronically infused (intracerebroventricularly) with Aβ40 or Aβ25-35 exhibited significantly reduced SRIF-like immunoreactivity in hippocampal CA1, temporal cortex, and frontoparietal cortex compared with controls (Nag et al., 1999; Nag and Tang, 2001; Aguado-Llera et al., 2005, 2018; Hervás-Aguilar et al., 2005; Burgos-Ramos et al., 2007). In senescence-accelerated mouse prone-8 (SAMP8) mice, a nontransgenic strain that shares many characteristics of human AD pathology (Morley et al., 2012), SRIF-expressing hippocampal CA1 interneurons showed decline at 12 months of age (M. J. Lagartos-Donate et al., preprint, DOI: https://doi.org/10.1101/598599). Correspondingly, at 12 months of age, SAMP8 mice display impairments in learning and memory behavior along with increased Aβ accumulation and oxidative stress in cortical tissues (Morley et al., 2012; Griñán-Ferré et al., 2018). In transgenic mice expressing mutant human APPswe and presenilin-1 dE9 (PS1dE9), an age-dependent increase in Aβ40 and Aβ42 coincided with deficits in memory behavior, aligning with decreased cortical SRIF levels and cholinergic markers (Savonenko et al., 2005). In mice over-expressing both the Swedish and London mutations of APP (APP751) and PS1 (PS1M146L), Aβ deposition starts as early as 2.5 months (Blanchard et al., 2003), with dystrophic neurites, a loss in SRIF mRNA expression, and a 50%–60% reduction in the numerical density of the SRIF-immunopositive cells in the CA1–CA3 stratum oriens and dentate gyrus at 6 months as compared with wild-type controls (Ramos et al., 2006). The SRIF loss at 6 months was relatively selective, with no significant changes in other neuronal markers respective to GABAergic, glutamatergic, and cholinergic systems. Additionally, there was a linear relationship between declining SRIF mRNA expression and increasing Aβ concentration. A subsequent evaluation by the same group reaffirmed the early SRIF loss with increased Aβ deposits along with heightened inflammatory activity (Moreno-Gonzalez et al., 2009). In an elegant study in which APP/PS1dE9 mice were crossbred with mice expressing enhanced green fluorescent protein under the control of the Gad1promoter in conjunction with in vivo two-photon imaging, Schmid et al., (2016) showed an age and Aβ plaque–dependent impairment of structural plasticity of dendritic spines of SRIF-expressing hippocampal O-LM interneurons compared with wild-type controls. A decreased axon survival and dendritic spine density, along with an increased turnover of spines of hippocampal inhibitory neurons, indicated a destabilized synaptic connectivity. These effects corresponded with an impairment of cholinergic input from the medial septum onto O-LM interneurons, consistent with SRIF-cholinergic interdependency.
Aβ-dependent impairments in learning and memory align with disruptions in hippocampal theta and gamma oscillations (Palop and Mucke, 2016; Mably and Colgin, 2018; Andrade-Talavera and Rodríguez-Moreno, 2021). Reports identify that the power of theta oscillations are reduced in several transgenic AD models associated with Aβ accumulation, including 3xTg (Akay et al., 2009; Mondragón-Rodríguez et al., 2018), CRND8 (Goutagny et al., 2013), APP/PS1 (Wang et al., 2002; Scott et al., 2012), and APP23 (Ittner et al., 2014) mice. Direct Aβ injection models also show theta oscillation dysfunction (Villette et al., 2010; Chung et al., 2020; Park et al., 2020). Reductions in gamma oscillations likewise occur in 3xTg (Mably et al., 2017) and 5xFAD mice (Iaccarino et al., 2016). Significantly, these investigations identify that oscillation dysfunctions occur prior to actual neuronal loss, with ramifications as a potential early AD pathology indicator. Moreover, studies using SRIF-Cre mice showed that the optogenetic activation of SRIF-expressing interneurons could selectively restore the power and synchronicity of the theta and gamma oscillation after injection of soluble AβOs (Chung et al., 2020; Park et al., 2020). Ex vivo voltage-clamp recordings from hippocampal slice CA1 pyramidal cells of the AβO-injected mice indicated that the optogenetic activation of SRIF interneurons enhanced inhibitory postsynaptic currents at these frequencies. This not only identifies a capacity to reverse AβO-related dysregulation through a SRIF interneuron activation but substantiates the impact of AβOs as to early hippocampal dysregulation independent of Aβ plaque formation.
Apolipoprotein E (apoE) evaluations support the relationship between SRIF and AD. ApoE regulates plasma lipid levels by increasing the degradation of particles rich in triglycerides and cholesterol and exists in three major isoforms: ε2, ε3, and ε4. Expression of APOE-ε4 is the greatest risk factor for LOAD (Jansen et al., 2019). Relative to APOE-ε3 homozygous carriers, individuals expressing one APOE-ε4 allele are 2.6–4.2 times more likely to develop AD, which increases to a 12.9–14.5-fold risk for APOE-ε4 homozygous carriers (Chai et al., 2021). Possession of the APOE-ε4 also lowers the mean age of disease onset and is associated with worse clinical outcomes (Mortensen and Høgh, 2001; Chai et al., 2021). Precisely how APOE-ε4 increases AD risk has not been fully determined, yet human and animal evidence shows that the presence of the ε4 genotype positively correlates with enhanced accumulation of interneuronal Aβ and AβOs (Christensen et al., 2010; Zepa et al., 2011). Several studies performed with APOE-ε4–knockin mice show enhanced age-dependent loss of SRIF-expressing interneurons, impairments in hippocampal neurogenesis, and deficits in learning and memory compared with controls (Li et al., 2009a; Andrews-Zwilling et al., 2010; Leung et al., 2012; Knoferle et al., 2014). These data correspond with post-mortem evaluations of AD tissue, with APOE-ε4 carriers showing substantially lower frontal cortex SRIF levels as compared with APOE-ε2/3 carriers (Grouselle et al., 1998). Sex differences further delineate the APOE-ε4 and SRIF link. Female APOE-ε4–knockin mice exhibit a significantly lower number of SRIF-expressing hilar GABAergic interneurons at 6 months of age when compared with APOE-ε3–knockins of the same age (Leung et al., 2012), whereas in male mice the number of SRIF-expressing hilar GABAergic interneurons is similar between APOE-ε4–knockin and APOE-ε3–knockin at all ages (1–16 months). These findings align with human studies showing that female APOE-ε4 carriers are more likely to convert to MCI and AD when compared with male APOE-ε4 carriers (Altmann et al., 2014), albeit whether the sex-specific findings in human APOE-ε4 carriers can be explained by dysregulations in SRIF still remains to be determined. It is also noteworthy that variations in the SRIF gene are implicated in APOE-ε4–related AD risk. In studies of Finnish (Vepsäläinen et al., 2007a) and Chinese (Xue et al., 2009) patients, APOE-ε4–positive individuals with the C allele carriers of the SRIF gene single-nucleotide polymorphism rs4988514 had increased AD risk. Conversely, in the Finish cohort study, a major haplotype TTG of SRIF was significantly under-represented among all of the AD patients, including APOE-ε4 carriers. Thus, genetic variations in the SRIF gene may serve as modifiers to AD risk.
The impact of AD on SSTs has been the focus of several investigations. Initial radioligand binding studies using radiolabeled SRIF-14 identify a general decrease of SSTs in post-mortem AD brains. Total SST density was reduced by ∼50% in both frontal cortex (Brodmann areas 6, 9, and 10) and temporal cortex (Brodmann area 21) compared with age-matched controls (Beal et al., 1985). Total SST density in the hippocampus was likewise reduced by 40%. Scatchard analyses supported that the reductions were due to receptor number rather than altered affinities. The postcentral gyrus, cingulate cortex, temporal pole, or superior temporal gyrus showed no significant changes. In another binding study using SRIF-14, a reduction in SSTs only occurred in the frontal cortex of AD tissue compared with controls (Bergström et al., 1991). In a third radioligand study, a reduced binding in the temporal cortex was identified using both SRIF-14 and the SST2,3,5 agonist SMS 204-090, with only SST2, SST3, and SST5 (based on the use of SMS 204-090) being substantially reduced in the frontal cortex compared with controls (Krantic et al., 1992). In immunohistochemical evaluations using SST subtype–specific antibodies, SST4 and SST5 expression significantly decreased in the frontal cortex of those with AD compared with controls, with no significant changes observed for either SST1, SST2 (moderate decrease), or SST3 (moderate increase) (Kumar, 2005). Lastly, SST1, SST3, and SST4 each showed decreased mRNA expression in the temporal lobe of those with AD compared with controls (Gahete et al., 2010), whereas SST2 mRNA expression decreased only in the inferior temporal lobe of those with AD compared with controls (Gahete et al., 2010). Several factors may explain the inconsistencies across these investigations. The most apparent is that the quality of post-mortem brain tissue can vary greatly across samples. Total tissue volumes and regional atrophy can only be compared with age-matched controls and not the “original” healthy brain affiliated with the AD tissue sample. When considering ligand binding evaluations, the use of either radiolabeled SRIF-14 or SST2,3,5 agonist limits the capacity to delineate the specific SST subtype. The immunological semiquantitative measures are relative to antibody adherence determined across a subset of tissue slices, with additional concern as to potential crossreactivities of antibodies. Moreover, mRNA expression is not actual membrane-associated protein nor an indicator of functional binding. Perhaps the most critical takeaway is that the radioligand studies identified a preservation of receptor-ligand binding in AD tissue, suggesting a maintenance of SST viability for pharmacologic targeting after AD develops.
In summary, substantial evidence identifies that the loss of SRIF and SRIF-expressing neurons is a pivotal pathological event in AD progression. Animal models validate Aβ accumulation in the brain, particularly hippocampal tissue, as a primary driver of SRIF-expressing neuron loss. This loss aligns with impairment of cholinergic transmission, theta and gamma oscillations, and learning and memory behavior. Studies in APOE-ε4 carriers with elevated rates of AD incidence and associated animal models further support the SRIF-AD link. SSTs generally decrease in AD post-mortem brain tissue compared with aged-matched non-AD controls, with variability in specific SST subtype expression measures depending on brain region and method of evaluation.
B. Somatostatin and Amyloid-β Peptide Catabolism
Aβ levels in the brain are maintained by a balance of anabolic and catabolic processes in coordination with the blood-brain barrier (BBB) transport mechanisms. Although Aβ overproduction is strongly associated with ADAD, the more gradual decline in Aβ clearance mechanisms is believed to be the driving force in LOAD (Mawuenyega et al., 2010). As LOAD is the most prevalent form of AD (https://pubmed.ncbi.nlm.nih.gov/38689398/), enhancing Aβ-mediated catabolism presents a strategy of significant therapeutic potential (Nalivaeva and Turner, 2019). Neprilysin (NEP) and insulin-degrading enzyme (IDE) are the major Aβ-degrading enzymes in the brain (Nalivaeva and Turner, 2019). Moreover, both NEP and IDE have a unique relationship with SRIF. SRIF is not only a substrate for NEP and IDE but further regulates the activity of both enzymes and the corresponding catabolism of Aβ (Fig. 3).

Schematic of SST-mediated Aβ catabolic mechanisms. The internalization of extracellular Aβ induces cell damage and proapoptotic mechanisms. (1) SST1,4 agonist activation induces NEP activity and corresponding extracellular Aβ degradation. (2) SST4 activation induces transcription of genes associated with generation of proteins involved in Aβ degradation. (3) Insulin-degrading enzyme acts intracellularly, capable of directly interacting with SRIF and Aβ to promote degradation in critical organelles (i.e., peroxisomes, mitochondria, endoplasmic reticulum).
NEP is a membrane-bound zinc metallopeptidase, with its active site facing the extracellular space. Acting primarily as an endopeptidase, NEP is found peripherally and centrally (Nalivaeva and Turner, 2019). Within the brain, NEP is expressed abundantly in areas associated with memory formation, particularly in the hippocampal formation and layers II/III and V of the neocortex (Fukami et al., 2002). NEP preferentially cleaves small peptides on the N‐terminal side of hydrophobic residues, including Aβ. NEP degrades monomeric Aβ (Iwata et al., 2000, 2001; Takaki et al., 2000; Hama et al., 2001; Shirotani et al., 2001; Leissring et al., 2003; Saito et al., 2005) and low-molecular-weight AβOs (Kanemitsu et al., 2003). Loss of NEP is associated with elevated brain Aβ levels and AD pathology. In Nep-knockout mice, Aβ40 and Aβ42 levels are twice as high as wild-type controls (Iwata et al., 2001). In human post-mortem studies of AD brain tissue, NEP activity, mRNA expression, and protein expression decreased compared with age-matched cognitively normal controls (Yasojima et al., 2001a,b; Russo et al., 2005; Miners et al., 2006; Carpenter et al., 2010; Wang et al., 2010; Zhou et al., 2013). A meta-analysis of these AD studies discerned that decreased NEP expression and activity progressed with increasing age, with the effect most pronounced in older individuals (Zhang et al., 2017). Nevertheless, individuals identified with prodromal AD also show reduced NEP levels in CSF relative to controls (Maruyama et al., 2005). Moreover, lower NEP expression levels are observed in APOE-ε4 carriers compared with noncarriers (Miners et al., 2006). This implies that reduced NEP activity is not secondary to tissue atrophy. NEP polymorphisms are also identified with increased susceptibility to LOAD (Sakai et al., 2004; Wood et al., 2007). Ultimately, the role of NEP in Aβ clearance and its associated decrease in the AD brain support a NEP-AD link.
SRIF plays a leading role in the NEP regulation of Aβ. Seminal work by Saito et al., (2005) first identified that SRIF increased brain NEP activity. When wild-type mouse-cultured primary neurons were treated with SRIF, NEP activity increased and Aβ42 expression decreased compared with controls. Conversely, when the primary neurons were prepared from Nep-knockout mice, SRIF treatment did not reduce Aβ42 expression relative to controls. Srif-knockout mice also exhibited a decrease in hippocampal NEP activity and increase in Aβ42 expression compared with wild-type controls, supporting SRIF as a regulator of NEP activity. When primary neurons were treated with either the SST antagonist BIM23056 or Gi-GPCR inhibitor pertussis toxin, NEP activation by SRIF was inhibited, indicating that activation of SSTs by SRIF enhanced downstream Gi-coupled GPCR signal transduction to increase NEP activity. The SRIF treatments had no significant impact on NEP mRNA or protein expression, suggesting that SRIF may regulate NEP through post-translational processes. This work led to the hypothesis that loss of brain SRIF initiates a decline in NEP activity with a corresponding elevation in steady-state Aβ levels (Iwata et al., 2005), driving a pathological feedback loop (Fig. 1). A study using SRIF fused with a linker to enhance BBB uptake (Rofo et al., 2021) substantiated the work by Saito and colleagues. The linker-fused SRIF administered intravenously over multiple doses was shown to increase hippocampal NEP activity and decrease membrane-bound Aβ42 expression levels. Thereby, loss of SRIF is implicated as a triggering event for Aβ accumulation leading to LOAD, whereas pharmacologically targeting brain SSTs presents a means to enhance NEP activity toward a disease-modifying AD treatment.
The recognition of SRIF’s effect on NEP activity led to the evaluation of SST subtype agonists and their influence on Aβ degradation. Given the significant alignment of brain SST4 distribution with areas of heavy NEP expression, a series of studies focused on SST4 mediation of NEP activity. In SAMP8 mice, single-dose intracerebroventricular administration of the SST4-selective agonist NNC 26-9100 increased cortical NEP activity, with enhanced learning and memory behavior compared with vehicle controls (Sandoval et al., 2012). NNC 26-9100 treatment correspondingly decreased protein expression of the AβO42 “trimer” in both extracellular and intracellular fractions of cortical tissue lysates compared with controls. The AβO42 trimer is notably implicated as an inhibitor of LTP (Townsend et al., 2006; Selkoe, 2008) and is capable of inducing conformational changes in tau protein, leading to disrupted axonal transport (Sherman et al., 2016). Additionally, when NEP-deficient mice were crossbred with APP23 transgenic mice, AβO42 trimer expression increased along with memory behavior impairments (Huang et al., 2006). NNC 26-9100 coadministered with the NEP inhibitor phosphoramidon (intracerebroventricularly) in SAMP8 mice inhibited the reduction in AβO42 protein expression, supporting a NEP-dependent mechanism (Sandoval et al., 2013). NNC 26-9100 likewise reduced AβO42 trimer protein expression in Tg2576 mice (Sandoval et al., 2013). In a third NNC 26-9100 evaluation, single-dose intracerebroventricular administration in 3xTg mice increased cortical NEP mRNA expression by approximately ninefold at 24 hours postinjection compared with vehicle controls (Sandoval et al., 2019). This corresponded with an approximately fivefold increase in SST4 mRNA expression without changes in any other SST subtype. This finding is consistent with an in vitro study in CHO-K1 cells treated with SRIF, which showed an upregulation of human SST4 at 24 hours (Hukovic et al., 1996). Nilsson et al., (2020) confirmed that NNC 26-9100 increased NEP activity in primary neurons while showing that the SST1 agonist CH275 was also capable of increasing NEP activity. Interestingly, primary neurons taken from Sst1 or Sst4 knockouts showed no difference in NEP activity compared with controls, whereas dual Sst1/Sst4 knockouts significantly decreased hippocampal NEP activity with increased Aβ40 and Aβ42 expression. This suggests that SST1 and SST4 may be redundant in maintaining NEP activity. Relatedly, SST1 and SST4 have shown to interact with the postsynaptic density proteins postsynaptic density protein (PSD)-93 and PSD-95, which target GPCRs to the membrane of dendritic postsynaptic terminals (Christenn et al., 2007). In APPswe/PS1dE9 mice, enhanced expression of PSD-93 upregulated SST4 on the cellular membrane, increased hippocampal NEP expression, and reduced Aβ40 and Aβ42 levels (Yu et al., 2017). This implies that enhanced expression of functional SST4 to the cellular membrane is itself sufficient to increase NEP activity and lower Aβ levels.
Originally named for its ability to metabolize insulin, IDE is a metallopeptidase found throughout the body with broad substrate specificity. IDE predominately exists within the cell cytosol, but is also present in endosomes (Hamel et al., 1991), peroxisomes (Morita et al., 2000), mitochondria (Leissring et al., 2004), and ER (Carpenter et al., 2010). The intracellular distribution of IDE is consistent with its role as a scavenging enzyme, metabolizing aggregation-prone peptides to maintain cellular homeostasis (Arbo et al., 2020). IDE has high affinity for β-structure–forming substates (Tundo et al., 2017; Kurochkin et al., 2018). In a study using a quenched Aβ40 peptide, which fluoresces upon cleavage inside the KLVFF region critical for aggregation, IDE catabolism capacity was evaluated in post-mortem hippocampal tissue from individuals at different stages of AD progression (Stargardt et al., 2013). IDE was shown to be the primary peptidase that degrades cytoplasmic monomeric Aβ40 in early-stage AD hippocampal tissue, whereas a decline in IDE corresponds with increased intraneuronal Aβ accumulation, leading to synaptic and neuronal dysfunction that precedes both extracellular plaque deposits and NFTs (Takahashi et al., 2017; Welikovitch et al., 2018). Ide-knockout mice likewise show a substantial increase in cerebral Aβ levels (Farris et al., 2003), whereas in transgenic mice overexpressing human IDE the levels of Aβ and plaques in the brain are significantly reduced compared with wild-type controls (Leissring et al., 2003). IDE may also reduce amyloidogenic fibrillization of Aβ42 in a nonproteolytic manner as a “dead-end” chaperone, preventing the formation of aggregates by the irreversible trapping of monomers (de Tullio et al., 2008, 2013). Interestingly, the association between type 2 diabetes mellitus and LOAD has been linked to IDE deficits (Farris et al., 2003; Wei et al., 2021), which align with dysregulation of brain glucose metabolism (Connolly et al., 2019; González et al., 2022). Multiple reports identify genetic variations in IDE as a risk factor for LOAD (Ertekin-Taner et al., 2004; Björk et al., 2007; Vepsäläinen et al., 2007b; Wang et al., 2012). Additionally, APOE-ε4 carriers exhibited reduced hippocampal IDE mRNA levels compared with noncarriers (Cook et al., 2003), providing another mechanism for how APOE-ε4 may increase the risk of LOAD.
SRIF is both a substrate and modulator of IDE. Although IDE can terminate the actions of SRIF, SRIF can also modulate the activity and function of IDE. Allosteric binding of SRIF to the active site of one IDE subunit was shown to increase IDE proteolytic activity, enhancing enzymatic cleavage of fluorogenic Aβ (Ciaccio et al., 2009). Docking evaluations show binding of SRIF to two additional sites on IDE, which can change the substrate specificity of IDE toward different substrates, including Aβ (Tundo et al., 2016). This identifies a complex interaction in which the effect of SRIF binding and modulation is dependent on the substrate as well as on the mode of substrate interaction with different allosteric sites. Although the cellular localization of IDE indicates that such an interaction would be isolated to the intracellular domain, extracellular Aβ binds to the neuronal plasma membrane (Johnson et al., 2011, 2013) and is internalized (Jin et al., 2016). Thus, extracellular Aβ content could be susceptible to such an SRIF-IDE-Aβ interaction. Lastly, IDE may also be regulated through SST activation by SRIF or SST agonists. In 3xTg mice administered the SST4 agonist NNC 26-9100, IDE mRNA expression increased by ∼15-fold compared with vehicle control (Sandoval et al., 2019). This finding aligns with previous data showing decreased intracellular AβO40 and AβO42 expression in cortical tissue of SAMP8 and 3xTg mice after an identical NNC 26-9100 treatment (Sandoval et al., 2012, 2013). Although the proposition of SST4 mediation of IDE expression adds to the complexity of the SRIF-IDE-Aβ interaction, it does so in a manner that further supports SRIF capacity to mitigate Aβ levels.
In summary, SRIF acting through SSTs in the brain promotes NEP activity and possibly IDE activity, with the capacity to enhance extracellular and intracellular Aβ catabolism. SST4 agonist activation in particular was shown to enhance NEP activation while also increasing mRNA expression of NEP and IDE.
C. Somatostatin and Amyloid-β Peptide Aggregation
A growing area in AD research focuses on Aβ cross-seeding. Cross-seeding is a process by which the amyloid structures of one type of protein act to “seed” and facilitate the aggregation of another amyloid protein, resulting in heterologous amyloids (Subedi et al., 2022). Such seeding may provide a mechanistic explanation for the presence of different misfolded proteins present in Aβ plaques (Jucker and Walker, 2018). Recent research suggests that heterotypic Aβ interactions facilitate amyloid assembly and modify amyloid structure between proteins via aggregation-prone regions (Konstantoulea et al., 2022). The aggregation-prone regions of various proteins interacted with AβOs and altered the Aβ aggregation kinetics and fibril morphology. This aligns with the theory that protein misfolding diseases are caused by seed polymerization and abnormal protein assemblies (Glenner, 1980; Prusiner, 1984). A feature of SRIF is its own capacity to self-aggregate and form amyloid-like structures (van Grondelle et al., 2007; Maji et al., 2009). Physiologically, this aggregative ability allows for high-density storage of inert peptides in secretory granules. Yet, it implicates the potential of SRIF to coaggregate with other proteins.
Recent in vitro research advances an intriguing hypothesis as to the potential of SRIF-AβO42 aggregation. A report showed SRIF binding to AβO42, forming “mixed assemblies” capable of interfering with Aβ fibrillization (Wang et al., 2017). The SRIF interaction did not occur between Aβ42 monomers or AβO40 forms. Another group similarly identified the capacity for SRIF to bind AβO42 tetramers in vitro (Puig et al., 2020). It was postulated that such SRIF-AβO42 assemblies could enhance AD pathogenesis by promoting and/or maintaining a soluble neurotoxic state, with SRIF influencing Aβ aggregation kinetics (Solarski et al., 2018). This would align with an increase in toxic AβOs and early neuron loss in brain regions of high SRIF concentration. Nevertheless, this SRIF-AβO42 aggregate assembly hypothesis is derived from in vitro data and comes with a number of qualifiers. The studies of these mixed assemblies used high concentrations of respective peptides to induce the observed effects [SRIF: 4 μM; Aβ42 and AβO42: 2.5, 5 μM (Wang et al., 2017); SRIF: 150 μM; AβO42 (tetramer): 35.5 and 57.5 μM (Puig et al., 2020)], increasing the likelihood of nonphysiologic outcomes. Moreover, proteolytic processes that are part of the normal in vivo environment were not accounted for in the evaluations. These considerations would foreseeably limit the in vivo manifestation of such assemblies, rendering them rare events in the brain. Yet, given the inherent aggregation of Aβ on cellular surfaces wherein a higher rate of interaction with SRIF may occur, such an interaction cannot be ruled out as a seed event. Relatedly, in a recent mouse study of chronic stress, a selective vulnerability of SRIF-expressing neurons of the prefrontal cortex occurred through an exacerbated unfolded protein response of the ER (Tomoda et al., 2022). There was a corresponding increase in SRIF protein aggregation, albeit the nature of the aggregated species was not determined. This would support a stressor-induced in vivo event capable of initiating a SRIF aggregation response. The extrapolation being the greater the intensity and timeframe of exposure to a given stressor or set of stressors the greater potential for such aggregative-prone species to accrue. Nevertheless, Aβ and AβO interactions are not unique to SRIF. The known list of proteins in which monomeric Aβ and AβO forms can bind is substantial and continues to grow (Wang et al., 2017; Konstantoulea et al., 2022). Thus, if SRIF-AβO42 assemblies are substantiated in vivo, it may well be one of many potential assemblies. In this context, a heterogeneity in amyloid assemblies could be indicative of differential pathological seeding events, aligning with a multifactorial AD etiology, or simply a result of an almost ubiquitous binding capacity of Aβ in its various forms.
In summary, based on in vitro observations, the formation of SRIF-AβO42 aggregate assemblies may serve as a seed event, promoting AD pathology. Further characterization of such mixed assemblies from brain tissue extracts across the stages of AD may provide a more complete picture. Focus on the early prodromal stage of AD in particular is necessary to substantiate any pathological seeding event unique to a specific protein or subset of proteins.
D. Somatostatin and Neuropsychiatric Symptoms
AD is associated with a number of neuropsychiatric symptoms, including depression, apathy, anxiety, fear, agitation, irritability, mood swings, changes in sleeping habits, and psychosis (Zhao et al., 2016; Wiels et al., 2021). These symptoms may also be early indicators to AD (Palmer et al., 2010; Spalletta et al., 2010, 2015; Dietlin et al., 2019; Agüera-Ortiz et al., 2021) (Fig. 2). Although the cause of these symptoms in AD is complex, both neurodegeneration and the body’s stress response contribute. In turn, both neurodegeneration and stress response connect SRIF to the neuropsychiatric symptoms. Much of our understanding of these interconnections comes from research in mood disorders, inclusive of major depressive disorder (MDD), schizophrenia, and bipolar disorder.
Mood disorders present with many of the neuropsychiatric symptoms observed in AD. A prominent neurologic feature of mood disorders is the loss of SRIF and SRIF-expressing neurons (Lin and Sibille, 2013; Robinson and Thiele, 2020). The loss of SRIF-expressing interneurons in these disorders is relatively distinct compared with other types of GABAergic interneurons (Duman et al., 2019; Fee et al., 2021; Prévot and Sibille, 2021). MDD, schizophrenia, and bipolar disorder each display marked SRIF declines in brain regions heavily impacted in AD (Table 1). Females show greater declines in SRIF levels across brain regions compared with males (Sibille et al., 2011; Tripp et al., 2011, 2012; Guilloux et al., 2012), which is consistent with females having a higher risk of developing mood disorders in general and in AD specifically (Spalletta et al., 2010; Lee et al., 2017). Changes in theta and gamma activity are likewise exhibited with mood disorders, in alignment with AD (Palop and Mucke, 2016; Mably and Colgin, 2018; Andrade-Talavera and Rodríguez-Moreno, 2021). Theta power during memory retrieval is reduced in MDD compared with healthy controls (Kane et al., 2019). Schizophrenic patients exhibit reduced θ and γ oscillatory activity along with impaired theta phase coupling between the hippocampus and medial prefrontal cortex during memory retrieval compared with control groups (Haenschel et al., 2009; Adams et al., 2020). Animal models of mood disorders likewise show impairment in theta and gamma oscillations, particularly tied to the hippocampus and the hippocampus-prefrontal cortex-amygdala circuit (Okonogi and Sasaki, 2021; Speers and Bilkey, 2021). A cholinergic interplay also exists, with cholinergic dysregulation being well documented in various neuropsychiatric conditions (Dulawa and Janowsky, 2019). The dysregulation of cholinergic projections from the NBM within the basal forebrain to the cerebral cortex notably coincides with a number of neuropsychiatric symptoms (van Dalen et al., 2017), consistent with AD and MCI (Mesulam, 1976; Whitehouse et al., 1981; Mesulam et al., 2004; Grothe et al., 2010). NBM damage correspondingly impacts SRIF-cholinergic interactions. NBM lesioning in rats results in cholinergic denervation associated with the loss of SRIF-immunoreactive neurons (Zhang et al., 1998) and decreased SRIF binding capacity (Epelbaum et al., 1986; Moyse et al., 1993).
SRIF decline in human mood disorders
The body’s stress response impacts brain health. Stress underpins anxiety, depression, and the risk of dementia (Justice, 2018; Franks et al., 2021). Stress activates the hypothalamic-pituitary-adrenal (HPA) axis, inducing the release of ACTH from the posterior pituitary and subsequent release of glucocorticoids from the adrenal glands. Increased brain glucocorticoid levels and receptor activity under heightened stress contribute to neuropsychiatric symptom severity (Spijker and van Rossum, 2012; van den Berg et al., 2020). Chronic glucocorticoid treatment in humans (Brown et al., 1999; Barrimi et al., 2013) and animal models of anxiety and depression (Ardayfio and Kim, 2006; David et al., 2009) corroborate the contributions of glucocorticoids to neuropsychiatric symptoms, whereas brain SRIF signaling counters the endocrine-mediated processes that induce glucocorticoid release under stress and blunts the broader stress effect (Stengel and Taché, 2017), opposing the stress-associated elevations in ACTH and epinephrine (Brown et al., 1984). Growing evidence reveals that the dysregulation of the HPA axis and increase in glucocorticoid levels contribute to cognitive decline in AD (Swaab et al., 2005; Milligan Armstrong et al., 2021). Glucocorticoid receptor activity plays an essential role in the regulation of the HPA axis and hippocampal-dependent spatial memory (McEwen et al., 2016), with high concentrations of glucocorticoid receptors present in the hippocampus (Morimoto et al., 1996; Wang et al., 2013). Moreover, increased glucocorticoid levels in animals subjected to chronic stress impair hippocampal GABAergic signaling, which is associated with the loss of SRIF-expressing interneurons in the hippocampus (Cullinan and Wolfe, 2000; Czéh et al., 2015). This aligns with other research showing that chronic stress reduces dendritic cell length within hippocampal CA3 and dentate gyrus and increases the loss of hippocampal CA1 neuronal spines compared with nonstressed animals (Magariños et al., 1997; McEwen, 1999). Chronic stress further results in the remodeling of synaptic connections in the hippocampus, amygdala, medial prefrontal, and orbitofrontal cortex (McEwen et al., 2016), aligning with the regional loss of SRIF and SRIF-expressing neurons observed in mood disorders (Table 1) and AD.
Investigations of anxiety and depression behavior in mice provide insight as to the impact of SRIF loss. In a study of how acute and chronic reduction of SRIF neuronal activity impact anxiety and depression behavior, SRIF-Cre mice were injected in the frontal cortex with either a Cre-dependent adeno-associated viral vector to inhibit SRIF neuronal activity or a control reporter (Soumier and Sibille, 2014). Anxiety [elevated plus maze (EPM)] behavior effects differed between acute and chronic inhibition states. Acute inhibition of SRIF neurons increased anxiety behavior compared with controls. Conversely, chronic SRIF neuron inhibition reduced anxiety behavior compared with controls. Selective ablation of the frontal cortex SRIF neurons likewise reduced anxiety behavior under baseline and chronic stress conditions (Soumier and Sibille, 2014). A subsequent evaluation was conducted in Srif-knockout mice to further delineate the chronic effects (Lin and Sibille, 2015). Under chronic stress, Srif-knockout mice displayed increased anxiety (EPM) and depression (novelty-suppressed feeding) behavior compared with wild-type mice. Methodological variables between Cre-dependent SRIF inhibition (Soumier and Sibille, 2014) and Srif-knockout mice (Lin and Sibille, 2015) may account for the difference in effect, with the Cre mice evaluations targeted to the frontal cortex and Srif-knockout mice having a whole-body and brain impact. Another variable is the impact of glucocorticoids. Under baseline conditions, plasma corticosterone levels were elevated in Srif-knockout mice compared with wild-type controls (Lin and Sibille, 2015), consistent with a loss of SRIF feedback inhibition on the HPA axis. Although Srif-heterozygous mice also exhibited elevated plasma corticosterone levels at baseline compared with controls, they did not exhibit enhanced anxiety or depressive behavior. Consequently, elevated corticosterone levels in the Srif-knockout mice could not be entirely responsible for the increase in anxiety or depression behavior. The Srif-knockout mice further showed a reduced gene expression of BDNF along with other genes related to GABAergic neuronal function compared with controls. This corresponds with a recent study in which ablation of dentate hilar SRIF-expressing interneurons in mice decreased BDNF and impaired learning and memory behavior compared with controls (Nagarajan et al., 2023). BDNF plays an important role in the maintenance and survival of SRIF-expressing interneurons (Grosse et al., 2005), increases SRIF gene expression in cortical tissue (Nawa et al., 1994; Villuendas et al., 2001; Sánchez-Muñoz et al., 2011), and enhances hippocampal neurogenesis (Scharfman et al., 2005). A downregulation of BDNF in the hippocampus and prefrontal cortex likewise occurs during chronic stress in rats (Roceri et al., 2004; Murakami et al., 2005). Moreover, several lines of evidence link BDNF deficits to depression (Castrén and Monteggia, 2021). It is notable that the associated stress-induced anxiety and depressive behavior response in mice is mitigated by eIF2 activation via inhibition of the eIF2 kinase (Lin and Sibille, 2015). Stress signals lead to the phosphorylation of eIF2 through kinase activation (Moon et al., 2018), which is in turn associated with AD neuronal degeneration (Chang et al., 2002; Oliveira and Klann, 2022) and AβO exposure in rodent neurons (Lourenco et al., 2013; Ma et al., 2013). These data demonstrate that anxiety and depression behavior associated with the loss of SRIF are contingent on signaling factors that are correspondingly impacted in AD.
Increased activity of SRIF-expressing neurons reduces anxiety and depression behavior. In an investigation of disinhibited SRIF-expressing interneurons using SRIF-Cre:γ2f/f mice versus nondisinhibited γ2f/f controls, increased activity of SRIF-expressing interneurons coincided with a reduction in anxiety (EPM) and depression [forced-swim test (FST), novelty-suppressed feeding] behavior (Fuchs et al., 2017). Stress-naïve SRIF-Cre:γ2f/f mice showed similar SRIF mRNA and protein compared with controls, suggesting that altered SRIF levels could not explain the reduction in anxiety and depression behavior. The reduction in anxiety behavior in SRIF-Cre:γ2f/f mice mimicked the response to benzodiazepines in wild-type controls (Löw et al., 2000). SRIF-Cre:γ2f/f mice also exhibited a reduction in depression (FST) behavior relative to γ2f/f controls. The behavioral changes in the SRIF-Cre:γ2f/f mice coincided with decreased phosphorylation of the mRNA translation factor eukaryotic translational elongation factor 2 (eEF2), consistent with antidepressant doses of ketamine (Li et al., 2010; Autry et al., 2011; Monteggia et al., 2013) and 5-HT2C receptor antagonists (Opal et al., 2014). Phosphorylation of eEF2 is also identified with AD-associated synaptic failure and cognitive impairment (Ma, 2023). Other studies suggest that the antidepressant actions of ketamine are mediated through the inhibition of eEF2 kinase signaling with an associated increase in BDNF (Nosyreva and Kavalali, 2010; Autry et al., 2011). The interplay between SRIF, SRIF-expressing neurons, and BDNF (Nawa et al., 1994; Villuendas et al., 2001; Grosse et al., 2005; Sánchez-Muñoz et al., 2011; Nagarajan et al., 2023) implicates a mechanism wherein SRIF inhibition of eEF2 kinase signaling with a corresponding BDNF elevation produces an antidepressant action. Altogether, the data indicate that chronically increased inhibitory synaptic input from SRIF-expressing interneurons results in behavior alterations and biochemical changes that are similar to established antidepressants (Fuchs et al., 2017).
Preclinical studies support the ability of SRIF administration to reduce anxiety and depression behaviors. SRIF intracerebroventricular dosing in rats reduced anxiety (EPM) and depression (FST) behavior compared with vehicle controls (Engin et al., 2008). The SRIF effect was similar to the benzodiazepine anxiolytic diazepam in reducing theta oscillation frequency while also increasing theta power. Moreover, coinfusion of subeffective doses of SRIF and diazepam significantly reduced anxiety (EPM) behavior, identifying an additive capacity to reduce anxiety. In a follow-up evaluation, intra-amygdalar and intraseptal administration of SRIF-14 or SRIF-28 reduced anxiety behavior (EPM, shock-probe test) in rats (Yeung et al., 2011), whereas intrastriatal administration of SRIF-14 or SRIF-28 did not reduce anxiety behavior, demonstrating that the anxiety-alleviating effects are site specific, consistent with benzodiazepine anxiolytics.
SST subtypes mediate the reduction of anxiety and depression behavior. In rats intracerebroventricularly administered SST2 agonist L-779,976, anxiety (EPM) and depression (FST) behavior were reduced compared with vehicle controls (Engin and Treit, 2009). The SST3 agonist L-796,778 likewise reduced depression behavior, but not anxiety behavior, compared with controls. However, the SST2 and SST3 agonist effects were at a high dose (27 μg). Although the SST1 agonist L-797,591, SST4 agonist L-803,087, and SST5 agonist L-817,818 did not produce a significant behavioral effect, neither the SST4 nor SST5 agonist were evaluated at a dose above 3 μg. Thus, the dyssynchronous dosing across the agonists rendered the determination of anxiety and depression behavioral actions relative to SST subtype involvement unclear. Nevertheless, a subsequent rat study showed that intra-amygdala or intraseptal administration of SST2 antagonist PRL2903 blocked the reduction of anxiety (EPM) behavior produced by SRIF, supporting the anxiolytic role of SST2 (Yeung and Treit, 2012). Mouse studies also support SST2 involvement in the reduction of anxiety and depression behavior while further showing an SST4 contribution. Intrahippocampal injection of the SST2 agonist L-054,264 reduced anxiety (EPM) and depression (FST) behavior in wild-type mice, yet only depression behavior was reduced with the SST4 agonist L-803,087 (Prévôt et al., 2017). Moreover, anxiety (EPM) behavior was increased in Sst2-knockout mice compared with wild-type controls, which is consistent with earlier work in Sst2-knockout mice (Viollet et al., 2000). Sst2-knockout mice exhibited high basal corticosterone plasma levels respective to wild-type controls (Prévôt et al., 2017), similar to Srif-knockout mice (Lin and Sibille, 2015), aligning with an SST2 mediation of SRIF action in inhibiting ACTH release (Strowski et al., 2002). Although basal plasma corticosterone levels in Sst4-knockout mice were similar to wild-type controls, the administration of either an SST2 or SST4 agonist reduced plasma corticosterone levels in wild-type mice under stressful conditions. In another study, anxiety (EPM) and depression (FST) behavior increased in Sst4-knockout mice compared with wild-type controls (Scheich et al., 2016), whereas intraperitoneal administration of the SST4 agonist J-2156 in wild-type mice decreased anxiety (EPM) and depression (tail-suspension test) behavior compared with vehicle controls. Overall, these animal model studies support SST2 and SST4 as primary mediators of SRIF’s depression and anxiety-reducing effects.
In summary, the loss of SRIF and SRIF-expressing neurons contribute to the neuropsychiatric symptoms observed in AD, with contributions from the body’s stress response. The brains regions impacted by this loss in AD are consistent with what is observed in mood disorders (Table 1). Preclinical data support SST2 and SST4 as the principal subtypes that mediate the actions of SRIF in reducing anxiety and depression behavior, consistent with their heightened densities in brain regions (Consortium, 2020; Sjöstedt et al., 2020) most impacted by mood disorders.
E. Somatostatin and Seizures
Seizures are characterized by abnormal and recurrent bursts of electrical activity in the brain, commonly involving the hippocampus, amygdala, frontal cortex, temporal cortex, and olfactory cortex (Chauhan et al., 2022). Although fundamentally distinct disorders, AD and seizures share key pathological hallmarks and regions of impact (Giorgi et al., 2020; Lehmann et al., 2021). In both seizures and AD, the balance in neuronal network excitatory-inhibitory activity is dysregulated, contributing to hyperexcitation. The excitatory-inhibitory balance dysregulation being the pivotal pathologic connection, traceable to Aβ accumulation (Romoli et al., 2021). Correspondingly, the loss of SRIF and SRIF-expressing interneurons in AD play a prominent role in this neuronal network excitatory-inhibitory imbalance, driving a positive feedback loop to neuronal death (Fig. 1).
Substantial clinical evidence identifies bidirectional influences between AD and seizures. Individuals with seizures have an increased risk of developing dementia (Breteler et al., 1995; Cordonnier et al., 2007; Costa et al., 2019; Gourmaud et al., 2020; Tsai et al., 2021), whereas individuals with AD exhibit greater levels of seizure activity (Imfeld et al., 2013; Vossel et al., 2013, 2016; Cheng et al., 2015; Vöglein et al., 2020; Zelano et al., 2020; Habeych et al., 2021). Moreover, those with AD or amnestic MCI that suffer seizure activity show earlier onset and quicker progression of cognitive decline than those without detectable seizure activity (Vossel et al., 2013, 2016; Vöglein et al., 2020). Individuals with ADAD are particularly prone to seizure activity. Seizure incidence is higher in those with PSEN1 (Janssen et al., 2003; Snider et al., 2005; Larner, 2010), PSEN2 (Marcon et al., 2004; Jayadev et al., 2010), or APP (Edwards-Lee et al., 2005; Cabrejo et al., 2006; Lindquist et al., 2008) mutations, linking seizures to Aβ over-production. Seizure activity further disrupts oscillatory networks governing theta and gamma rhythms critical in information processing and memory (Lopez-Pigozzi et al., 2016; Malkov et al., 2022). This disruption in oscillatory activity with seizures is analogous to oscillatory disruptions in AD (Palop and Mucke, 2016; Mably and Colgin, 2018; Andrade-Talavera and Rodríguez-Moreno, 2021) and with the loss of SRIF in conjunction with Aβ accumulation (Villette et al., 2010; Chung et al., 2020; Park et al., 2020). Importantly, neuronal hyperexcitation has the capacity to increase Aβ levels. Individuals with late-onset epilepsy of unknown origin show heightened Aβ42 levels in their CSF, with 17.5% of patients progressing to AD (Costa et al., 2019). Drug-resistant temporal lobe epilepsy (TLE) increases Aβ42 expression in the hippocampus, with the rise of hippocampal phosphorylated-APP correlated with impaired cognitive function (Gourmaud et al., 2020). These findings align with studies in Tg2576 mice showing enhanced neuronal activity resulted in the release of Aβ with corresponding regional vulnerability to Aβ deposition (Cirrito et al., 2005, 2008; Bero et al., 2011), whereas stress-induced elevations of Aβ in the hippocampal interstitial fluid of Tg2576 mice are blocked in the absence of neuronal activity (Kang et al., 2007).
Preclinical studies verify that Aβ can induce seizure activity. APP/PS1 mice with elevated Aβ levels display increased neuronal hyperexcitability compared with age-matched wild-type controls (Minkeviciene et al., 2009). In recording episodes at the onset of Aβ pathogenesis, at least one unprovoked seizure was detected in 65% of the mice, of which 46% had multiple seizures and 38% had a generalized seizure. In FAD mice (lines hAPP-J20, hAPP-J9, APP23/PS45, and APP/PSEN1dE9), elevated levels of Aβ were associated with increased spontaneous seizure activity in the cortex and hippocampus, with associated remodeling of inhibitory circuits (Palop et al., 2007). A series of studies by Busche and colleagues mechanistically linked Aβ to neuronal hyperexcitation and cognitive dysfuntion. Initial investigations in 6- to 7-month-old APP23/PS45 mice showed that cortical neuron hyperexcitability was affiliated with high Aβ plaque burden (Busche et al., 2008). The hyperactivity was associated with a decrease in GABAergic inhibition. A subsequent evaluation in 6- to 7-month-old APP23/PS45 mice also identified hyperactivity in the plaque-bearing hippocampal CA1 neurons (Busche et al., 2012). Yet, in 1- to 2-month-old APP23/PS45 mice, a selective increase in hyperactive neurons occurred before plaque formation, indicating that soluble Aβ underlies early dysregulation (Busche et al., 2012). In a third investigation, a reduction of soluble AβOs through inhibition of β-secretase activity rescued APP23/PS45 mice from neuronal hyperactivity, long-range circuit dysfunction as related diminished slow-wave activity, and memory defects (Keskin et al., 2017). A recent study by Zott et al., (2019) further demonstrated that when soluble AβOs extracts from AD patient brains were applied to hippocampal CA1 mouse neurons, marked increases in hyperactivity resulted both in vivo and in vitro. These evaluations support both soluble AβOs and Aβ plaques as contributors to neuronal hyperexcitation.
SRIF and SRIF-expressing neuron cellular interactions provide an additional framework of understanding seizure activity in AD (Fig. 4). In the hippocampus, activity-dependent release of SRIF occurs during and after seizures to reduce neuronal hyperexcitation both presynaptically and postsynaptically (Vezzani et al., 1992; Marti et al., 2000a,b). In slice preparations, SRIF administration inhibited excitatory transmission in hippocampal CA1 and CA3 neurons (Tallent and Siggins, 1997, 1999). The SRIF-induced depression of excitatory postsynaptic currents was particularly robust during hyperexcited states, supporting the role of SRIF in mitigating active seizures. Furthermore, SRIF suppressed presynaptic glutamate release from Schaeffer collateral-CA1 synapses (Tallent and Siggins, 1997; Kozhemyakin et al., 2013). As glutamate homeostasis is impaired in AD patients (Masliah et al., 1996; Scott et al., 2011), with soluble AβOs potentiating glutamate-induced neuronal excitation (Li et al., 2011; Zott et al., 2019), the loss of SRIF and SRIF-expressing neurons further impairs the “brake” to glutamate-induced hyperexcitation. Astrocytes also play a role in SRIF-mediated mechanisms, influencing both the input and output SRIF-expressing interneurons of the hippocampus (Honoré et al., 2021). Astrocytes are able to regulate transmission at inhibitory synapses of SRIF-expressing interneurons (Fig. 4), whereas Aβ-induced astrogliosis and dysregulation of astrocyte functions contribute to glutamate dysregulation and hyperexcitability, with astrocyte-mediated inflammation further promoting seizure activity (Dejakaisaya et al., 2021; Vezzani et al., 2022).

Schematic of SRIF-associated cellular interactions governing neuronal excitatory-inhibitory actions in the neocortex and hippocampus with AD impact. SRIF-expressing GABAergic neurons release SRIF and GABA in a calcium-dependent manner, producing primarily inhibitory actions on neurons both presynaptically and postsynaptically through SSTs. Presynaptic glutamatergic neurons release glutamate in a calcium-dependent manner, producing primarily excitatory actions through receptor activation [reviews: Reiner and Levitz, (2018), Negrete-Díaz et al., (2022), and Chen et al., (2023)]. Released glutamate can be taken up by astrocytes via the excitatory amino acid transporter (EAAT1/2), which can be converted to glutamine. Glutamine is released via astrocyte sodium-coupled amino acid transporter (SNAT 3/5). Astrocytes actively modulate hippocampal circuits; as part of the “tripartite synapse,” they regulate glutamate, GABA, and ATP. SRIF-expressing interneuron inhibition of excitatory transmission is, in part, mediated by astrocytic GABAB, which regulates gliotransmitter release (Matos et al., 2018; Shen et al., 2022) [astrocyte release of gliotransmitters and corresponding autoregulatory mechanisms in AD and epilepsy reviewed elsewhere: (Dejakaisaya et al., 2021; Price et al., 2021)]. Astrocytic endfeet wrap around the BBB endothelium and pericytes, maintaining BBB integrity. Microglia surveil the brain, removing debris. * The cellular dynamic is dysregulated in AD (numbering is not necessarily indicative of sequence of events): (1) Elevated Aβ induces the loss of presynaptic GABAergic SRIF-expressing neurons, reducing inhibitory control. (2) Hyperexcitation increases glutamate and Aβ release. Elevated Aβ and loss of SRIF-expressing neuron feedback control contributes to dysregulated glutamate release. (3) BBB breakdown associated with inflammation and altered astrocyte endfeet interaction increases uptake of toxic blood components into the brain, including Aβ. (4) Aβ and inflammatory cytokines activate astrocytes and microglia. Astrocytes undergo astrogliosis, decreasing glutamate uptake transport and conversion to glutamine while increasing release of proinflammatory cytokines and glutamate. Microglial activation increases proinflammatory cytokine release and generation of reactive oxygen species (ROS). (5) Loss of inhibitory signaling and enhanced glutamate levels drive postsynaptic hyperexcitation.
Seizure activity itself results in the loss of SRIF-expressing neurons (Tallent and Qiu, 2008). The loss of SRIF-expressing interneurons was first identified in the hilus of the dentate gyrus after repeated seizures in rats (Sloviter, 1987). SRIF-expressing interneuron loss in hippocampal CA1 and CA3 has since been confirmed across numerous seizure models (Houser, 2014). Moreover, the seizure vulnerability that comes about with the loss of SRIF-expressing neurons may be exacerbated by concurrent dysregulation of cholinergic circuitry in AD. In a study of TLE, the cholinergic medial-septum hippocampal circuit that plays a primary antiseizure role was dependent on downstream SRIF effector action (Wang et al., 2020). In rats, deafferentation of hippocampal cholinergic neurons resulted in the loss of hilar SRIF-expressing neurons, worsening the seizure-induced loss of SRIF-expressing neurons (Jolkkonen et al., 1997). Interestingly, an interrelated effect is observed with language processing. Language difficulties are well recognized in patients with epileptic seizures (Unterberger et al., 2021) and AD (Szatloczki et al., 2015). It has been proposed that excitatory-inhibitory imbalances foster language impairment and evolve largely from conjoined impairment of cholinergic and SRIF processes aligning with Aβ load in AD (Almeida and Radanovic, 2022).
Several studies have attempted to delineate the roles SST subtypes in seizure activity. In an early investigation, the SST2,3,5 agonist octreotide injected into the hippocampus of rats significantly reduced the number and duration of kainite-induced seizures (Vezzani et al., 1991). Similarly, intracerebroventricular administration of octreotide or its analog lanreotide prevented or attenuated pilocarpine-induced status epilepticus in more than 65% of rats (Kozhemyakin et al., 2013). In associated slice evaluations, the SRIF inhibition of glutamate release at Schaffer collateral-CA1 pyramidal neuron synapses was mediated through presynaptic SST2 (Kozhemyakin et al., 2013). In a study using a series of selective SST subtype agonists, intrahippocampal administration of the SST2 agonist L-779,976, SST3 agonist L-796,778, or SST4 agonist L-803,087 were each found to protect rats against pilocarpine-induced seizures (Aourz et al., 2011). Moreover, a cooperative interaction between subtypes was identified. The antagonism of SST3 with SST3-ODN-8 blocked the anticonvulsant effect of an SST4 agonist, whereas the antagonism of SST2 with cyanamid-154806 inhibited the anticonvulsants effect of SST3 and SST4 agonists. Functional interactions between SST1/SST2 (Cammalleri et al., 2004), SST1/SST4 (Cammalleri et al., 2009), and SST2/SST4 (Moneta et al., 2002) have likewise been observed in mouse hippocampal slice evaluations of epileptiform activity. Furthermore, SST2, SST3, and SST4 each have shown to moderate cortical excitability and seizure severity in mice (Qiu et al., 2008). In Sst2-, Sst3-, and Sst4-knockout mice treated with the GABAA receptor antagonist pentylenetetrazole to induce seizures, all mice exhibited shorter latencies to different seizure stages with increased seizure severity when compared with wild-type mice. However, when systemically injected with kainite to induce seizures, only Sst4 knockouts showed increased seizure sensitivity. Additionally, SST4 coupled to M-channels was shown to be essential to its inhibition of epileptiform activity the hippocampal CA1. These data support the differential impact of SST subtype activity relative to the form of seizure induction in association with distinctive regions of effect and cellular processes. Although data support SST4 playing a major role in mitigating seizure activity in mice, an earlier study showed that treatment with the SST4 agonist L-803,087 induced excitatory effects in hippocampal CA1 slices of wild-type mice (Moneta et al., 2002). The excitatory effect was diminished by coadministration of octreotide. Yet, the same study showed that intrahippocampal injection of the SST2 agonists octreotide, BIM23120, or L-779976 did not affect kainite-induced seizures. Differences in SST subtype agonists used, dosing ranges, routes of administration, timeframes, manners of seizure induction, and in vivo versus in vitro evaluations each play a role in the discrepancies between studies. Slice evaluations notably impact neuron connectivity. Dyssynchronous regional and temporal changes in SST subtypes following seizures in rats (Csaba et al., 2004; Kwak et al., 2008) and mice (Iwasawa et al., 2019) further implicate differences in receptor regulation. For example, SST2 has shown to be sensitive to SRIF-mediated downregulation, resulting in a sustained loss of surface expression in the dentate gyrus after seizures (Dournaud et al., 1998; Csaba et al., 2004). Resected human hippocampal tissue from patients with intractable seizures correspondingly shows evidence of SST2 downregulation (Csaba et al., 2005). It is also notable that the decline in hippocampal SST2 with acute seizure activity is consistent with investigations of Aβ infusion (Aβ25–35 and Aβ1–42) (Aguado-Llera et al., 2018).
In summary, the interconnection between seizure occurrence and AD is traceable to increased brain Aβ levels and loss of SRIF-expressing neuronal function. The excitatory-inhibitory imbalance underlying seizure activity involves the dysregulation of multiple cellular processes, key of which is the interplay between glutamatergic and GABAergic SRIF-expressing neurons. Despite a lack of definitive delineation of SST subtype effect in seizure activity, most studies support SST2 and SST4 activation in the mitigation of hippocampal hyperexcitability, with likely interactions between SST subtypes.
F. Somatostatin and Inflammation
Neuroinflammation plays a prominent role in AD pathogenesis, marked by production of proinflammatory cytokines and reactive oxygen species (Leng and Edison, 2021). Neuroinflammation accelerates neurodegeneration and is associated with many AD risk factors, including seizures (Rana and Musto, 2018; Komoltsev et al., 2021; Vezzani et al., 2022) and neuropsychiatric symptoms (Benedetti et al., 2020; Milligan Armstrong et al., 2021; Troubat et al., 2021). Preclinical studies show that the loss of SRIF-expressing entorhinal and hippocampal neurons is heightened under inflammatory conditions (Gavilán et al., 2007; Moreno-Gonzalez et al., 2009), whereas SRIF can mitigate CNS inflammation, modulate microglial cell activation states, and help maintain BBB integrity under inflammatory conditions.
Neuroinflammation is accompanied by the activation and proliferation of microglia, the resident macrophage of the brain. Microglia continuously survey the CNS microenvironment, responding to inflammatory mediators, such as Aβ and damage signals (Leng and Edison, 2021). The activation states of microglia are typically classified as proinflammatory “M1” and anti-inflammatory “M2” based on cytokine release profiles and functional outcomes. This classification generally conveys a protective M2 phenotype with the removal of pathogens, cellular debris, and Aβ and a cytotoxic M1 phenotype affiliated with enhanced neuroinflammation and worsening AD pathology (Leng and Edison, 2021). Thus, the microglial response can be beneficial or detrimental depending on the level and timeframe of activation. Microglia also have extensive reciprocal communication with neurons, capable of actively modulating neuronal signaling (Li et al., 2012; Szepesi et al., 2018). Recent work in APP/PS1 mice identified that 98% of SRIF-expressing interneurons in the hippocampal CA1 receive putative microglia contacts (Gervais et al., 2022). The microglia displayed enhanced contact onto interneuronal somata in APP/PS1 mice compared with wild-type controls, capable of controlling neuronal activity.
Reactive microglia closely colocalize with Aβ in AD brain tissue (McGeer et al., 1987; Tooyama et al., 1990). In an evaluation of post-mortem AD human brain tissue, smaller AβOs more robustly activate microglia and increased neurotoxicity compared with larger AβO species (Yang et al., 2017). An age-dependent accumulation of soluble AβOs in APP/PS1 mice showed to be a primary factor in the microglia switch to the cytotoxic M1 form (Jimenez et al., 2008), consistent with other preclinical studies (Sondag et al., 2009; Maezawa et al., 2011; Ferretti et al., 2012). Aβ-induced microglia activation increases the release of proinflammatory cytokines, inclusive of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, interferon-γ, and monocyte chemoattractant protein-1 (Combs et al., 2001; Floden et al., 2005; Martin et al., 2017). In turn, these cytokines lead to AD-associated synaptic loss (Leng and Edison, 2021) and BBB dysregulation (Gullotta et al., 2023). These cytokines also act on their receptors to enhance signaling through the nuclear factor κB pathway, which upregulates β-secretase activity with a corresponding increase in Aβ generation (Chen et al., 2012). Notably, Aβ-activated microglia directly contribute to the significant early loss of SRIF-expressing neurons in the entorhinal cortex of PS1/APP mice associated with elevated proinflammatory cytokines (Moreno-Gonzalez et al., 2009), which is consistent with early neurodegeneration in the entorhinal cortex in human AD tissue studies (Gómez-Isla et al., 1996).
SRIF modulation of microglia response was first evaluated in BV2, N9, and primary rodent microglia cell lines, each expressing SST2, SST3, and SST4 (Fleisher-Berkovich et al., 2010). SRIF treatment enhanced microglia migration and increased Aβ42 phagocytosis across cell lines in a concentration-dependent manner. However, immunostimulation of microglia by bacterial lipopolysaccharide (LPS) prior to treatment almost completely abolished the Aβ phagocytosis effect, indicating an inflammatory-sensitive SRIF effect. A study of the SST4 agonist NNC 26-9100 treatment in BV2 cells likewise showed an increase in the uptake of fluorescent-tagged Aβ42 under noninflammatory conditions compared with vehicle controls (Schober et al., 2021). Nonetheless, another study found that SRIF treatment inhibited LPS-induced microglial activity and reactive oxygen species production as well as decreased TNF-α, IL-1β, and prostaglandin-E2 levels (Bai et al., 2015). Investigations further identify the ability of SRIF to inhibit LPS-induction of inducible nitric oxide synthase expression and neuronal degeneration associated with the suppression of microglia inflammatory activation (Bai et al., 2015; Hernández et al., 2020). NNC 26-9100 similarly inhibits LPS-induced nitric oxide in BV2 cells (Schober et al., 2021). Moreover, in response to LPS-induced inflammation, treatment of BV2 cells with the SST4 agonist SM-I-26 decreased mRNA expression of proinflammatory cytokines Tnf-α and Il-6 while increasing mRNA expression of the anti-inflammatory cytokine Il-10 and the antioxidant catalase (Silwal et al., 2021). It was hypothesized that SST4 agonist treatment may switch activated microglia from a proinflammatory M1 to an anti-inflammatory M2 phenotype (Silwal et al., 2021), which has been proposed as a mechanism to treat neurodegenerative diseases (Guo et al., 2022).
Transgenic AD mouse modeling studies support an SST4 mediation of Aβ clearance by microglia. The intracerebroventricular administration of NNC 26-9100 in 3xTg mice decreased cortical Cd33 and increased of macrophage scavenger receptor-1 (Msr1) mRNA expression (Sandoval et al., 2019). Cd33 is predominately expressed in microglia (Jiang et al., 2014), positively correlating to amyloid burden and LOAD (Griciuc et al., 2013). Cd33 protein expression is elevated in post-mortem AD brain tissue as compared with controls, indicating that a reduction in Cd33 may be beneficial (Griciuc et al., 2013). Msr1 is a microglia cell surface receptor involved in Aβ phagocytosis, with increased expression identified with enhanced Aβ phagocytosis (Chung et al., 2001; Frenkel et al., 2013). Knockout of Msr1 in APPswe/PS1dE9 mice increased Aβ levels as well as mortality rate (Frenkel et al., 2013). Interestingly, Nep and Ide mRNA expression were also significantly lower in the brains of APPswe/PS1dE9 Msr1-knockouts compared with APPswe/PS1dE9 controls, indicating Msr1 involvement in Aβ peptidase regulation. Correspondingly, SRIF treatment of BV2 cells is reported to increase IDE protein and mRNA expression, IDE secretion, and Aβ degradation (Tundo et al., 2012). Although the roles of other SST subtypes have not been ascertained, the results of these studies indicate that SST4 plays a key role in microglia function, albeit human microglia studies remain to confirm these observations.
The BBB is a semipermeable barrier that tightly regulates the movement of substances between the systemic circulation and CNS (Liebner et al., 2018). The endothelial cells of the BBB lack fenestrations and form tight junctions that greatly reduce paracellular permeability. The tight junctions are maintained by several specialized proteins, principle of which are claudin-5, occludin, and zonula occludens (ZO)-1. BBB integrity is further maintained by surrounding pericytes, astrocytes, and neurons, which, with the endothelium, are formally defined as the neurovascular unit (Fig. 4). BBB breakdown attributed to Aβ and inflammation occurs in both human AD and preclinical AD models (Montagne et al., 2017). BBB in vitro modeling shows that AβOs upregulate matrix metalloproteinases and downregulate tight junction proteins ZO-1, claudin-5, and occludin, with corresponding increases in paracellular permeability (Kook et al., 2012; Wan et al., 2015). These models likewise show an AβO-induced increase in protein expression of the receptor for advanced glycation end-products, which transports Aβ into the brain. A number of proinflammatory cytokines, including IL-1β, IL-6, IL-17, INF-γ, and TNF-α, reduce claudin-5, ZO-1, and occludin expression, with corresponding disruption of BBB tight junctions, and enhance paracellular permeability (Takata et al., 2021). Additionally, in an in vitro model using human-derived BBB endothelial cells, IL-1β and TNF-α decreased Aβ efflux associated with a decrease in efflux transporter protein expression (Versele et al., 2022). The encompassing data support a positive feedback loop in which elevated Aβ and inflammation drive BBB dysregulation, reducing the capacity to clear brain Aβ and furthering inflammation and BBB dysfunction (Fig. 1). Inflammation likewise impacts other cells of the neurovascular unit, contributing to neurovascular uncoupling and diminished cerebral blood flow (Nelson et al., 2016). Reduced cerebral blood flow and corresponding hypoperfusion contribute to the chain of events that ultimately contribute to neuronal death. In another interlinking of AD-related conditions, it is significant that inflammation-induced BBB dysfunction also plays a role in epileptic activity (Fattorusso et al., 2021; van Vliet and Marchi, 2022) and neuropsychiatric disorders (Welcome, 2020; Medina-Rodriguez and Beurel, 2022).
SRIF reduces the inflammation-induced dysregulation of the BBB. In human brain CMEC/D3 endothelial cells, treatment with LPS, TNF-α, or interferon-γ resulted in a loss of tight junction cellular integrity (Basivireddy et al., 2013). The LPS and cytokine treatment decreased SRIF expression and secretion from the endothelium, associated with the loss of ZO-1 protein expression at the tight junctions. Treatment with SRIF, the SST2 agonist L-779,976, or the SST4 agonist L-803,087 each reversed the ZO-1 expression loss. Interestingly, the inflammatory treatment increased the endothelial protein expression of SST2 and SST4, suggesting that the increased expression of SST2 and SST4 may serve to counter BBB tight junctional disruption. SRIF treatment further inhibited inflammation-mediated changes in expression of extracellular signal-regulated kinases and inducible nitric oxide synthase. Caco-2 endothelial cells show similar effects, with SRIF treatment increasing occludin and ZO-1 protein expression while inhibiting LPS-induced redistribution of tight junction proteins (Lei et al., 2014). Moreover, in CMEC/D3 cells treated with Aβ, SRIF treatment decreased tight junction protein disruption, paracellular permeability, and matrix metalloproteinase-2 (MMP2) protein expression compared with vehicle controls (Paik et al., 2019). MMP2 notably contributes to BBB breakdown in AD and is regulated through proinflammatory cytokines (Weekman and Wilcock, 2016).
Inflammatory effects that extend to other cells of the neurovascular unit are impacted by SRIF. Pericytes are a type of mural cell of the microcirculation that wrap around endothelial cells, maintaining the structural and functional integrity of the BBB and blood-retinal barrier. Pericyte degeneration leads to neurovascular uncoupling, reduces oxygen to the brain, and induces metabolic stress (Kisler et al., 2017). In an AD post-mortem tissue examination compared with cognitively normal controls, pericyte number and coverage in the cortex and hippocampus were reduced by 59% and 60%, respectively (Sengillo et al., 2013). This reduction correlated with the magnitude of BBB breakdown to plasma‐derived proteins. There is also accelerated pericyte loss in the post-mortem brain tissue of APOE-ε4 compared with APOE-ε3 carriers, aligning with a heightened degree of BBB breakdown in APOE-ε4 carriers (Halliday et al., 2016). In an evaluation of isolated human pericytes, proinflammatory and proapoptotic actions result when exposed to conditioned media from LPS-stimulated BV-2 microglia (Mazzeo et al., 2017), whereas the addition of SRIF to the LPS treatment of microglia resulted in media that stopped the inflammation-induced damage of the pericytes by reducing the proinflammatory mediators and counteracting the imbalance between apoptotic and survival intermediates.
In summary, inflammation contributes to AD pathology in alignment with the loss of SRIF and SRIF-expressing neurons, whereas SRIF acting through SSTs has an anti-inflammatory effect. SST4 agonist activation has been shown to lessen inflammatory activation of microglia in in vitro rodent cell studies. SRIF mitigation of inflammation-induced BBB dysregulation in human cell line studies implicates SST4 and SST2 as the primary mediators of this effect. Although more extensive in vivo characterization of anti-inflammatory activities is required, evidence supports a SRIF and specific SSTs capable of mitigating AD-associated inflammatory processes.
VI. Therapeutic Implications of Somatostatin Receptor Agonists in Alzheimer Disease
The decline of SRIF and loss of SRIF-expressing neurons in the brains of AD patients is linked to pathological progression and cognitive decline. This supports the targeting of SRIF-associated mechanisms for AD treatment. From a practical perspective, the use of SRIF as a means of treatment is problematic as it is a large peptide that does not readily cross the BBB and has an extremely short half-life. Moreover, concern as to the potential of toxic SRIF-Aβ aggregate assemblies arises with an approach that substantially elevates SRIF levels in the brain (Solarski et al., 2018). Correspondingly, the SRIF secretogues FK960 and FK962, which showed early promise in preclinical studies (Matsuoka and Aigner, 1997; Matsuoka and Satoh, 1998; Tokita et al., 2002, 2005; McCarthy et al., 2011), never advanced through a completed clinical trial. The most viable route is the advancement of an enzymatically stable small-molecule SST subtype–selective agonist with effective brain uptake. With the additional consideration as to the targeted SST subtype, a selective agonist would help to limit the side-effect profile. Yet, despite the known potential of SST agonists for AD treatment, the few clinical evaluations conducted provide little tangible insight (Table 2).
Clinical trials of SST agonist analogs in cognition performance testing
A. Clinical Evaluations
The first human investigation of an SRIF-focused treatment relative to cognitive decline in AD evaluated the SST2,3,5 agonist seglitide using a double-blinded, placebo-controlled crossover study design (Cutler et al., 1985). Seglitide was infused at a variable dose for 5.5 hours in 10 patients “presumed” to have mild AD based solely on the outcomes of a Mini-Mental State Examination. There was no benefit in arithmetic or pair-associated or serial learning compared with receiving placebo. However, testing was conducted only 3–5 hours after infusion. There was also no detectable seglitide in the CSF, indicating a lack of CNS uptake. Another double-blinded, placebo-controlled study evaluated the SST2,3,5 agonist octreotide in 14 patients meeting the National Institute of Neurologic and Communicative Disorders and Stroke (NINCDS) Alzheimer’s Disease and Related Disorders Association (ADRDA) criteria for AD (Mouradian et al., 1991). This study used a variable dosing regimen with infusions lasting 20 minutes, 3 hours, or 8 hours, with one 8-hour patient also receiving an infusion of the cholinesterase inhibitor physostigmine (0.3 mg/h). An extensive battery of learning and memory performance tests was conducted, with no significant improvement in any treatment paradigm compared with receiving placebo. Similar to the first study, this evaluation was limited by the testing timeframe, with evaluations performed either directly following infusion (20-minute group) or carried out during the last hour of infusion (3- and 8-hour groups). In another placebo-controlled trial performed in 23 AD patients meeting NINCDS-ADRDA criteria, octreotide was infused (150 μg/h) for 90 minutes followed by a 30-minute stabilization period and then a 30-minute cognitive testing period (Craft et al., 1999). AD patients showed improved declarative story-recall memory but not in selective attention using the Stroop interference test compared with receiving placebo. AD patients receiving octreotide under conditions of hyperglycemia did not show improvement in declarative story-recall memory when compared with receiving placebo, suggesting that hyperglycemia negatively impacted the effect of octreotide. In similarly aged healthy controls, cognitive tests were similar across all treatments. Furthermore, octreotide reduced plasma cortisol, corticotropin, and epinephrine levels in those with AD when compared with receiving placebo, implicating peripherally mediated actions in the memory effect. A follow-up double-blind crossover study with octreotide infusion (150 μg/h) using a similar assessment paradigm was performed in 16 memory-impaired patients (7 AD patients meeting NINCDS-ADRDA criteria for probable AD and 9 amnestic-MCI patients), along with 19 similarly aged cognitively intact patients (Watson et al., 2009). The effect of octreotide on delayed memory was dependent both on the presence or absence of APOE-ε4 and presence or absence of memory impairment. In the cognitively intact older adults, octreotide significantly improved delayed story recall when compared with receiving placebo. Within those that were memory impaired, APOE-ε4–negative patients with the least amount of cognitive impairment showed greater octreotide-induced memory facilitation. Nevertheless, the improvements in the APOE-ε4–negative patients were marginal.
The limited cognitive benefit in the clinical studies of SRIF analogs comes with several qualifiers. First, the SST agonists used are problematic. The physiochemical characteristics of seglitide and octreotide are unsuitable for traversing the BBB (Banks et al., 1990; Jaehde et al., 1994; Fricker et al., 2002), and actual receptor engagement in cortical or hippocampal tissue was not confirmed. Additionally, although both seglitide and octreotide are most often noted for their high binding affinity to SST2, they also have high binding affinity for SST3 and SST5. This negates the ability to delineate specific SST subtype effects. Critically, no selective SST1 or SST4 analog has ever been tested in AD or amnestic MCI patients. Secondly, testing parameters limit the ability to draw meaningful conclusions. The low patient numbers, variable dosing levels, mixing of AD and MCI patient populations, and different diagnostic determinations reduce the capacity to delineate outcomes or crosscompare between studies. Most significant are the short timeframes, with cognitive assessments either being conducted over the actual injection period or within hours after injection. This restricts interpretation to an acute window of effect when numerous peripheral variables are more likely to impact results. Most critically, no long-term treatment studies have been conducted as to the capacity of a BBB-permeable selective SST subtype agonist to mitigate AD symptomology or pathology.
B. Moving Forward
The advancement of an SST agonist for AD treatment must first determine which subtype to focus energies. As addressed in this review, both SST2 and SST4 are heavily involved in higher cognitive processes with the potential to enhance cognition. However, SST2 targeting presents greater liability. Although SST2 is expressed throughout the brain, it has significant peripheral distribution with pronounced pituitary expression (Consortium, 2020; Sjöstedt et al., 2020) and extensive endocrine actions (Günther et al., 2018). This suggests a higher likelihood of SST2-related side effects, whereas SST4 targeting presents several advantages. SST4 is not heavily expressed in the periphery and has no pituitary expression (Consortium, 2020; Sjöstedt et al., 2020). SST4 agonist evaluations show no effect on glucagon, growth hormone, or insulin release (Rohrer and Schaeffer, 2000), indicating limited endocrine-associated actions. Moreover, the SST4 agonist peripheral actions that have been identified are inflammatory-state dependent, with the capacity to mitigate inflammatory associated pain (Helyes et al., 2001, 2006, 2009, Lei et al., 2014; Elekes et al., 2008; Van Op den Bosch et al., 2009). Such peripheral effects are not innately problematic and may provide an added value to the AD population. The low level of receptor internalization following agonist treatment (Kreienkamp et al., 1998; Schreff et al., 2000) and a relatively rapid rate of receptor recycling (Smalley et al., 2001) further support SST4 as a reliable and sustainable target. Lastly, SST4 agonists have shown to increase brain NEP activity with associated reduction of Aβ/AβOs levels (Sandoval et al., 2012, 2013, 2019; Yu et al., 2017; Nilsson et al., 2020), suggesting an SST4 agonist as disease modifying. Given the high expression of SST4 in the neocortex and hippocampus (Consortium, 2020; Sjöstedt et al., 2020), regions heavily impacted by Aβ accumulation in AD, enhancement of NEP activity would be focused to the regions of greatest need. Such a brain-targeted activation likewise reduces concerns related to peripheral NEP activity. Furthermore, as decline in Aβ clearance is the primary cause of elevated Aβ/AβO levels in LOAD (Mawuenyega et al., 2010), enhancement of Aβ/AβO clearance through the SST4-NEP pathway could be of particular benefit for the majority of the AD population.
The compatibility of new therapies with established AD drug treatments is another important consideration. Given the multifactorial pathogenesis of AD, combination treatments geared to different targets may provide the greatest benefit (Cummings et al., 2019). Established symptomatic treatments, ChEIs and memantine, may work well when combined with an SST4 agonist. ChEIs have been a mainstay of AD treatment of over 20 years, maximizing the availability of endogenous acetylcholine through inhibition of its catabolism. A number of studies identify long-term use of ChEIs produce cognitive benefits in AD patients (Rogers et al., 2000; Doody et al., 2001; Doraiswamy et al., 2002; Courtney et al., 2004; Farlow et al., 2005; Xu et al., 2021). Moreover, discontinuation of ChEIs in those with moderate-to-severe AD significantly increases the probability of nursing home placement within the first year (Howard et al., 2015), suggesting that the use of ChEIs plays an important role in reducing caregiver burden. Yet, the effectiveness of ChEIs in AD continues to be debated, with the benefits often being quite modest and accompanied by numerous side effects (Ruangritchankul et al., 2021). Adverse events with ChEIs notably increase in older populations with dementia (Kröger et al., 2015). The high degree of the operational interdependency of the cholinergic system with SRIF and SRIF-expressing neurons in the regulation of cognition, along with the ability of SRIF to mitigate cholinergic deficits and facilitate cholinergic activity (Mancillas et al., 1986; Araujo et al., 1990; Matsuoka et al., 1994; Nakata et al., 1996; Matsuoka and Aigner, 1997; Tokita et al., 2002, 2005), espouses a combined treatment approach. Foreseeably, such a combination could facilitate lower dosing of ChEIs, reducing the ChEI side effect profile and potentially extending the viable window of ChEI use. There is also the possibility of an additive disease-modifying effect, with evidence that ChEIs may inhibit brain tissue atrophy (Hashimoto et al., 2005; Dubois et al., 2015; Cavedo et al., 2016) along with the SST4 agonist’s capacity to enhance Aβ catabolism within the brain (Sandoval et al., 2012, 2013, 2019; Nilsson et al., 2020).
Memantine, a low-affinity noncompetitive voltage-dependent NMDA receptor antagonist blocks the excitotoxic effects of glutamate that can lead to neuronal dysfunction. It was the first drug approved by the FDA to treat moderate-to-severe AD. Early memantine studies identified cognitive improvements in moderate-to-severe AD patients, with and without coadministration of a ChEI (Reisberg et al., 2003; Tariot et al., 2004). Yet, recent evaluations of memantine in those with moderate-to-severe AD across more comprehensive data sets show only a small clinical benefit while being ineffective in mild AD (McShane et al., 2019). Although the use of a more potent NMDA antagonists has been suggested (Selkoe, 2019), the side effects of memantine are already extensive (Rossom et al., 2004), rendering such an approach problematic, whereas SRIF mitigates glutamate release and associated neuronal hyperexcitability through receptor activation (Boehm and Betz, 1997; Tallent and Siggins, 1997; Kozhemyakin et al., 2013), with evidence supporting SST4 mediation of this effect (Qiu et al., 2008; Aourz et al., 2011; Hou and Yu, 2013). Thus, a combination of memantine and an SST4 agonist may allow for lower effective dosing of memantine in AD treatment, reducing side effects and prolonging beneficial effects.
Alleviation of AD comorbid neuropsychiatric symptoms and seizures present another promising aspect of an SST4 agonist approach. Unsurprisingly, these conditions share neurologic and neurochemical characteristics, with the loss of SRIF and SRIF-expressing neurons being a prominent feature in alignment with AD. Individuals with epilepsy are more likely to develop certain neuropsychiatric disorders, whereas those with neuropsychiatric disorders are more likely to develop epilepsy (Tolchin et al., 2020). Upwards of 50% of individuals with epilepsy present a neuropsychiatric comorbidities (Salpekar and Mula, 2019). A poorer AD prognosis is also associated with the presentation of neuropsychiatric symptoms (Palmer et al., 2010; Spalletta et al., 2012, 2015; Agüera-Ortiz et al., 2021) and seizures (Breteler et al., 1995; Cordonnier et al., 2007; Costa et al., 2019; Gourmaud et al., 2020; Tsai et al., 2021). These conditions, which are already difficult to treat independent of AD, can be particularly difficult to treat in those with AD. A recent study observed that the use of atypical antipsychotics for treating neuropsychiatric symptoms in those with AD had no effect on improving neuropsychiatric symptoms and was associated with a decline in cognitive and global function (Oh et al., 2021). A systematic literature review further demonstrated that atypical antipsychotics for the treatment of dementia-related psychosis in older adults are associated with a small numerical symptom improvement but with a high risk of adverse events, including cognitive decline and potentially higher mortality (Yunusa et al., 2021). Antiseizure medication outcomes fair little better. Antiseizure medication side effects range from reduced cognition and enhanced agitation to hyponatremia and decreased bone density (Vossel et al., 2017). Preclinical studies support SST4 agonist use for the mitigation of both neuropsychiatric symptoms (Scheich et al., 2016; Prévôt et al., 2017) and seizure severity (Qiu et al., 2008; Cammalleri et al., 2009; Aourz et al., 2011; Hou and Yu, 2013). Although the effectiveness of an SST4 agonist approach for these conditions in humans requires additional research, current data provide a sound footing for human investigation that could run in tandem with larger AD clinical trials. There is also a strong argument for the evaluation of an SST4 agonist for the treatment of neuropsychiatric symptoms and seizures independent of AD given the high rate of drug resistance to many currently used medications (Bystritsky, 2006; Voineskos et al., 2020; Fattorusso et al., 2021).
Beyond direct SST agonist targeting, pathways involved in SRIF and SRIF-expressing neuron regulation provide additional avenues for therapeutic development. BDNF induces SRIF gene expression (Villuendas et al., 2001; Sánchez-Muñoz et al., 2011), supports SRIF-expressing interneuron survival (Grosse et al., 2005), and is associated with a reduction in Aβ aggregation, Aβ-induced neurotoxicity, and synaptic dysfunction (Caffino et al., 2020). IGF1 produces a protective effect against Aβ respective to enhanced SRIF tone and SST expression (Aguado-Llera et al., 2005, 2018), with hippocampal IGF1 protein expression linked to the maintenance of neuronal integrity and cognitive function (Sun et al., 2005). Estrogens protect from Aβ-induced cell death and prevent the depletion of hippocampal SRIF through an IGF1-mediated mechanism (Perianes-Cachero et al., 2015) as well as improve synaptic plasticity, diminish brain inflammation, and reduce Aβ-associated injury (Uddin et al., 2020). Although eIF2α phosphorylation is involved in inflammation (Lourenco et al., 2013), epilepsy (Carnevalli et al., 2004, 2006), and neuronal degeneration in AD tissue (Chang et al., 2002; Oliveira and Klann, 2022), dephosphorylation of eIF2α in SRIF-expressing interneurons promotes memory formation (Sharma et al., 2020). Phosphorylation of eEF2 is also identified with AD synaptic failure and cognitive impairments (Ma, 2023), impacting BDNF and depression (Nosyreva and Kavalali, 2010; Autry et al., 2011) as well as the excitatory-inhibitory balance of GABAergic neurons (Heise et al., 2017). Each of these molecules present pharmacological targets with advantages and disadvantages that reach beyond the SRIF and SRIF-expressing neurons yet highlight the underlying theory as to SRIF-mediated mechanisms capable of enhancing cognition and/or reducing the pathological influence of Aβ.
VII. Conclusions
Over 4 decades of research across cellular, animal, and human studies establish the substantial interconnections between SRIF and AD. The loss of SRIF and SRIF-expressing neurons in the brains of AD patients drives a series of pathologic changes linked to nearly every AD symptom (Fig. 1). Although research into the mechanisms that underlie the vulnerability of SRIF-expressing neurons is ongoing, the considerable evidence presented in this review supports SSTs as valid targets for AD treatment. Among the SSTs, SST4 presents ideal attributes. Moving forward, careful consideration needs to be given to small-molecule design. A small-molecule program must not only focus on receptor affinity and selectivity but optimization of pharmacokinetics and BBB permeability. A successful drug candidate will not only depend on the effective exploitation of the encompassing knowledge of SRIF analog design but a holistic understanding of AD pathological progression toward appropriate clinical trial design and outcomes assessment. Although there remains more to be learned of the SRIF-AD interconnection, drug discovery and development must advance to the testing of our most promising targets.
Data Availability
This review article contains no datasets generated or analyzed during the present study.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Sandoval, Witt.
Footnotes
- Received February 13, 2024.
- Revision received July 1, 2024.
- Accepted July 8, 2024.
This review was supported by National Institutes of Health National Institute on Aging [Grants R01AG047858 and R41AG080914] (to K.A.W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Drs. Witt and Sandoval are co-inventors on patents pertaining to discovery of somatostatin agonists. Dr. Witt is founder of Somatolynk Inc., a company focused on somatostatin directed small molecule therapeutics.
Abbreviations
- Aβ
- amyloid-β peptide
- ACTH
- adrenocorticotropic hormone
- AD
- Alzheimer disease
- ADAD
- autosomal dominant Alzheimer disease
- ADRDA
- Alzheimer Disease and Related Disorders Association
- AβO
- amyloid-β peptide oligomer
- ApoE
- apolipoproein E
- APP
- amyloid precursor protein
- BBB
- blood-brain barrier
- BDNF
- brain-derived neurotrophic factor
- ChEI
- cholinesterase inhibitor
- CNS
- central nervous system
- CSF
- cerebrospinal fluid
- CST
- cortistatin
- eEF2
- eukaryotic translational elongation factor 2
- eIF2α
- eukaryotic initiation factor 2α
- EPM
- elevated plus maze
- ER
- endoplasmic reticulum
- FDA
- Food and Drug Administration
- FST
- forced swim test
- GPCR
- G-protein–coupled receptor
- HPA
- hypothalamic-pituitary-adrenal
- IDE
- insulin-degrading enzyme
- IGF
- insulin-like growth factor
- IL
- interleukin
- LOAD
- late-onset Alzheimer disease
- LPS
- lipopolysaccharide
- LTP
- long-term potentiation
- MCI
- mild cognitive impairment
- MDD
- major depressive disorder
- MSR1
- macrophage scavenger receptor-1
- NBM
- nucleus basalis of Maynert
- NEP
- neprilysin
- NFT
- neurofibrillary tangle
- NINCDS
- National Institute of Neurological and Communicative Disorders and Stroke
- NMDA
- N-methyl-D-aspartate
- O-LM
- oreins-lacunosum moleculare
- PS1
- presenilin-1
- PS1dE9
- presenilin-1 dE9
- PSD
- postsynaptic density protein
- SAMP8
- senescence-accelerated mouse prone 8
- SRIF
- somatostatin
- SST
- somatostatin receptor
- TLE
- temporal lobe epilepsy
- TNF
- tumor necrosis factor
- ZO
- zonula occludens
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




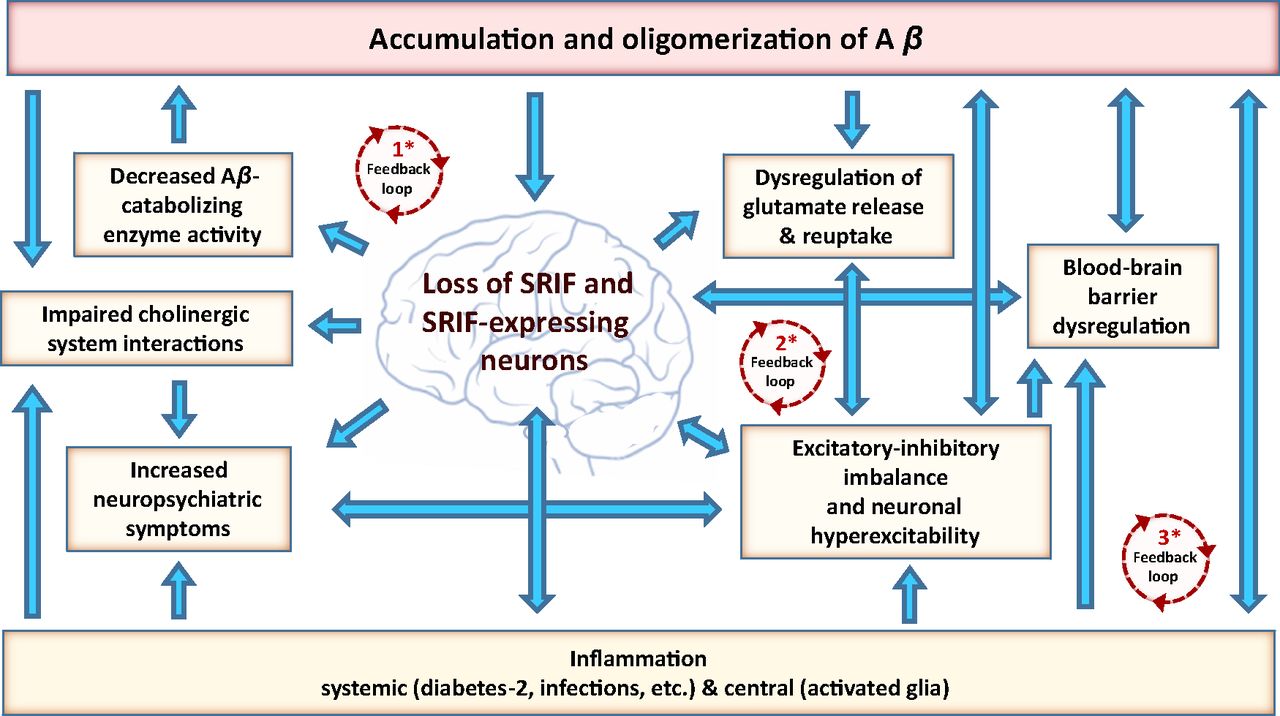{kind=link}
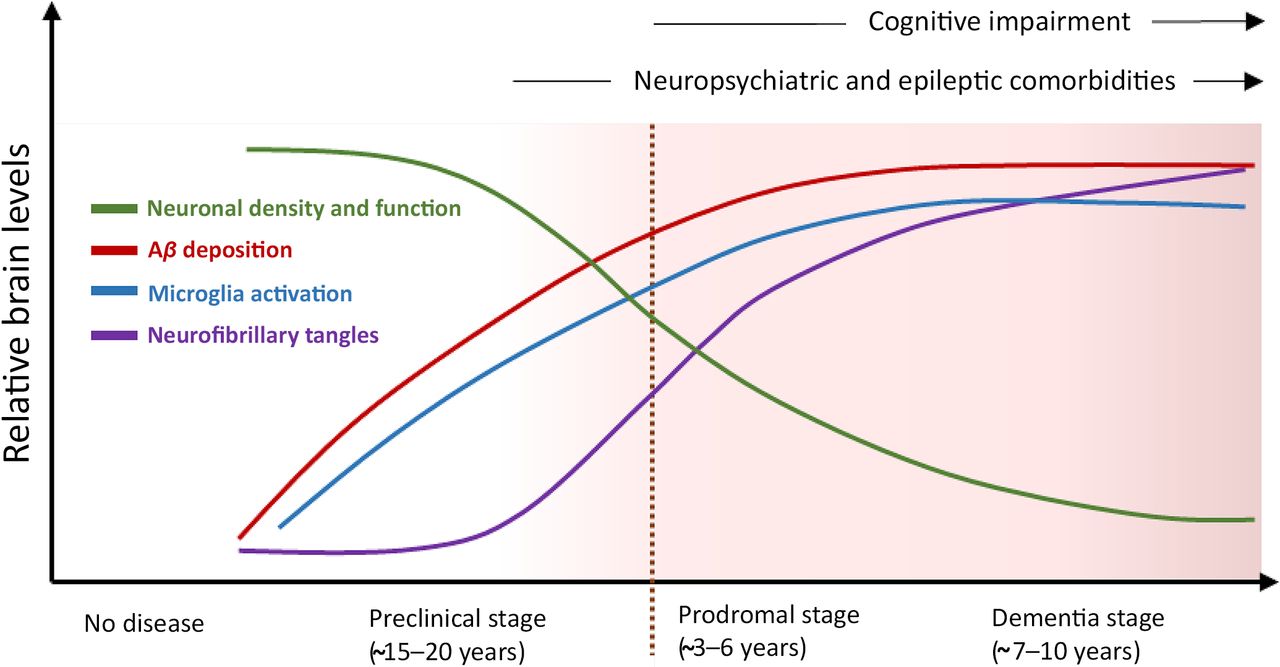{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Somatostatin and Somatostatin Receptors
- III. Somatostatin-Expressing Neurons
- IV. Somatostatin in Learning and Memory
- V. Somatostatin Links to Alzheimer Disease
- VI. Therapeutic Implications of Somatostatin Receptor Agonists in Alzheimer Disease
- VII. Conclusions
- Data Availability
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters